
Abstract
Background
Crisaborole ointment, 2%, is a nonsteroidal phosphodiesterase 4 inhibitor for the treatment of mild-to-moderate atopic dermatitis (AD).
Objectives
The aim of this study was to evaluate the safety, effectiveness, and pharmacokinetics (PK) of crisaborole in infants aged 3 to < 24 months with mild-to-moderate AD in an open-label study.
Methods
Infants (3 to < 24 months) with Investigator’s Static Global Assessment (ISGA) of mild (2) or moderate (3) and percentage of treatable body surface area (%BSA) ≥ 5 received crisaborole twice daily for 28 days; a cohort with moderate AD per ISGA and %BSA ≥ 35 were included in a PK analysis. Endpoints included safety (primary), efficacy, and PK (exploratory).
Results
Included were 137 infants total (mean age [SD], 13.6 months [6.42]), with 21 in the PK cohort (12.7 months [6.58]). Treatment-emergent adverse events (TEAEs) were reported for 88 (64.2%) patients (98.9% rated as mild/moderate). TEAEs were considered treatment-related for 22 patients (16.1%); most frequently reported were application site pain (3.6%), application site discomfort (2.9%), and erythema (2.9%). ISGA clear/almost clear with ≥ 2-grade improvement at day 29 was achieved by 30.2% of patients. From baseline to day 29, mean percentage change in Eczema Area and Severity Index score was − 57.5%, and mean change in Patient-Oriented Eczema Measure total score was − 8.5. Crisaborole systemic exposures in infants were characterized and, based on nonlinear regression analysis, were comparable with that in patients aged ≥ 2 years.
Conclusions
In this open-label study, crisaborole was well tolerated and effective in infants (3 to < 24 months) with mild-to-moderate AD with systemic exposures similar to patients aged ≥ 2 years.
Clinical Trial Registration
NCT03356977.
Atopic dermatitis (AD) is a skin disease that causes inflamed and itchy skin. Crisaborole is an ointment that is approved to treat patients aged 2 years and older with mild-to-moderate AD. This clinical trial studied crisaborole in infants with mild-to-moderate AD who were 3 to under 24 months old. These infants were treated with crisaborole twice a day for 28 days. The trial studied crisaborole’s safety, effectiveness, and absorption into the bloodstream. In total, 137 infants were treated. Although side effects of some sort occurred in about two-thirds of patients, only 1 in 6 patients experienced side effects that were attributed to crisaborole. When these side effects did occur, these were mainly pain, discomfort, or redness where crisaborole was applied. Fewer than 1 in 25 patients experienced each side effect where crisaborole was applied. The doctors saw improvement in the AD symptoms of some patients at day 29 of the study compared to the beginning of the study. Crisaborole blood-level measurements in this age group were consistent with those seen in patients aged 2 years and older. Overall, crisaborole was considered well tolerated and effective in infants (3 to under 24 months old) with mild-to-moderate AD.
AbstractSection Video abstractSafety, Effectiveness, and Pharmacokinetics of Crisaborole in Infants Aged 3 to < 24 Months with Mild-to-Moderate Atopic Dermatitis: An Open-Label, Phase 4 Study (MP4 40891 MB)
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Digital Features for this article can be found at https://doi.org/10.6084/m9.figshare.15109695. |
Crisaborole ointment, 2%, was well tolerated in this open-label study of infants aged 3 to <24 months with mild-to-moderate AD. Application site pain/discomfort was reported at a rate similar to those in crisaborole studies of patients aged ≥2 years; no new safety signals were identified. |
Improvements from baseline in exploratory efficacy endpoints (including Investigator’s Static Global Assessment, Eczema Area and Severity Index, percentage of treatable body surface area, and Patient-Oriented Eczema Measure outcomes) were observed at the first postbaseline assessment (day 8 or 15, depending on outcome measure) and continued through the end of treatment (day 29). |
Based on nonlinear regression analysis accounting for dose and age differences, crisaborole systemic exposure in infants aged 3 to <24 months was comparable with that observed in crisaborole studies of patients aged ≥2 years. |
1 Introduction
Atopic dermatitis (AD) is a chronic, highly pruritic, inflammatory skin disease that affects an increasing number of patients worldwide, especially in industrialized, temperate countries, such as the United States [1,2,3]. AD affects patients of all ages and is one of the most common, chronic, relapsing childhood dermatoses, impacting 15–30% of the pediatric population in the United States [1, 3]. Approximately 60% of patients experience first signs and symptoms before 1 year of age, and disease activity for many persists well into adulthood [4]. AD has a profoundly negative effect on the health-related quality of life of pediatric patients—including sleep disturbance, mood changes, and impaired psychosocial functioning—and their caregivers—including psychological distress [2, 5].
There are a limited number of prescription agents approved to treat patients aged < 2 years with AD, some of which have safety concerns that include boxed warnings, resulting in a high unmet need in this patient population. Crisaborole ointment, 2%, is a nonsteroidal phosphodiesterase 4 inhibitor for the treatment of mild-to-moderate AD. As of January 2020, it is approved in the United States, Australia, Canada, and Israel for patients aged ≥ 2 years. In two vehicle-controlled phase III clinical studies of 1522 patients aged ≥ 2 years with mild-to-moderate AD (AD-301: NCT02118766; AD-302: NCT02118792), significantly more crisaborole-treated than vehicle-treated patients achieved Investigator’s Static Global Assessment (ISGA) success (defined as clear [0] or almost clear [1] with ≥ 2-grade improvement from baseline); 32.8% versus 25.4% achieved ISGA success in AD-301 (p = 0.038) and 31.4% versus 18.0% achieved ISGA success in AD-302 (p < 0.001) [6]. In addition, ISGA clear or almost clear was achieved in a significantly greater proportion of patients in the crisaborole group than the vehicle group; 51.7% versus 40.6% of patients achieved ISGA clear or almost clear in AD-301 (p = 0.005) and 48.5% versus 29.7% achieved ISGA clear or almost clear in AD-302 (p < 0.001) [6]. ‘Application site pain’ was the most common treatment-related adverse event (AE) in pooled data of the two studies (4.4% vs 1.2%; p = 0.001) [6]. In a long-term (48 weeks), single-arm, open-label safety extension study of the phase III trials (AD-303), the most frequently reported treatment-related AEs were ‘dermatitis atopic’ (3.1%), ‘application site pain’ (2.3%), and ‘application site infection’ (1.2%) [7].
The CrisADe CARE 1 study (NCT03356977), a multicenter, open-label, single-arm, phase IV trial, is the first trial designed to evaluate the safety, effectiveness, and pharmacokinetics (PK) of crisaborole applied twice daily to infants aged 3 to < 24 months with mild-to-moderate AD.
In addition, propylene glycol systemic exposure was assessed. Propylene glycol, an additive in many foods and an excipient in many commonly used medications, cosmetics, and topical products, including infant wipes, is a component of the crisaborole formulation. Propylene glycol is designated by the US Food and Drug Administration as a “generally recognized as safe” substance [8], but exposure to high systemic concentrations from administration or absorption of propylene glycol (especially in infants administered products containing propylene glycol by intravenous route), have resulted in AEs, including central nervous system toxicity, hyperosmolarity, hemolysis, cardiac arrhythmia, and lactic acidosis [9]. The measurement of propylene glycol systemic concentrations during treatment with crisaborole has not previously been studied.
2 Methods
2.1 Study Design
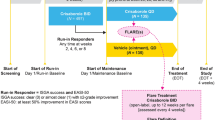
CrisADe CARE 1 was a multicenter, open-label, single-arm, phase IV study designed to evaluate the safety, efficacy, and PK of crisaborole in infants aged 3 to < 24 months with mild-to-moderate AD. The institutional review board at each study site approved the study protocol, and written informed consent was provided by parents or legal guardians. The study was conducted in accordance with the protocol, local legal and regulatory requirements, and the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Subjects, International Conference on Harmonisation Guideline for Good Clinical Practice, and the Declaration of Helsinki.
2.2 Patients and Treatment
Patients were recruited from 30 study sites across three countries (United States, Canada, and Australia). Enrolled patients were aged 3 to < 24 months with a diagnosis of AD per Hanifin and Rajka criteria [10], mild (2) or moderate (3) AD per ISGA [6], and a percentage of treatable body surface area (%BSA) ≥ 5, excluding the scalp. Patients were excluded if they had received systemic corticosteroids or immunosuppressive agents within 28 days of the first dose of study drug; used high- or medium-potency topical corticosteroids, topical calcineurin inhibitors (TCIs), topical antibiotics, light therapy, antibacterial soaps (for bathing), bleach baths, or topical sodium hypochlorite-based products on treatable AD lesions within 7 days of the first dose of study drug; used systemic antihistamines or low-potency topical corticosteroids within 3 days of the first dose of study drug; or used emollients, topical antihistamines, or topical hydrocortisone < 1% on treatable AD lesions within 8 h of the first dose of study drug. Patients in the PK cohort (of whom at least three patients were to be aged 3 to < 9 months) were required to have moderate AD per ISGA with a treatable %BSA ≥ 35, excluding the scalp, at baseline and have adequate venous access. Patients were excluded from the PK cohort if they had lesions on the extremities (i.e., below wrists or ankles) or within 2 cm of the mouth to prevent inadvertent ingestion of crisaborole.
Open-label crisaborole was applied twice daily to all AD-affected areas of the body throughout the 28-day duration of the study, even if the AD lesions resolved. Patients in the PK cohort had twice daily visits on days 1 through the morning of day 8 for application of investigational product at the site. Caregivers were instructed to avoid applying crisaborole to mucous membranes and the scalp to avoid potential patient dissatisfaction with ointment application to scalp hair. In the PK cohort, application to the hands, feet, and perioral areas was also avoided to prevent inadvertent ingestion of crisaborole. Crisaborole could also be applied to any new treatable AD-involved areas that appeared outside of these areas following baseline (day 1) after consultation with the investigator at the next visit. Therapies not permitted during the study were systemic corticosteroids, antihistamines, leukotriene receptor antagonists, and immunosuppressants. Additionally, topical agents, such as low-to-high-potency topical corticosteroids, TCIs, topical antihistamines, topical antibiotics, topical sodium hypochlorite-based products, antibacterial soaps, bleach baths, diaper rash creams, lotions, ointments, and powders, and light therapy, were not permitted. Use of bland emollients was permitted to manage dry skin in areas surrounding but not on or overlapping with treatable AD-involved areas or on AD-involved areas where crisaborole was not applied.
For the non-PK cohort, treatment adherence was recorded by site staff for the day 1 morning dose and by parent/guardian at home in a dosing diary for subsequent doses. For the PK cohort, the day 1 morning dose through the day 8 morning dose were recorded by site staff, while all subsequent doses were recorded by parent/guardian at home in the dosing diary. Patients were considered adherent to treatment if 80–120% of the expected number of doses were received.
2.3 Study Outcomes and Assessments
Site visits occurred at screening, baseline (day 1), day 8, day 15, day 22 (telephone visit), and day 29 during the treatment period, and telephone follow-up visits occurred at days 36 and 57.
Primary endpoints were the incidence of treatment-emergent adverse events (TEAEs), which were assessed at all study visits and follow-up visits; clinically significant changes in vital signs, such as temperature, blood pressure, pulse, and respiratory rate (assessed at screening, baseline, day 8, day 15, and day 29); height, weight, and physical examination (assessed at baseline and day 29); electrocardiography (assessed at baseline, day 8, and day 29); and laboratory assessments, including metabolic panels, serum chemistry, and hematology panels (assessed at screening and day 29, and also at day 8 for the PK cohort). Blood pressure, electrocardiography, and cardiovascular and neurologic physical examination were included to monitor for potential cardiovascular and neurologic effects of propylene glycol. The investigator recorded all directly observed TEAEs and all TEAEs spontaneously reported by the patient’s parent/legal guardian. Each patient’s parent/legal guardian was to be questioned about the occurrence of AEs in a non-leading manner. Verbatim AE terms reported by the investigator were coded to Medical Dictionary for Regulatory Activities (MedDRA) preferred terms.
Exploratory efficacy endpoints included the proportion of patients who achieved ISGA success (defined as clear [0] or almost clear [1] with ≥ 2-grade improvement from baseline; ISGA was assessed at day 8, day 15, and day 29), proportion of patients who achieved ISGA clear or almost clear, percentage change from baseline in Eczema Area and Severity Index (EASI [11, 12]; assessed at days 15 and 29), change from baseline in %BSA (assessed at days 15 and 29), and change from baseline in Patient-Oriented Eczema Measure (POEM proxy version [13]; assessed at days 8, 15, and 29). Efficacy outcome measures are further described in Supplemental Table 1 (in Electronic Supplementary Material [ESM]).
Exploratory PK endpoints for crisaborole were maximum observed plasma concentration (Cmax) at day 8, time to reach maximum observed plasma concentration (Tmax) at day 8, and area under the plasma concentration-time profile for the dosing interval (AUCtau) at day 8. PK parameters of crisaborole and its metabolites were assessed for the patients in the PK cohort using blood samples collected before the day 8 morning dose, 3 h after the day 8 morning dose, and 12 h after the day 8 morning dose. A 3-h sample time was chosen for Cmax based on previous data obtained from patients with AD and healthy volunteers (data on file). In addition, plasma concentrations of propylene glycol were assessed for all patients at screening and day 29 (12 h after the last dose). For patients in the PK cohort, propylene glycol concentrations were also assessed before the day 8 morning dose. Plasma samples were analyzed using validated analytical methods.
2.4 Study Statistical Considerations
All safety, efficacy, PK, and propylene glycol concentration data were summarized using descriptive statistics. The 95% CIs for binary endpoints were obtained by Clopper–Pearson exact method. A target of 125 enrolled patients was chosen to ensure that approximately 100 patients completed the study, assuming a 20% dropout rate. The sample size was determined by clinical judgment based on experience with other clinical studies of the investigational product. Statistical power was not calculated because there was no comparator group within the trial. A sample size of 16 patients was selected for the PK cohort to provide 96.0% overall power for 90% one-sided CIs for Cmax and AUCtau on day 8 to be less than the previously observed 85th percentiles of 166 ng/mL and 1281 ng h/mL, respectively, under maximum-use conditions in patients aged 2–17 years [14]. For the propylene glycol analysis, unplanned postbaseline visits were excluded.
3 Results
3.1 Patients
A total of 137 patients (non-PK cohort, n = 116; PK cohort, n = 21) from the United States (n = 112; 81.8%), Australia (n = 15; 10.9%), and Canada (n = 10; 7.3%) were evaluated in the study from January 16, 2018 (first patient first visit), to April 12, 2019 (last patient last visit). In total, nine patients (6.6%) discontinued treatment. Among these, four (2.9%) discontinued treatment because of a TEAE. Other reasons for discontinuing treatment were lack of efficacy (n = 1; 0.7%), lost to follow-up (n = 1; 0.7%), and withdrawal by parent or guardian (n = 3; 2.2%) (Supplemental Fig. 1 in ESM).
Patient demographics and baseline disease characteristics are summarized in Table 1. Overall, the median age was 13 months with median ISGA of 3.0 and mean %BSA of 28.1. One patient with severe AD (ISGA 4) was enrolled in and completed the trial but was considered a protocol deviation due to the severe baseline ISGA. Generally, baseline characteristics were similar between the patients included in the non-PK cohort and the PK cohort; however, %BSA involvement and disease severity (per ISGA and EASI scores) were greater in the PK cohort, consistent with inclusion criteria. A total of 22 patients (16.1%) had a medical history that included at least one other atopic condition, such as asthma, food allergies, and/or allergic/seasonal rhinitis. Approximately half of patients (n = 72; 52.6%) had used topical corticosteroids prior to entering the study, and two patients (1.5%) had used TCIs. Overall, 126 patients (92%) were considered adherent to the treatment regimen (received 80–120% of the expected number of doses).
3.2 Safety (Primary Endpoint)
In the overall study population (N = 137), all-cause TEAEs were reported for 88 (64.2%) patients; 98.9% of which were rated as mild or moderate. Treatment-related AEs were reported for 22 patients (16.1%). Four (2.9%) patients discontinued treatment because of a TEAE and remained in the study, including one patient who experienced a serious TEAE of ‘febrile convulsion’ (not related to treatment). The other three patients who discontinued treatment because of a TEAE were one patient with ‘dermatitis infected’ (not related to treatment), one patient with ‘application site pain’ (treatment-related), and one patient with ‘application site discomfort’ (treatment-related). The most frequently reported (≥ 5%) all-cause TEAEs were ‘pyrexia’ (9.5%), ‘upper respiratory tract infection’ (7.3%), ‘diarrhea’ (7.3%), ‘dermatitis atopic’ (6.6%), ‘dermatitis diaper’ (6.6%), and ‘cough’ (5.1%) (Table 2). The most frequently reported (≥ 2.5%) treatment-related treatment area AEs were ‘application site pain’ (n = 5; 3.6%), ‘application site discomfort’ (n = 4; 2.9%), and ‘erythema’ (n = 4; 2.9%) (Table 3). Treatment-related ‘application site pain’ or ‘application site discomfort’ (occurring in nine patients with no overlap between each AE term) were reported on the face for four patients, on the leg for four patients, on the arm for three patients, on the chest for two patients, on the back for one patient, on the neck for one patient, and on the abdomen for one patient. (Note: each patient could report more than one body location for ‘application site pain’ or ‘application site discomfort’ AEs.) Patients with an ‘application site erythema’ AE did not overlap with any patients with an ‘erythema’ AE. No safety signals were identified in clinical laboratory findings, weight, height, electrocardiograms, or vital signs (data not shown).
3.3 Efficacy
In the overall study population, 20.0% of patients achieved ISGA success at day 8 (first postbaseline assessment) and 30.2% achieved ISGA success at day 29 (Fig. 1). In addition, 40.7% of patients achieved ISGA of clear or almost clear at day 8 and 47.3% achieved ISGA of clear or almost clear at day 29 (Fig. 1). Mean (standard error [SE]) EASI score decreased from 11.8 (0.72) at baseline to 5.0 (0.50) for a mean percentage change (SE) from baseline of − 57.5% (3.27) at day 29 (Fig. 2). Mean (SE) %BSA was reduced from 28.1 (1.88) at baseline to 12.4 (1.18) for a mean change (SE) from baseline of − 15.2 (1.51) at day 29 (Supplemental Fig. 2 in ESM). Mean (SE) POEM total score improved from 14.8 (0.52) at baseline to 6.1 (0.48) for a mean change (SE) from baseline of − 8.5 (0.5) at day 29 (Fig. 3). When POEM total score was analyzed by domain, mean changes from baseline ranged from approximately − 1 to − 2 across subscales at day 29, including improvements in itching and sleep loss (Fig. 4).

ISGA response with crisaborole. 95% CIs were obtained by Clopper–Pearson exact method. ISGA success was defined as ISGA clear (0) or almost clear (1) with ≥ 2-grade improvement from baseline. ISGA Investigator’s Static Global Assessment

Mean percentage change from baseline in EASI score with crisaborole. EASI Eczema Area and Severity Index, SE standard error

Mean change from baseline in POEM total score with crisaborole. POEM Patient-Oriented Eczema Measure, SE standard error

Mean change from baseline in POEM subscale scores with crisaborole. BL baseline, POEM Patient-Oriented Eczema Measure, SE standard error
3.4 Pharmacokinetics
Eighteen patients were evaluated for PK parameters, and all had quantifiable concentrations of crisaborole (Table 4) and the inactive metabolites (data on file). Based on nonlinear regression analysis, which accounts for dose differences (due to differences in %BSA) and age differences (accounting for the difference in clearance of crisaborole), crisaborole exposure was comparable with that in patients aged ≥ 2 years observed in previous studies [14,15,16].
3.5 Propylene Glycol Concentrations
A majority of the patients had measurable concentrations of propylene glycol at screening covering a wide range (0–30,000 ng/mL) (Supplemental Table 2 in ESM). After treatment with crisaborole, propylene glycol concentrations were still measurable and had a similarly wide range (0–44,000 ng/mL). Based on the wide overlapping ranges observed between screening and end of treatment visits and examination of individual patient profiles, no systematic trend for increased propylene glycol plasma concentrations following treatment with crisaborole was identified. In addition, no propylene glycol toxicity effects were identified, including no changes in electrocardiogram, clinical laboratory, or vital sign parameters.
4 Discussion
AD is relatively common in patients aged < 2 years, with an Australian study reporting the cumulative prevalence for parent-reported atopic eczema as 28.0% in the first 12 months of life [17]. Topical corticosteroids and TCIs are commonly recommended topical prescription medications to treat AD; however, most are not approved to treat infants aged < 2 years in most countries, including the United States. Topical corticosteroids may be used intermittently for flares but are not recommended for prolonged use because of concerns about local and systemic AEs [18, 19]. Meanwhile, TCIs have been associated with application site burning and stinging, and they currently have a boxed warning in the United States for risk for malignancy [19]. As a result, additional options to treat AD signs and symptoms and improve health-related quality of life are needed, specifically for infants with AD.
This report describes the first clinical study of crisaborole in infants aged < 2 years with AD. Crisaborole was well tolerated with a low incidence of treatment-related AEs. The most frequently reported TEAEs were similar to those typically associated with childhood illnesses (e.g., pyrexia, upper respiratory tract infection, diarrhea, cough). ‘Application site pain’ and ‘application site discomfort’ were reported at rates (3.6% and 2.9%, respectively) similar to those of ‘application site pain’ in patients aged ≥ 2 years treated with crisaborole in the phase III studies (AD-301/AD-302 pooled: 4.4%) [6]. Although application site discomfort was not among the most frequently reported TEAEs in the phase III studies, this comparison is apt because ‘application site discomfort’ and ‘application site pain’ were often reported from an observer/parent/caregiver perspective in this study and not directly from the patient.
In this open-label study, approximately 30% and 47% of patients achieved ISGA success and/or ISGA clear or almost clear at day 29, respectively, which is consistent with the rates reported for crisaborole-treated patients aged ≥ 2 years in the pivotal phase III studies for the same endpoints (ISGA success: 32.8% [AD-301] and 31.4% [AD-302]; ISGA clear/almost clear: 51.7% [AD-301] and 48.5% [AD-302]) [6]. After 1 week of treatment with crisaborole, up to 20% or 40% of patients had clinical improvements in their AD as defined by ISGA success and/or ISGA clear or almost clear, respectively. Additionally, improvements in EASI score and %BSA involvement were observed as early as the first postbaseline assessment (day 15). It should be noted that effectiveness was an exploratory endpoint in this study and there was no comparator group. However, these results are consistent with a recently published vehicle-controlled phase IIa study, which demonstrated improvements in crisaborole-treated target lesion EASI score by day 15 in adults with mild-to-moderate AD [20].
Additionally, patient-reported outcomes improved at the first postbaseline assessment (day 8) and were sustained at day 29 with mean changes from baseline in POEM total scores of − 6.9 and − 8.5, respectively. At both timepoints, improvements exceeded the minimal clinically important difference (change of 3.4) [21, 22]. In addition, the POEM subscale data show an association between crisaborole use and improvements in sleep and itch, which are important factors in the quality of life of patients with AD and their caregivers. This is the first crisaborole study to include POEM, which is recommended as a patient/caregiver-reported clinical endpoint in AD trials by several international bodies, including the Harmonising Outcome Measures for Eczema (HOME) initiative [23].
Generally, PK parameters were comparable to those seen in phase I/II studies in patients aged ≥ 2 years [14,15,16], indicating that bioavailability (or amount of crisaborole absorbed per unit dose) does not vary for patients aged > 3 months. With regard to propylene glycol exposure, no systematic trend for increased propylene glycol concentrations after treatment with crisaborole was identified based on examination of individual patient concentration profiles. The maximum serum concentration observed in this study (44,000 ng/mL) was well below levels that have been reported in case studies to be toxic (above 180,000–250,000 ng/mL [9]). Furthermore, based on extensive assessments, including physical examinations, clinical laboratory tests, vital signs, blood pressure, and electrocardiography, no safety concerns attributable to propylene glycol exposure (i.e., central nervous system toxicity, hyperosmolarity, hemolysis, cardiac arrhythmia, or lactic acidosis) were identified. Although a wide range of propylene glycol concentrations was observed at screening (before prior AD treatment washout and before crisaborole treatment), specific sources were not identified since propylene glycol is found ubiquitously in many products, including medications, foods, and moisturizers [9].
Limitations of this study include its open-label nature and lack of a comparator group, as well as the exploratory nature of the efficacy analyses. In addition, as with any study in patients of this age range, the study relied on parents/observers/caregivers to report sensory AEs, such as ‘application site pain.’ Additionally, because propylene glycol is found in numerous products, it was not possible to capture all potential sources of propylene glycol exposure, nor the frequency or quantity of exposure (particularly food/diet sources).
5 Conclusions
Crisaborole was well tolerated in this open-label study of infants aged 3 to < 24 months with mild-to-moderate AD. Safety and efficacy were consistent with that observed in previous studies of crisaborole in patients aged ≥ 2 years. In particular, ‘application site pain’ and ‘application site discomfort’ were reported at rates similar to those seen in crisaborole studies in patients aged ≥ 2 years, and no new safety signals were identified. Improvements from baseline in signs and symptoms of AD and patient-reported outcomes were observed at the first postbaseline assessment (day 8 or 15) and continued to improve through day 29. Overall, the results of this study show that crisaborole may be a safe and efficacious treatment option for infants aged 3 to < 24 months.
Data Sharing Statement
Upon request, and subject to certain criteria, conditions, and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines, and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.
Change history
17 September 2021
A peer-reviewed video abstract, plain language summary, and digital features link was retrospectively added to this publication
References
Bieber T. Atopic dermatitis. N Engl J Med. 2008;358(14):1483–94.
Lewis-Jones S. Quality of life and childhood atopic dermatitis: the misery of living with childhood eczema. Int J Clin Pract. 2006;60(8):984–92.
Eichenfield LF, Ellis CN, Mancini AJ, Paller AS, Simpson EL. Atopic dermatitis: epidemiology and pathogenesis update. Semin Cutan Med Surg. 2012;31(3 Suppl):S3–5.
Eichenfield LF, Tom WL, Chamlin SL, Feldman SR, Hanifin JM, Simpson EL, et al. Guidelines of care for the management of atopic dermatitis: section 1. Diagnosis and assessment of atopic dermatitis. J Am Acad Dermatol. 2014;70(2):338–51.
Kim SH, Hur J, Jang JY, Park HS, Hong CH, Son SJ, et al. Psychological distress in young adult males with atopic dermatitis: a cross-sectional study. Medicine (Baltimore). 2015;94(23):e949.
Paller AS, Tom WL, Lebwohl MG, Blumenthal RL, Boguniewicz M, Call RS, et al. Efficacy and safety of crisaborole ointment, a novel, nonsteroidal phosphodiesterase 4 (PDE4) inhibitor for the topical treatment of atopic dermatitis (AD) in children and adults. J Am Acad Dermatol. 2016;75(3):494–503.e6.
Eichenfield LF, Call RS, Forsha DW, Fowler J Jr, Hebert AA, Spellman M, et al. Long-term safety of crisaborole ointment 2% in children and adults with mild to moderate atopic dermatitis. J Am Acad Dermatol. 2017;77(4):641–649 e5.
US Food & Drug Administration. GRAS Substances (SCOGS) database. https://www.fda.gov/food/generally-recognized-safe-gras/gras-substances-scogs-database. Accessed 13 Nov 2019.
Lim TY, Poole RL, Pageler NM. Propylene glycol toxicity in children. J Pediatr Pharmacol Ther. 2014;19(4):277–82.
Hanifin JM, Rajka G. Diagnostic features of atopic dermatitis. Acta Derm Venereol Suppl (Stockh). 1980;92(suppl):44–7.
Bissonnette R, Papp KA, Poulin Y, Gooderham M, Raman M, Mallbris L, et al. Topical tofacitinib for atopic dermatitis: a phase IIa randomized trial. Br J Dermatol. 2016;175(5):902–11.
Barbier N, Paul C, Luger T, Allen R, De Prost Y, Papp K, et al. Validation of the Eczema Area and Severity Index for atopic dermatitis in a cohort of 1550 patients from the pimecrolimus cream 1% randomized controlled clinical trials programme. Br J Dermatol. 2004;150(1):96–102.
Charman CR, Venn AJ, Williams HC. The patient-oriented eczema measure: development and initial validation of a new tool for measuring atopic eczema severity from the patients’ perspective. Arch Dermatol. 2004;140(12):1513–9.
Zane LT, Kircik L, Call R, Tschen E, Draelos ZD, Chanda S, et al. Crisaborole topical ointment, 2% in patients 2 to 17 years of age with atopic dermatitis: a phase 1b, open-label, maximal-use systemic exposure (MUSE) study. Pediatr Dermatol. 2016;33(4):380–7.
Purohit V, Riley S, Tan H, Ports WC. Patient and disease characteristic predictors of systemic exposure to crisaborole. Presented at: 2019 ACCP Annual Meeting (American College of Clinical Pharmacology); September 15–17, 2019; Chicago, Illinois; Poster 080.
Tom WL, Van Syoc M, Chanda S, Zane LT. Pharmacokinetic profile, safety, and tolerability of crisaborole topical ointment, 2% in adolescents with atopic dermatitis: an open-label phase 2a study. Pediatr Dermatol. 2016;33(2):150–9.
Martin PE, Koplin JJ, Eckert JK, Lowe AJ, Ponsonby AL, Osborne NJ, et al. The prevalence and socio-demographic risk factors of clinical eczema in infancy: a population-based observational study. Clin Exp Allergy. 2013;43(6):642–51.
Hengge UR, Ruzicka T, Schwartz RA, Cork MJ. Adverse effects of topical glucocorticosteroids. J Am Acad Dermatol. 2006;54(1):1–15 (quiz 6–8).
Luger T, Boguniewicz M, Carr W, Cork M, Deleuran M, Eichenfield L, et al. Pimecrolimus in atopic dermatitis: consensus on safety and the need to allow use in infants. Pediatr Allergy Immunol. 2015;26(4):306–15.
Bissonnette R, Pavel AB, Diaz A, Werth JL, Zang C, Vranic I, et al. Crisaborole and atopic dermatitis skin biomarkers: an intrapatient randomized trial. J Allergy Clin Immunol. 2019;144(5):1274–89.
Schram ME, Spuls PI, Leeflang MM, Lindeboom R, Bos JD, Schmitt J. EASI, (objective) SCORAD and POEM for atopic eczema: responsiveness and minimal clinically important difference. Allergy. 2012;67(1):99–106.
Gaunt DM, Metcalfe C, Ridd M. The Patient-Oriented Eczema Measure in young children: responsiveness and minimal clinically important difference. Allergy. 2016;71(11):1620–5.
Barrett A, Hahn-Pedersen J, Kragh N, Evans E, Gnanasakthy A. Patient-reported outcome measures in atopic dermatitis and chronic hand eczema in adults. Patient. 2019;12(5):445–59.
Acknowledgements
The authors thank the study patients, parents/guardians/caregivers, investigators, and investigational sites whose participation made this study possible. Medical writing and editorial assistance under the guidance of the authors was provided by Robert J. Schoen, PharmD, and Jennifer C. Jaworski, MS, of ApotheCom, San Francisco, CA, USA, and was funded by Pfizer Inc. in accordance with Good Publication Practice (GPP3) guidelines (Ann Intern Med. 2015;163:461–4).
Funding
This study was funded by Pfizer Inc. Open Access for this article was funded by Pfizer Inc.
Author information
Authors and Affiliations
Consortia
Corresponding author
Ethics declarations
Conflict of interest
J. Schlessinger has no conflicts of interest to declare. J. S. Shepard was compensated as a principal investigator for this study and was also compensated to serve on an advisory board for Pfizer Inc. R. Gower has received research grants for Pfizer crisaborole clinical trials, and consulting fees for participating in the Pfizer Crisaborole Advisory Board. J. C. Su has been an investigator, speaker, and/or advisor for Pfizer Inc., Amgen, AbbVie, Ego Pharmaceuticals, Eli Lilly, GlaxoSmithKline, Janssen/Johnson & Johnson, L’Oréal, Meda, Novartis, Pierre Fabre, and Sanofi. C. Lynde has served as a principal investigator, consultant, and/or speaker for Pfizer Inc., AbbVie, Bausch, Celgene, Eli Lilly, Janssen, LEO Pharma, Novartis, and Valeant. A. Cha, V. Purohit, L. Takiya, J. L. Werth, C. Zang, and B. Vlahos are employees and stockholders of Pfizer Inc. W. C. Ports was an employee of Pfizer Inc. at the time of this study and is a stockholder of Pfizer Inc.
Research involving human participants and/or animals
The institutional review board at each study site approved the study protocol, and written informed consent was provided by parents/legal guardians. The study was conducted in accordance with the protocol, local legal and regulatory requirements, and the general principles set forth in the International Ethical Guidelines for Biomedical Research Involving Human Subjects, International Conference on Harmonisation Guideline for Good Clinical Practice, and the Declaration of Helsinki.
Informed consent
Written informed consent was provided by parents or legal guardians of participants of the study.
Additional information
William C. Ports: Affiliation at the time of this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Schlessinger, J., Shepard, J.S., Gower, R. et al. Safety, Effectiveness, and Pharmacokinetics of Crisaborole in Infants Aged 3 to < 24 Months with Mild-to-Moderate Atopic Dermatitis: A Phase IV Open-Label Study (CrisADe CARE 1). Am J Clin Dermatol 21, 275–284 (2020). https://doi.org/10.1007/s40257-020-00510-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-020-00510-6