
Abstract
Autoimmune bullous diseases are a group of chronic inflammatory disorders caused by autoantibodies targeted against structural proteins of the desmosomal and hemidesmosomal plaques in the skin and mucosa, leading to intra-epithelial or subepithelial blistering. The oral mucosa is frequently affected in these diseases, in particular, in mucous membrane pemphigoid, pemphigus vulgaris, and paraneoplastic pemphigus. The clinical symptoms are heterogeneous and may present with erythema, blisters, erosions, and ulcers localized anywhere on the oral mucosa, and lead to severe complaints for the patients including pain, dysphagia, and foetor. Therefore, a quick and proper diagnosis with adequate treatment is needed. Clinical presentations of autoimmune bullous diseases often overlap and diagnosis cannot be made based on clinical features alone. Immunodiagnostic tests are of great importance in differentiating between the different diseases. Direct immunofluorescence microscopy shows depositions of autoantibodies along the epithelial basement membrane zone in mucous membrane pemphigoid subtypes, or depositions on the epithelial cell surface in pemphigus variants. Additional immunoserological tests are useful to discriminate between the different subtypes of pemphigoid, and are essential to differentiate between pemphigus and paraneoplastic pemphigus. This review gives an overview of the clinical characteristics of oral lesions and the diagnostic procedures in autoimmune blistering diseases, and provides a diagnostic algorithm for daily practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Autoimmune bullous diseases (AIBDs) are characterized by autoantibody-mediated blistering of the skin and/or mucous membranes [1]. These diseases can be subdivided into two groups based on the level of blistering; pemphigoid diseases characterized by subepithelial blistering and pemphigus diseases characterized by intra-epithelial blistering [2, 3]. Several AIBD subtypes exist within these two major groups, with distinct clinical and diagnostic features [4,5,6]. This review focusses on AIBD subtypes with involvement of the oral mucosa.
Mucous membrane pemphigoid (MMP) is a group of AIBDs that predominantly affects the mucous membranes, but may mildly involve the skin [2, 7]. Autoantibodies are directed against structural proteins of the hemidesmosome in the epithelial basement membrane zone (EBMZ), or proteins that are closely related. Hemidesmosomal dysfunction leads to a loss of connection between basal epithelial cells and the dermis, resulting in subepithelial blistering. Mucous membrane pemphigoid includes different pemphigoid subtypes, such as anti-BP180 MMP (classic MMP) and anti-laminin-332 MMP. Epidermolysis bullosa acquisita (EBA) with predominant mucous membrane involvement is also classified as a subtype of MMP. Linear IgA disease (LAD) is a subtype of pemphigoid and can present with predominant mucous membrane involvement. Anti-p200 pemphigoid is an extremely rare disease and may also have mucosal involvement [8].
Pemphigus diseases comprise mucocutaneous intra-epithelial blistering diseases that target desmosomal proteins, resulting in loss of cell adhesion between keratinocytes [3]. The two main variants are pemphigus vulgaris (PV) with autoantibodies targeting desmoglein 3 (DSG3) and sometimes desmoglein 1 (DSG1), and pemphigus foliaceus with autoantibodies reactive to DSG1 only. Pemphigus vulgaris presents with mucosal lesions, and the skin may be involved, while in pemphigus foliaceus only the skin is affected. Other rare variants of pemphigus include pemphigus vegetans, pemphigus erythematosus, and fogo selvagem (endemic pemphigus foliaceus). The last two are clinical variants of pemphigus foliaceus with reactivity to DSG1 and present with only skin lesions. Paraneoplastic pemphigus (PNP) is a different disease entity related to malignancies, especially hematological malignancies and Castleman disease, and is often life threatening [1]. The clinical hallmark is painful oral mucosal lesions accompanied with morphologically heterogenous skin lesions [9]. The pathomechanism of PNP is complex with involvement of both humoral and cellular autoimmunity.
Chronic oral lesions can be painful, and can seriously influence the quality of life, nutrition status, and dental health of patients [10, 11]. Poor dental cleaning due to painful lesions may result in periodontitis, a chronic inflammatory disease of the gingiva [11]. If not managed adequately, patients are at risk of losing the surrounding teeth. Oral manifestations of MMP, PV, and PNP may seem identical; however, the health impact, treatment, and prognosis of the diseases differ substantially. Therefore, it is important that clinicians make a quick and correct diagnosis. In addition to clinical assessment of the patient, immunodiagnostic tests are essential to differentiate between the AIBD subtypes. The aim of this review is to provide clinicians with a complete overview of the clinical features of AIBDs predominantly involving the oral mucosa, and to describe the diagnostic process.
2 Mucous Membrane Pemphigoid
Mucous membrane pemphigoid is a group of chronic autoimmune diseases characterized by subepithelial blistering and affects mucosal surfaces of various sites. The oral mucosa is predominantly affected (85%), followed by the ocular (65%), nasal (20–40%), anogenital (20%), pharyngeal (20%), laryngeal (5–10%), and esophageal mucosa (5–15%) [2]. The skin may also be involved in 25–30% of the cases but in lesser extent than the mucous membranes [12]. Clinical severity is highly variable in MMP [2].
2.1 Epidemiology and Etiology
Mucous membrane pemphigoid occurs predominantly in the elderly with a mean age of onset in the mid-60 s and an annual incidence of 1.3 and 2.0 newly diagnosed cases per million inhabitants in France and Germany [13,14,15]. Women are more often affected than men.
The etiology of MMP is unknown. A genetic association in patients with MMP with HLA-DBQ1*0301 has been described, suggesting a role for T-lymphocyte recognition of antigens in the EBMZ [16,17,18]. The formation of subepithelial blisters is caused by IgG and/or IgA autoantibodies directed against several components of the hemidesmosome, in particular BP180 and BP230 [12, 19]. In two thirds of the MMP cases, autoantibodies are directed against BP180, mainly against the C-terminal domain and/or the NC16A domain, whereas in bullous pemphigoid, autoantibodies are primarily directed against the NC16A domain [20,21,22,23]. Different epitope profiles of BP180 may be associated with the presence of mucosal involvement with or without skin lesions. Other antigens in MMP include laminin 332, p200, type VII collagen, and α6ß4 integrin. In MMP, the presence of both IgG and IgA in serum is associated with the need for long-term systemic treatment [24].
2.2 Clinical Features
Oral lesions are usually the initial manifestation in MMP and may present anywhere on the oral mucosa (Fig. 1a–c) [25, 26]. The gingiva is most often affected in MMP and may represent the onset of the disease [14, 15, 27,28,29]. Desquamation of the gingiva appears as patches or widespread erythema [26]. Patients with oral MMP present with tense serous or hemorrhagic bullae that easily rupture as a result of mechanical forces, leading to irregularly shaped erosions or ulcers with yellowish slough surrounded by an erythematous halo [19, 26]. Symptoms may consist of pain, dysphagia, soreness, foetor, bleeding and/or peeling of the mucosa [30, 31]. Other commonly affected areas are the palatal mucosa and less often the buccal mucosa and the tongue [27]. Oral lesions often heal without scarring unlike other subtypes of MMP such as ocular MMP [19]. Anti-laminin 332 MMP is a rare subtype often leading to cicatrization and tissue destruction, with possible involvement of pharyngeal and laryngeal mucosa, and a risk of airway obstruction [32]. There are conflicting reports about an increased relative risk for malignancy in this subtype [33, 34]. In some cases, the clinical presentation of oral MMP may resemble that of oral lichen planus [35, 36].

Overlapping features in autoimmune bullous diseases, Stevens–Johnson syndrome, and oral lichen planus demonstrate that oral blistering diseases cannot be differentiated based on clinical presentation. Lesions include desquamative gingivitis (a, d, m), cheilitis (e, f, g, h, j, k), erythema and erosions and blistering of the buccal mucosa (b, i, l, n), palatum (c), and tongue (e, o)
2.3 Other Pemphigoids with Oral Involvement
Other pemphigoid subtypes may also present with oral lesions; however, the difference between MMP and cutaneous pemphigoids relies on the predominant affected site. Bullous pemphigoid primarily affects the skin, with occasionally mild mucosal lesions, whereas MMP predominantly affects the mucous membranes [7]. Epidermolysis bullosa acquisita and LAD are pemphigoid subtypes that also can have predominant mucosal involvement; however, they are not restricted to the oral mucosa alone.
Epidermolysis bullosa acquisita is a subtype of pemphigoid characterized by tissue-bound and circulating autoantibodies against type VII collagen, the main component of the anchoring fibrils below the lamina densa [2]. The incidence ranges from 0.25 and 0.5 new cases per million per year in central Europe and the disease may develop at any age [13]. Two major forms of EBA can be distinguished: the non-inflammatory variant (also termed classical or mechanobullous EBA) and the inflammatory variant [2, 37]. The first form presents with fragile skin, nail loss, and tense vesiculobullous eruptions on trauma-prone areas followed by scarring, milia, and hypo- or hyperpigmentation. These manifestations are also seen in the genetic skin blistering disorder, dystrophic epidermolysis bullosa with a mutation in the same protein, type VII collagen. The inflammatory variant resembles bullous pemphigoid, and blisters heal without scarring. Involvement of mucous membranes is present in more than half of the cases in both variants [38]. The oral mucosa may show erosions, bullae, gingivitis, loss of teeth, and reduced ability to open the mouth. Oral lesions heal with scar formation, and are often therapy resistant [29, 39].
Linear IgA disease is a rare disease characterized by linear deposition of solely IgA along the EBMZ. The incidence ranges between 0.23 and 0.57 per million per year in Europe [40]. Both children and adults can be affected, in particular, before the age of 5 years and after the age of 60 years [41]. Linear IgA disease in childhood seems to be self-limiting, whereas the adult type is characterized by a more chronic disease course. Patients with LAD present with bullae on urticarial plaques in a ring-shaped distribution, also called the ‘crown of jewels’, on the trunk and extremities. Involvement of mucous membranes is seen in 80% of the cases [41]. Oral involvement may consist of painful erosions and ulceration, and in some cases, erosive cheilitis or desquamative gingivitis [42]. Other affected areas in LAD include ocular, nasal, and genital mucous membranes [41]. The IgA autoantibodies in LAD are mainly directed against BP180 and its ectodomain cleavage products, the 120-kDa (LAD-1) and the 97-kDa (LABD97) antigen [2].
Anti-p200 pemphigoid was first described in 1996 and is caused by autoantibodies against a 200-kDa (p200) protein of unknown identity located in the lamina lucida [43, 44]. The clinical presentation is heterogeneous and consists of itchy tense bullae and erythematous or urticarial plaques, which are mainly present on the hands, feet, extremities, and trunk [45]. Oral and genital mucous membranes are involved in 50% of the cases [8].
3 Pemphigus Vulgaris
3.1 Epidemiology
Pemphigus vulgaris is a rare disease with an estimated prevalence of 94.8 per million inhabitants in Germany in 2014 [46]. The estimated annual incidence in European countries lies within the range of 0.7–8 cases per million inhabitants, and is the highest among patients living in South-Eastern European countries [47,48,49,50,51,52,53,54,55]. On average, patients develop PV in the fifth decade of life, with a slight female predominance [46, 54, 55]. The observed mortality rates are increased 2.3- to 3.3-fold compared with the general population, with 1-, 5-, and 10-year survival rates of 95%, 93%, and 90%, respectively [55,56,57,58,59]. Infections are the most frequent cause of death, particularly pneumonia and sepsis [56, 57].
3.2 Pathogenesis
The pathogenesis of PV relies on IgG-targeting desmosomal proteins DSG1 and DSG3 [60].
Genetics seems to contribute in the disease pathogenesis, as population-specific associations between PV and several HLA class II genes have been described, and also associations with non-HLA genes were reported [61, 62]. Overall, the most commonly reported PV-associated HLA class II alleles are HLA-DQB1*0503 and DRB1*0402, which are expressed on antigen-presenting cells.
Most patients with PV with only DSG3 autoantibodies develop a mucosal-dominant phenotype, whereas patients with both DSG1 and DSG3 reactivity display mucocutaneous lesions [63]. This is explained by the desmoglein compensation hypothesis [63, 64]. In the skin, DSG3 is only expressed at the basal layers, while DSG1 is expressed throughout the whole epithelium, and therefore can compensate for DSG3 function loss. However, in the mucosa, DSG3 is expressed in the whole epithelium, whereas DSG1 expression is high in the superficial layers, and significantly lower in the basal layers and therefore loss of DSG3 cannot be adequately compensated by DSG1. This explains why patients with exclusive DSG3 reactivity display mucosal lesions only. Interestingly, seroconversion from DSG3 to DSG1 reactivity and vice versa may occur during the disease course, with corresponding conversion of the clinical pemphigus phenotype.
Several theories concerning the blistering mechanism in PV exist. One theory suggests the binding of autoantibodies may interfere with the cell adhesion ability of desmogleins by steric hindrance [65, 66]. Others observed that autoantibodies induce clustering of the desmogleins, leading to non-assembly of the desmosomes and depletion of desmogleins by internalization, which can weaken the desmosomes’ adhesion strength [67,68,69]. Last, there is evidence that signaling pathways play a role in acantholysis in PV [70, 71]. Some authors hypothesize that signaling pathways in PV may lead to alteration of the cytoskeleton, leading to shrinkage of the basal keratinocytes, and eventually to programmed cell death [72, 73]. Animal studies and in vitro experiments have shown that the IgG Fab fragments purified from the sera of patients with PV directly lead to a pemphigus disease phenotype, implying that Fc-mediated effects of IgG are not necessarily involved in the pathogenesis [74, 75].
3.3 Clinical Features
Patients with PV often present with oral mucosal involvement, with or without skin lesions (Fig. 1d–f). The oral mucosa is the first site to be involved in 50–80% of patients with PV [25]. Mucocutaneous PV is more common than mucosal dominant PV [76]. Oral involvement in pemphigus vegetans, a clinical variant of PV, is reported in 60–80% of the cases [77]. Skin lesions typically consist of flaccid blisters that easily rupture, leaving erosions and crusts, and are commonly distributed on the face, scalp, and upper chest [3]. The oral mucosa displays erosions more often than intact blistering. The palatal mucosa, buccal mucosa, labial mucosa, and the tongue are most commonly affected. Desquamative gingivitis is seen in approximately one fourth of patients with PV, whereas it is the most common (and often the sole) manifestation in patients with MMP [28, 76]. Mucous membranes other than the oral mucosa may be involved, including the pharyngeal and nasal mucosa, and less common, the genital, ocular, laryngeal, and esophageal mucosa. Oral lesions heal without scarring.
4 Paraneoplastic Pemphigus
4.1 Epidemiology
Paraneoplastic pemphigus, first described by Anhalt et al., is a distinct, extremely rare, and often lethal variant of pemphigus associated with malignancy [78]. In most cases, underlying B-cell lymphoproliferative diseases can be found, such as non-Hodgkin lymphomas, chronic lymphatic leukemia, or Castleman disease. Paraneoplastic pemphigus is rarely associated with other solid tumors [79]. However, 10% of cases have no malignancy at the time of the diagnosis and data on follow-up are lacking [80].
The diagnosis of PNP is based on three criteria: (1) initial mucosal involvement; (2) detection of circulating autoantibodies against envoplakin, periplakin, and/or a2-macroglobulin-like 1 (A2ML1); and (3) the detection of an underlying neoplasm [9]. Paraneoplastic pemphigus may occur at any age without sex predominance, although most of the cases are diagnosed at approximately 60 years of age [80, 81]. The disease may also develop in children, frequently associated with Castleman disease [9].
The underlying malignancies in PNP can be difficult to control, which makes the prognosis very poor with a reported mortality rate of 75–90% [82, 83]. The survival rate in a French cohort after 1, 2, and 5 years was 49%, 41%, and 38%, respectively [84]. The main causes of death include infections, the progression of the neoplasm, and bronchiolitis obliterans [80, 84, 85].
4.2 Pathogenesis
The pathogenesis of PNP is complex, with involvement of both the humoral and cellular immune systems [79]. Some data confirm the presence of certain genetic predispositions in PNP. A Chinese and a French cohort show a clear association between PNP and HLA-Cw*14, HLA-DRB1*04, and HLA-DRB1*14 [86, 87]. Currently, different mechanisms are assumed to contribute to the development of PNP [9]. Direct effects of the associated tumor were found, such as production of autoantibodies targeting epithelial antigens, and high interleukin-6 secretion, resulting in reactive autoimmunity [82, 88]. Indirectly, autoreactive cellular cytotoxicity against the underlying tumor has been described, showing natural killer cells, macrophages, and cytotoxic CD8 + T lymphocytes, the latter producing high levels of interferon-γ and tumor necrosis factor-α in PNP skin [89]. The cellular response may result in an interface dermatitis revealing new antigens (epitope spreading). Moreover, a humoral immune response against the tumor may lead to cross-reactivity with epithelial antigens, also called antigen mimicry [82, 90, 91].
A heterogeneous autoantibody repertoire can be found in PNP; envoplakin, periplakin, BP230, desmoplakin, and plectin are all plakin family proteins that may be targeted. Recently, epiplakin was described as an autoantigen in PNP associated with bronchiolitis obliterans in Japanese patients [92]. Autoantibodies against several desmosomal cadherins such as DSG1 and DSG3, and desmocollin-1, -2, and -3 have also been identified [93]. The latest discovered protein targeted by PNP autoantibodies is the A2ML1 protein, an epithelial-expressed protease inhibitor [94]. In some cases, autoantibodies against the desmosomal armadillo protein plakophilin-3, BP180, laminin 332, and the neuromuscular junction were detected [95, 96]. Although PNP and PV share some cadherin autoantigens, the targeted epitopes and the IgG isotypes differ in the two diseases [97]. The exact pathogenic role of autoantibodies targeting different antigens and their influence on the disease phenotype are uncertain.
4.3 Clinical Features
The most pronounced clinical sign is involvement of the mucosa, predominantly the oral mucosa with painful stomatitis, gingivitis, hemorrhagic cheilitis, erythema, blistering, and erosions (Fig. 1g–i) [80, 98]. The involvement of the mucosa may extend to the upper gastrointestinal tract and upper airways. The conjunctiva is involved in 70% of the cases, with the presence of corneal ulceration, perforation, melting, or symblepharon [99].
Moreover, PNP shows a wide spectrum of cutaneous eruptions such as macules, papules, plaques, and blistering erosions. Five clinical phenotypes of PNP can be distinguished; PV-like, bullous pemphigoid-like, erythema exsudativum multiforme-like, graft vs. host disease-like, and lichen planus-like [9, 100, 101].
In addition, PNP can affect other organs and tissues besides the mucosa and skin, suggesting the use of the name paraneoplastic autoimmune multi-organ syndrome according to the latest literature [79]. Yet, no international consensus exists on the use of the term paraneoplastic autoimmune multi-organ syndrome. Pulmonary involvement in the form of bronchiolitis obliterans occurs in 30% of the PNP cases, which is fatal in 90% of the cases [85]. Bronchiolitis obliterans is very common in Castleman disease-associated PNP (71%) and a DSG3-dependent mechanism is assumed [80, 85, 102]. Other affected organs besides the lungs include the thyroid gland, kidneys, and smooth muscle tissue [9]. Muscle weakness (39%) and myasthenia gravis can also be seen in PNP and are associated with a certain antibody repertoire [96].
5 Diagnostic Algorithm in Patients with Oral Blisters
The diagnostic algorithm in patients with oral blisters includes the performance of two biopsies for histopathology and direct immunofluorescence microscopy (DIF), collecting a serum sample for immunoserological tests, and the performance of microbiological diagnostics to test for infectious causes (Fig. 2).

Flowchart of the diagnostic pathway in patients with oral blistering, with the focus on autoimmune bullous diseases affecting the oral mucosa
5.1 Main Differential Diagnosis
The differential diagnosis of erosive oral lesions include infectious agents, due to viruses and bacteria, drug-induced reactions, inflammatory conditions, and systemic diseases (Table 1). These conditions have some specific morphological features and overlapping clinical features, therefore histology is required for the diagnosis.
Oral lesions caused by infectious agents are usually transient. Primary infection with herpes simplex is the most common viral cause of erosions and blisters in the oral mucosa [103]. Stevens–Johnson syndrome and toxic epidermal necrolysis are severe, mostly drug-induced reactions with an abrupt onset of erosive stomatitis and hemorrhagic cheilitis followed by widespread skin lesions (Fig. 1j–l) [104]. The gingiva is rarely affected in Stevens–Johnson syndrome and toxic epidermal necrolysis. Other mucosal involvement is frequently seen, such as ocular and genital involvement.
Recurrent aphthous stomatitis is the most frequent inflammatory condition of the oral mucosa with recurrent self-limiting ulcers affecting non keratinized mucosa. Multiple factors are involved in the etiology of recurrent aphthous stomatitis [103]. Behçet syndrome is a chronic multi-system condition characterized by diffuse aphthous erosions and ulcers on the labial, buccal, and lateral tongue mucosa [31]. Ocular and genital involvement is also frequently seen.
Lupus erythematosus is a systemic autoimmune disorder with a wide variety of clinical features. Oral lesions in lupus erythematosus are usually multiple, located on the buccal mucosa, hard palate, lips, and gingiva. The distribution is asymmetrical with different morphological features ranging from plaques, erythema, and ulcerations [19].
Oral lichen planus is a chronic immune mediated disease with highly variable clinical features ranging from subtle white reticular patterns to erosions and ulcerations (Fig. 1m–o). Lesions are usually symmetrically distributed in the oral cavity. Affected sites of the oral cavity include the buccal mucosa, tongue, and gingiva. Histopathologic biopsies of oral lichen planus often show sub-epithelial blistering without preservation of the basal cells, in contrast to pemphigoid [19].
5.2 Nikolsky Sign
When encountering a patient with a blistering disease, clinicians may test the positivity of the Nikolsky sign consisting of two variants. The Nikolsky sign I (or direct Nikolsky sign) can be tested by sideward friction of healthy-looking skin or mucosa, and is positive when the mechanical pressure induces an erosion. The Nikolsky sign II (or indirect Nikolsky sign) tests the ability to split the epidermis by pulling the blister remnant, and is positive when the blister extends far beyond the existing erosion [105, 106].
In patients with a clinical phenotype compatible with AIBD, the Nikolsky sign on the skin may be a useful diagnostic tool with moderate sensitivity; however, a very high specificity for the diagnosis of pemphigus diseases [107]. The Nikolsky sign can also be tested on the gingiva in patients with only oral involvement, however, it is often positive in both MMP and pemphigus, and therefore not a useful tool to differentiate between these diseases [108]. Moreover, the Nikolsky sign on the oral mucosa may cause unnecessary injury to the tissue and pain to the patient.
5.3 Histopathological Findings
Histopathology alone is not sufficient to diagnose AIBD because it often is non-specific, but is useful to differentiate between non-autoimmune bullous diseases in the differential diagnosis (Table 1). A skin biopsy for histopathological examination should be taken at the edge of the blister, including one third blister and two thirds perilesional skin [4, 6]. Mucosal biopsies for histopathology are usually taken from lesional mucosa. In the presence of a blister, a biopsy for histopathology should be taken at the edge of the blister to determine the level of the blister in AIBD. In the presence of an ulceration, a biopsy for histopathology should be taken from the perilesional erythema. One of the differential diagnoses of an ulcerative process is oral lichen planus. In this respect, the presence of an interface dermatitis can only be determined in lesional tissue. Moreover, another differential diagnosis is spinocellular carcinoma and this also can only be excluded with a lesional biopsy.
In MMP, histopathological features may include subepithelial blistering with an infiltrate consisting of eosinophils, lymphocytes, and/or neutrophils [25]. In EBA, histopathological findings are dependent on the disease phase, as in early stages, vacuolar changes along the BMZ accompanied with edema in the papillary dermis are shown. In a later stage, a subepithelial blister is formed with a mixed infiltrate consisting of neutrophils, eosinophils, and mononuclear cells [37]. In LAD, an anti-p200 pemphigoid histopathological examination of lesional skin may show subepithelial blister formation with mainly neutrophilic infiltrate in the papillary dermis, in anti-p200 pemphigoid, eosinophils are sometimes present [109, 110].
Characteristic histopathological findings of PV are acantholysis with rounding up of keratinocytes, and a suprabasilar cleft [111]. The basal keratinocytes remain attached to the basement membrane, and line the blister floor, resulting in a typical tombstone appearance. Eosinophils may be present, infiltrating the epidermis [6]. The histopathology of PNP is not exclusive and different patterns can be seen, with vacuolar interface dermatitis or keratinocyte necrosis in most cases, and rarely intra-epithelial acantholysis [112].
5.4 Direct Immunofluorescence Microscopy
Direct immunofluorescence plays a very important role in differentiating between the different AIBDs. Autoantibodies bound in the skin are visualized by adding a fluorescent-labeled antibody against human IgG to a frozen skin section [113]. Skin biopsies should measure 4 mm in diameter, and are preferably taken perilesional, 1–2 cm adjacent to a blister, to yield the highest sensitivity [114,115,116]. Mucous membrane biopsies should be at least 3 mm in diameter. Oral biopsies are recommended to be taken from the buccal mucosa, localized one third of the distance from the mouth corner to the last molar, to avoid the parotid duct [117]. A DIF biopsy of non-lesional buccal mucosa in patients with solely desquamative gingivitis is recommended [118]. A recent retrospective analysis showed comparable sensitivity of a DIF biopsy in normal and perilesional mucosa [119]. Biopsies for DIF should preferably be stored in 0.9% sodium chloride for 24 hours to reduce IgG background staining [120, 121]. For a transportation of several days, the biopsy can be stored in Michel’s medium.
The location of a DIF biopsy depends on the distribution of lesions. In patients with both skin and mucosal lesions, a skin biopsy is usually performed. In cases with only mucosal lesions, a mucosal biopsy is taken from the affected site. If the DIF result is negative, repeated biopsies, or biopsies from other mucosal sites or the skin can be taken [122, 123].
Direct immunofluorescence of intact perilesional mucosa in MMP shows deposition of IgG, in some cases, IgA autoantibodies and/or complement C3c along the EBMZ. The sensitivity of DIF microscopy for the diagnosis of MMP lies between 69 and 83%, and increases when multiple and repeated biopsies are performed [123,124,125,126]. Similar to MMP, EBA and anti-p200 pemphigoid show linear depositions of IgG and/or IgA and C3c along the EBMZ. In LAD, DIF is the reference standard for diagnosis, and is characterized by a linear deposition of exclusively IgA along the EBMZ [116].
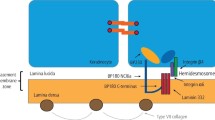
Analysis of the serration pattern by DIF discriminates between MMP subtypes, but can be indeterminable on mucosal biopsies, and performance of an additional skin biopsy may be considered [116, 127]. The classic MMP, anti-p200 pemphigoid, anti-laminin 332 pemphigoid, and LAD show a n-serrated pattern, due to binding of autoantibodies against hemidesmosomal proteins above the lamina densa (Fig. 3a). Epidermolysis bullosa acquisita-type MMP can be diagnosed by recognition of an u-serrated pattern by binding of autoantibodies in the sublamina densa (Fig. 3b) [116, 120]. Exclusive linear IgA depositions with an u-serrated pattern in patients with oral lesions without skin involvement is diagnosed as IgA MMP [128].

Immunofluorescence results compatible with the diagnosis of pemphigoid diseases. a Direct immunofluorescence microscopy shows linear IgG along the epithelial basement membrane zone in an n-serrated pattern. b Direct immunofluorescence microscopy shows linear IgG along the epithelial basement membrane zone in a u-serrated pattern. c Indirect immunofluorescence microscopy on a substrate of salt-split skin (indirect immunofluorescence on salt-split skin), showing IgG bound to the epithelial side of the artificial split, wherein BP180 and BP230 are located. d Indirect immunofluorescence on salt-split skin showing IgG bound to the dermal side of the artificial split, wherein laminin-332, p200, and type VII collagen are located
Direct immunofluorescence microscopy is the reference standard in pemphigus diagnosis, and shows binding of IgG on the epithelial cell surface (ECS) [114]. The deposition pattern might be smooth or granular, in line with the theory on clustering of desmogleins (Fig. 4a, b). Complement C3 deposits may be found in approximately 61% of PV cases [28].

Direct immunofluorescence microscopy staining of a pemphigus patient. a IgG staining in a smooth epithelial cell surface pattern. b IgG staining in a granular epithelial cell surface pattern
In PNP, the DIF on skin and mucosa may show deposition of IgG and C3c in an ECS pattern, in some cases with additional linear-granular depositions of mainly complement and sometimes IgG along the EBMZ (Fig. 5a) [129]. The combined ECS and anti-EBMZ pattern is seen in 27% of the cases; however, it has a very high specificity of 97%. Rarely, a linear anti-EMBZ pattern may be the only finding with DIF microscopy [130].

Diagnostic results in paraneoplastic pemphigus. a Direct immunofluorescence microscopy with an epithelial cell surface and anti-basement membrane zone pattern. b Indirect immunofluorescence microscopy on a rat bladder with IgG staining in an epithelial cell surface pattern, compatible with the diagnosis of paraneoplastic pemphigus. c Serum analyzed for IgG against paraneoplastic pemphigus antigens by immunoblotting (1) and immunoprecipitation (2). A2ML1 alpha-2-macroglobulin-like 1, EPL epiplakin, PPL periplakin
5.5 Immunoserology
Several immunoserologic tests are used to detect circulating autoantibodies against hemidesmosomal and desmosomal proteins, and may be valuable for diagnosing and discriminating between AIBDs. Circulating autoantibodies in serum of MMP are usually low in concentration and therefore not always detectable [27]. This is also the case in EBA, which lacks detectable circulating autoantibodies in more than half of the cases. In contrast, serological tests are essential for the diagnosis of PNP.
5.5.1 Indirect Immunofluorescence
With indirect immunofluorescence (IIF) microscopy, the patients’ serum is applied to a substrate, and after incubation and washing away unbound antibodies, a fluorescent-labeled antibody visualizes autoantibodies bound to the substrate. Monkey esophagus is frequently used as a substrate because it is widely commercially available. Indirect immunofluorescence on monkey esophagus shows the highest sensitivity for the diagnosis of pemphigus diseases, and circulating autoantibodies in a smooth ECS pattern can be identified in approximately 86–90% of patients with PV [114, 131].
A second commonly used substrate is salt-split skin (SSS), which can discriminate between pemphigoid subtypes [8]. Salt-split skin is obtained by incubation of human skin in sodium for 2–3 days, resulting in a reproducible artificial split in the lamina lucida, with antigens located either at the epithelial or dermal side. Indirect immunofluorescence on SSS in classic MMP with autoantibodies directed against BP180 or BP230 shows binding to the epithelial side (Fig. 3c). Other subtypes of MMP show antibodies directed against p200, laminin-332, and type VII collagen, which bind to the dermal side of the artificial split (Fig. 3d). Anti-laminin-332 MMP with epithelial binding has been reported. This is explained by the co-existence of antibodies to BP180 or BP230 and the low sensitivity of the technique, which leads to a negative result of indirect immunofluorescence on the dermal side of the split skin [34]. In LAD, IIF on SSS exclusively shows IgA, which most often binds to the epithelial side, or the dermal side in the case of IgA-EBA-type MMP [116].
Indirect immunofluorescence on a substrate of rat bladder is one of the few serological tests that can diagnose PNP, and therefore is highly important. Rat bladder is a perfect substrate as it does not contain DSG1 and DSG3, but expresses desmoplakin, epiplakin, and periplakin [93]. Positive IIF on a rat bladder is very specific for PNP, and may show different patterns, such as urothelial cell surface staining and cytoplasmic staining (Fig. 5b) [130].
5.5.2 Other Immunoserological Tests
Enzyme-linked immunosorbent assay (ELISA) and immunoblot are commonly used to specify targeted antigens. The ELISA technique is performed using a 64-well plate, of which each well is coated with a specific AIBD-related antigen. Serum is added to enable autoantibodies to bind the specific antigen on the sides of the well. After washing away non-specific unbound antibodies, an enzyme-linked IgG conjugate is added. The enzyme substrate is applied in the next step, resulting in a measurable color reaction, of which the intensity reflects on the amount of autoantibodies. To perform the immunoblot technique, denatured proteins of the skin are first sorted based on molecular size by gel electrophoresis, and are transferred onto a membrane [132, 133]. Autoantibodies directed against skin proteins can bind the membrane, and are visualized by staining. The molecular size of the stained protein identifies which antigen is targeted.
In MMP, the NC16A ELISA is frequently used for the detection of circulating autoantibodies; however, a retrospective study showed a high number of false-positives (11.3%) in non-pemphigoid controls [115]. Izumi et al. reported the detection of BP180 by full-length BP180 ELISA because MMP autoantibodies may target different regions, especially the C-terminal domain [134]. However, the full-length BP180 ELISA is not commercially available.
Enzyme-linked immunosorbent assay kits are also commercially available for BP230 and type VII collagen [135]. The BP230 ELISA is less sensitive with an additional diagnostic added value of only 5% compared to NC16A ELISA in bullous pemphigoid [135]. The type VII collagen ELISA has a low sensitivity of 45%, and when combined with IIF on SSS the sensitivity rises to 50% [127]. The immunoblot assay in MMP has a highly variable sensitivity. Autoantibodies targeting BP180, LABD97, LAD-1, and p200 can be detected by immunoblotting, although the latter requires a sophisticated preparation of a human dermal extract, which is only performed in highly specialized laboratories [45]. In LAD, immunoblot can detect circulating IgA autoantibodies against BP180, LABD97, or LAD-1.
Antibodies against DSG1 and/or DSG3 can be measured by the ELISA technique in approximately 90% of patients with pemphigus [6]. Antibody titers against DSG1 measured by ELISA correlate well with the disease activity of PV, whereas DSG3 antibody titers correlate with the disease course in only two thirds of the patients [136, 137]. In several patients with PV, high levels of DSG3 antibodies remain measurable after achieving remission, and it is hypothesized that they might be non-pathogenic [138]. Immunoblot is not useful for the diagnosis of PV because anti-DSG1 and DSG3 autoantibodies are often directed against conformation epitopes, meaning that the denaturation process changes the epitopes’ composition, and therefore immunoblot will be negative [133].
The diagnosis of PNP is based on the detection of autoantibodies against envoplakin and periplakin, or A2ML1. Radioactive and nonradioactive immunoprecipitation is the most sensitive (95%) and specific (100%) method, but these tests are not widely available (Fig. 5c). Immunoblot can show reactivity against the 210-kDa envoplakin, 190-kDa periplakin, 180-kDa BP180, 230-kDa BP230, 250 and 210-kDa desmoplakin I and II, and 500-KDa plectin (Fig. 5c). Immunoblot in combination with IIF on the rat bladder has shown to be an excellent alternative for immunoprecipitation, with very high sensitivity and specificity. This alternative provides a valuable, relative easy method to perform PNP diagnostic in most laboratories [93].
6 Conclusions
Clinicians should be aware that oral lesions in AIBD may look similar, and might be the only manifestation of disease. A quick and proper diagnosis is necessary for adequate treatment, which may take place in a multi-disciplinary setting. Direct immunofluorescence on a biopsy from skin or mucosa combined with IIF and additional serological tests are the cornerstones for the diagnosis of AIBD. By following the described diagnostic algorithm, clinicians should be able to recognize and accurately diagnose AIBD affecting the oral mucosa.
References
Jonkman MF. Autoimmune bullous diseases. London: Springer; 2016.
Schmidt E, Zillikens D. Pemphigoid diseases. Lancet. 2013;381:320–32.
Bystryn J-C, Rudolph JL. Pemphigus. Lancet. 2005;366:61–73.
Murrell DF, Pena S, Joly P, Marinovic B, Hashimoto T, Diaz LA, et al. Diagnosis and management of pemphigus: recommendations by an international panel of experts. J Am Acad Dermatol. 2018. https://doi.org/10.1016/j.jaad.2018.02.021.
Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867–77.
Hertl M, Jedlickova H, Karpati S, Marinovic B, Uzun S, Yayli S, et al. Pemphigus: S2 guideline for diagnosis and treatment—guided by the European Dermatology Forum (EDF) in cooperation with the European Academy of Dermatology and Venereology (EADV). J Eur Acad Dermatol Venereol. 2015;29:405–14.
Chan LS, Ahmed AR, Anhalt GJ, Bernauer W, Cooper KD, Elder MJ, et al. The first international consensus on mucous membrane pemphigoid: definition, diagnostic criteria, pathogenic factors, medical treatment, and prognostic indicators. Arch Dermatol. 2002;138:370–9.
Meijer JM, Diercks GFH, Schmidt E, Pas HH, Jonkman MF. Laboratory diagnosis and clinical profile of anti-p200 pemphigoid. JAMA Dermatol. 2016;152:897–904.
Frew JW, Murrell DF. Paraneoplastic pemphigus (paraneoplastic autoimmune multiorgan syndrome): clinical presentations and pathogenesis. Dermatol Clin. 2011;29(3):419–25.
Suliman NM, Johannessen AC, Ali RW, Salman H, Astrom AN. Influence of oral mucosal lesions and oral symptoms on oral health related quality of life in dermatological patients: a cross sectional study in Sudan. BMC Oral Health. 2012;12:19.
Jascholt I, Lai O, Zillikens D, Kasperkiewicz M. Periodontitis in oral pemphigus and pemphigoid: a systematic review of published studies. J Am Acad Dermatol. 2017;76(975–978):e3.
Chan LS, Wojnarowska F, Fine J-D, Zone JJ, Kirtschig G, Lozada-Nur F, et al. The first international consensus on mucous membrane pemphigoid. Arch Dermatol. 2004;138:370–9.
Bertram F, Bröcker EB, Zillikens D, Schmidt E. Prospektive Untersuchung der Inzidenz blasenbildender Autoimmundermatosen in Unterfranken. J Dtsch Dermatol Ges. 2009;7:434–40.
Arduino PG, Broccoletti R, Carbone M, Conrotto D, Pettigiani E, Giacometti S, et al. Describing the gingival involvement in a sample of 182 Italian predominantly oral mucous membrane pemphigoid patients: a retrospective series. Med Oral Patol Oral Cir Bucal. 2017;22:e149–52.
Bagan J, Jiménez Y, Murillo J, Bagan L. Oral mucous membrane pemphigoid: a clinical study of 100 low-risk cases. Oral Dis. 2018;24:132–4.
Welsh KI, Challacombe SJ, Black MM, Theron J, Setterfield J, Wojnarowska F, et al. Mucous membrane pemphigoid: HLA-DQB1*0301 is associated with all clinical sites of involvement and may be linked to antibasement membrane IgG production. Br J Dermatol. 2010;145:406–14.
Carrozzo M, Fasano ME, Broccoletti R, Carbone M, Cozzani E, Rendine S, et al. HLA-DQB1 alleles in Italian patients with mucous membrane pemphigoid predominantly affecting the oral cavity. Br J Dermatol. 2003;145:805–8.
Delgado JC, Turbay D, Yunis EJ, Yunis JJ, Morton ED, Bhol K, et al. A common major histocompatibility complex class II allele HLA-DQB1*0301 is present in clinical variants of pemphigoid. Proc Natl Acad Sci USA. 1996;93:8569–71.
Mustafa MB, Porter SR, Smoller BR, Sitaru C. Oral mucosal manifestations of autoimmune skin diseases. Autoimmun Rev. 2015;14:930–51.
Balding SD, Prost C, Diaz LA, Bernard P, Bedane C, Aberdam D, et al. Cicatricial pemphigoid auto antibodies react with multiple sites on the BP180 extracellular domain. J Investig Dermatol. 1996;106:141–6.
Oyama N, Setterfield JF, Powell AM, Sakuma-Oyama Y, Albert S, Bhogal BS, et al. Bullous pemphigoid antigen II (BP180) and its soluble extracellular domains are major autoantigens in mucous membrane pemphigoid: the pathogenic relevance to HLA class II alleles and disease severity. Br J Dermatol. 2006;154:90–8.
Lee JB, Liu Y, Hashimoto T. Cicatricial pemphigoid sera specifically react with the most C-terminal portion of BP180. J Dermatol Sci. 2003;32:59–64.
Calabresi V, Arduino P, Tirone F, Parodi A, Zambruno G, Bertolusso G, et al. Oral pemphigoid autoantibodies preferentially target BP180 ectodomain. Clin Immunol. 2006;122:207–13.
Setterfield J, Shirlaw PJ, Kerr-Muir M, Neill S, Bhogal BS, Morgan P, et al. Mucous membrane pemphigoid: a dual circulating antibody response with IgG and IgA signifies a more severe and persistent disease. Br J Dermatol. 1998;138:602–10.
Buonavoglia A, Leone P, Dammacco R, Di Lernia G, Petruzzi M, Bonamonte D, et al. Pemphigus and mucous membrane pemphigoid: an update from diagnosis to therapy. Autoimmun Rev. 2019;18(4):349–58.
Scully C, Lo Muzio L. Oral mucosal diseases: mucous membrane pemphigoid. Br J Oral Maxillofac Surg. 2008;46:358–66.
Petruzzi M. Mucous membrane pemphigoid affecting the oral cavity: short review on etiopathogenesis, diagnosis and treatment. Immunopharmacol Immunotoxicol. 2012;34:363–7.
Villa A, Treister N, Woo S-B, Saavedra A, Sultan A. Oral mucous membrane pemphigoid and pemphigus vulgaris: a retrospective two-center cohort study. Oral Dis. 2017;23:498–504.
Ramos-e-Silva M, de-Moura-Ferreira A, Castro Jacques C. Oral involvement in autoimmune bullous diseases. Clin Dermatol. 2011;29:443–54.
Perschbacher K. Mucocutaneous diseases of the oral cavity. Diagn Histopathol. 2018;24:166–71.
Baglama Š, Trčko K, Rebol J, Miljković J. Oral manifestations of autoinflammatory and autoimmune diseases. Acta Dermatovenerol Alp Pannonica Adriat. 2018;27:9–16.
Chin-Che Hsu R, Lazarova Z, Lee HG, Tung YC, Yu HS. Antiepiligrin cicatricial pemphigoid. J Am Acad Dermatol. 2000;42:841–4.
Egan CA, Lazarova Z, Darling TN, Yee C, Coté T, Yancey KB. Anti-epiligrin cicatricial pemphigoid and relative risk for cancer. Lancet. 2001;357:1850–1.
Bernard P, Antonicelli F, Bedane C, Joly P, Le Roux-Villet C, Duvert-Lehembre S, et al. Prevalence and clinical significance of anti-laminin 332 autoantibodies detected by a novel enzyme-linked immunosorbent assay in mucous membrane pemphigoid. JAMA Dermatol. 2013;149:533–40.
Benzaquen M, Suter VGA, Borradori L, Gschwend M, Feldmeyer L. Mucous membrane pemphigoid of the oral lichen type: a retrospective analysis of 16 cases. J Eur Acad Dermatol Venereol. 2019;33:e205–7.
Fukuda A, Himejima A, Tsuruta D, Koga H, Ohyama B, Morita S, et al. Four cases of mucous membrane pemphigoid with clinical features of oral lichen planus. Int J Dermatol. 2016;55:657–65.
Chen M, Kim GH, Prakash L, Woodley DT. Epidermolysis bullosa acquisita: autoimmunity to anchoring fibril collagen. Autoimmunity. 2012;45:91–101.
Buijsrogge JJA, Diercks GFH, Pas HH, Jonkman MF. The many faces of epidermolysis bullosa acquisita after serration pattern analysis by direct immunofluorescence microscopy. Br J Dermatol. 2011;165:92–8.
Bascones-Martínez A, García-García V, Meurman JH, Requena-Caballero L. Immune-mediated diseases: what can be found in the oral cavity? Int J Dermatol. 2015;54:258–70.
Lings K, Bygum A. Linear IgA bullous dermatosis: a retrospective study of 23 patients in Denmark. Acta Derm Venereol. 2015;95:466–71.
Wojnarowska F, Marsden RA, Bhogal B, Black MM. Chronic bullous disease of childhood, childhood cicatricial pemphigoid, and linear IgA disease of adults: a comparative study demonstrating clinical and immunopathologic overlap. J Am Acad Dermatol. 1988;19:792–805.
Sánchez AR, Rogers RS, Kupp LI, Sheridan PJ. Desquamative gingivitis associated with IgG/IgA pemphigoid presents a challenging diagnosis and treatment: a case report. J Periodontol. 2005;75:1714–9.
Chen KR, Shimizu S, Miyakawa S, Ishiko A, Shimizu H, Hashimoto T. Coexistence of psoriasis and an unusual IgG-mediated subepidermal bullous dermatosis: identification of a novel 200-kDa lower lamina lucida target antigen. Br J Dermatol. 1996;134:340–6.
Zillikens D, Kawahara Y, Ishiko A, Shimizu H, Mayer J, Rank CV, et al. A novel subepidermal blistering disease with autoantibodies to a 200-kDa antigen of the basement membrane zone. J Investig Dermatol. 1996;106:1333–8.
Goletz S, Hashimoto T, Zillikens D, Schmidt E. Anti-p200 pemphigoid. J Am Acad Dermatol. 2014;71:185–91.
Hubner F, Recke A, Zillikens D, Linder R, Schmidt E. Prevalence and age distribution of pemphigus and pemphigoid diseases in Germany. J Investig Dermatol. 2016;136(12):2495–8.
Tsankov N, Vassileva S, Kamarashev J, Kazandjieva J, Kuzeva V. Epidemiology of pemphigus in Sofia, Bulgaria: a 16-year retrospective study (1980–1995). Int J Dermatol. 2000;39:104–8.
Hahn-Ristic K, Rzany B, Amagai M, Brocker EB, Zillikens D. Increased incidence of pemphigus vulgaris in southern Europeans living in Germany compared with native Germans. J Eur Acad Dermatol Venereol. 2002;16:68–71.
V’lckova-Laskoska MT, Laskoski DS, Kamberova S, Caca-Biljanovska N, Volckova N. Epidemiology of pemphigus in Macedonia: a 15-year retrospective study (1990–2004). Int J Dermatol. 2007;46:253–8.
Micali G, Musumeci ML, Nasca MR. Epidemiologic analysis and clinical course of 84 consecutive cases of pemphigus in eastern Sicily. Int J Dermatol. 1998;37:197–200.
Baican A, Baican C, Chiriac G, Chiriac MT, Macovei V, Zillikens D, et al. Pemphigus vulgaris is the most common autoimmune bullous disease in Northwestern Romania. Int J Dermatol. 2010;49:768–74.
Michailidou EZ, Belazi MA, Markopoulos AK, Tsatsos MI, Mourellou ON, Antoniades DZ. Epidemiologic survey of pemphigus vulgaris with oral manifestations in northern Greece: retrospective study of 129 patients. Int J Dermatol. 2007;46:356–61.
Serwin AB, Koper M, Flisiak I. Incidence of pemphigus vulgaris and pemphigus foliaceus in North-East Poland (Podlaskie Province): a 15-year (2001–2015) bicentric retrospective study. Int J Dermatol. 2018;57:933–7.
Marazza G, Pham HC, Scharer L, Pedrazzetti PP, Hunziker T, Trueb RM, et al. Incidence of bullous pemphigoid and pemphigus in Switzerland: a 2-year prospective study. Br J Dermatol. 2009;161:861–8.
Langan SM, Smeeth L, Hubbard R, Fleming KM, Smith CJP, West J. Bullous pemphigoid and pemphigus vulgaris: incidence and mortality in the UK: population based cohort study. BMJ. 2008;337:a180.
Kridin K, Sagi SZ, Bergman R. Mortality and cause of death in patients with pemphigus. Acta Derm Venereol. 2017;97:607–11.
Huang Y-H, Kuo C-F, Chen Y-H, Yang Y-W. Incidence, mortality, and causes of death of patients with pemphigus in Taiwan: a nationwide population-based study. J Investig Dermatol. 2012;132:92–7.
Kridin K. Pemphigus group: overview, epidemiology, mortality, and comorbidities. Immunol Res. 2018;66:255–70.
Kridin K, Zelber-Sagi S, Bergman R. Pemphigus vulgaris and pemphigus foliaceus: differences in epidemiology and mortality. Acta Derm Venereol. 2017;97:1095–9.
Hammers CM, Stanley JR. Mechanisms of disease: pemphigus and bullous pemphigoid. Annu Rev Pathol. 2016;11:175–97.
Vodo D, Sarig O, Sprecher E. The genetics of pemphigus vulgaris. Front Med (Lausanne). 2018;5:226.
Sinha AA. The genetics of pemphigus. Dermatol Clin. 2011;29(381–91):vii.
Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Investig. 1999;103:461–8.
Hanakawa Y, Matsuyoshi N, Stanley JR. Expression of desmoglein 1 compensates for genetic loss of desmoglein 3 in keratinocyte adhesion. J Investig Dermatol. 2002;119:27–31.
Heupel W-M, Zillikens D, Drenckhahn D, Waschke J. Pemphigus vulgaris IgG directly inhibit desmoglein 3-mediated transinteraction. J Immunol. 2008;181:1825–34.
Sekiguchi M, Futei Y, Fujii Y, Iwasaki T, Nishikawa T, Amagai M. Dominant autoimmune epitopes recognized by pemphigus antibodies map to the N-terminal adhesive region of desmogleins. J Immunol. 2001;167:5439–48.
Oktarina DAM, van der Wier G, Diercks GFH, Jonkman MF, Pas HH. IgG-induced clustering of desmogleins 1 and 3 in skin of patients with pemphigus fits with the desmoglein nonassembly depletion hypothesis. Br J Dermatol. 2011;165:552–62.
van der Wier G, Pas HH, Kramer D, Diercks GFH, Jonkman MF. Smaller desmosomes are seen in the skin of pemphigus patients with anti-desmoglein 1 antibodies but not in patients with anti-desmoglein 3 antibodies. J Investig Dermatol. 2014;134(8):2287–90.
Aoyama Y, Kitajima Y. Pemphigus vulgaris-IgG causes a rapid depletion of desmoglein 3 (Dsg3) from the Triton X-100 soluble pools, leading to the formation of Dsg3-depleted desmosomes in a human squamous carcinoma cell line, DJM-1 cells. J Investig Dermatol. 1999;112:67–71.
Li X, Ishii N, Ohata C, Furumura M, Hashimoto T. Signalling pathways in pemphigus vulgaris. Exp Dermatol. 2014;23:155–6.
Saito M, Stahley SN, Caughman CY, Mao X, Tucker DK, Payne AS, et al. Signaling dependent and independent mechanisms in pemphigus vulgaris blister formation. PLoS One. 2012;7:e50696.
Grando SA. Pemphigus autoimmunity: hypotheses and realities. Autoimmunity. 2012;45:7–35.
Bystryn J-C, Grando SA. A novel explanation for acantholysis in pemphigus vulgaris: the basal cell shrinkage hypothesis. J Am Acad Dermatol. 2006;54:513–6.
Rock B, Labib RS, Diaz LA. Monovalent Fab’ immunoglobulin fragments from endemic pemphigus foliaceus autoantibodies reproduce the human disease in neonatal Balb/c mice. J Clin Investig. 1990;85:296–9.
Yamagami J, Payne AS, Kacir S, Ishii K, Siegel DL, Stanley JR. Homologous regions of autoantibody heavy chain complementarity-determining region 3 (H-CDR3) in patients with pemphigus cause pathogenicity. J Clin Investig. 2010;120:4111–7.
Laskaris G, Sklavounou A, Stratigos J. Bullous pemphigoid, cicatricial pemphigoid, and pemphigus vulgaris: a comparative clinical survey of 278 cases. Oral Surg Oral Med Oral Pathol. 1982;54:656–62.
Ahmed AR, Blose DA. Pemphigus vegetans: Neumann type and Hallopeau type. Int J Dermatol. 1984;23:135–41.
Anhalt GJ, Kim SC, Stanley JR, Korman NJ, Jabs DA, Kory M, et al. Paraneoplastic pemphigus: an autoimmune mucocutaneous disease associated with neoplasia. N Engl J Med. 1990;323:1729–35.
Amber KT, Valdebran M, Grando SA. Paraneoplastic autoimmune multiorgan syndrome (PAMS): beyond the single phenotype of paraneoplastic pemphigus. Autoimmun Rev. 2018;17:1002–10.
Ohzono A, Sogame R, Li X, Teye K, Tsuchisaka A, Numata S, et al. Clinical and immunological findings in 104 cases of paraneoplastic pemphigus. Br J Dermatol. 2015;173:1447–52.
Czernik A, Camilleri M, Pittelkow MR, Grando SA. Paraneoplastic autoimmune multiorgan syndrome: 20 years after. Int J Dermatol. 2011;50(8):905–14.
Nousari HC, Kimyai-Asadi A, Anhalt GJ. Elevated serum levels of interleukin-6 in paraneoplastic pemphigus. J Investig Dermatol. 1999;112(3):396–8.
Nguyen VT, Ndoye A, Bassler KD, Shultz LD, Shields MC, Ruben BS, et al. Classification, clinical manifestations, and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome: a reappraisal of paraneoplastic pemphigus. Arch Dermatol. 2001;137:193–206.
Leger S, Picard D, Ingen-Housz-Oro S, Arnault JP, Aubin F, Carsuzaa F, et al. Prognostic factors of paraneoplastic pemphigus. Arch Dermatol. 2012;148:1165–72.
Lee J, Bloom R, Amber KT. A systematic review of patients with mucocutaneous and respiratory complications in paraneoplastic autoimmune Mmltiorgan syndrome: Castleman’s disease is the predominant malignancy. Lung. 2015;193(4):593–6.
Liu Q, Bu D-F, Li D, Zhu X-J. Genotyping of HLA-I and HLA-II alleles in Chinese patients with paraneoplastic pemphigus. Br J Dermatol. 2008;158:587–91.
Martel P, Loiseau P, Joly P, Busson M, Lepage V, Mouquet H, et al. Paraneoplastic pemphigus is associated with the DRB1*03 allele. J Autoimmun. 2003;20(1):91–5.
Zhu X, Zhang B. Paraneoplastic pemphigus. J Dermatol. 2007;34(8):503–11.
Reich K, Brinck U, Letschert M, Blaschke V, Dames K, Braess J, et al. Graft-versus-host disease-like immunophenotype and apoptotic keratinocyte death in paraneoplastic pemphigus. Br J Dermatol. 1999;141(4):739–46.
Wang L, Bu D, Yang Y, Chen X, Zhu X. Mechanisms of disease Castleman’s tumours and production of autoantibody in paraneoplastic pemphigus. Lancet. 2004;363(9408):525–31.
Vezzoli P, Berti E, Marzano AV. Rationale and efficacy for the use of rituximab in paraneoplastic pemphigus. Expert Rev Clin Immunol. 2008;4(3):351–63.
Tsuchisaka A, Numata S, Teye K, Natsuaki Y, Kawakami T, Takeda Y, et al. Epiplakin is a paraneoplastic pemphigus autoantigen and related to bronchiolitis obliterans in Japanese patients. J Investig Dermatol. 2016;136(2):399–408.
Poot AM, Diercks GFH, Kramer D, Schepens I, Klunder G, Hashimoto T, et al. Laboratory diagnosis of paraneoplastic pemphigus. Br J Dermatol. 2013;169:1016–24.
Numata S, Teye K, Tsuruta D, Sogame R, Ishii N, Koga H, et al. Anti-alpha-2-macroglobulin-like-1 autoantibodies are detected frequently and may be pathogenic in paraneoplastic pemphigus. J Investig Dermatol. 2013;133:1785–93.
Lambert J, Bracke S, Van Roy F, Pas HH, Bonné S, De Schepper S. Serum plakophilin-3 autoreactivity in paraneoplastic pemphigus. Br J Dermatol. 2010;163:630–2.
Wang R, Li J, Wang M, Hao H, Chen X, Li R, et al. Prevalence of myasthenia gravis and associated autoantibodies in paraneoplastic pemphigus and their correlations with symptoms and prognosis. Br J Dermatol. 2015;172:968–75.
Futei Y, Amagai M, Hashimoto T, Nishikawa T. Conformational epitope mapping and IgG subclass distribution of desmoglein 3 in paraneoplastic pemphigus. J Am Acad Dermatol. 2003;49(6):1023–8.
Silva JA, de Mesquita KC, Igreja AC, Lucas IC, Freitas AF, Oliveira SM, et al. Paraneoplastic cutaneous manifestations: concepts and updates. An Bras Dermatol. 2013;88(1):9–22.
Broussard KC, Leung TG, Moradi A, Thorne JE, Fine J. Autoimmune bullous diseases with skin and eye involvement: cicatricial pemphigoid, pemphigus vulgaris, and pemphigus paraneoplastica. Clin Dermatol. 2016;34:205–13.
Sehgal VN, Srivastava G. Paraneoplastic pemphigus/paraneoplastic autoimmune multiorgan syndrome. Int J Dermatol. 2009;48(2):162–9.
Nguyen VT, Ndoye A, Basseler KD, Shultz LD, Shields MC, Ruben BS, et al. Classification, clinical manifestations, and immunopathological mechanisms of the epithelial variant of paraneoplastic autoimmune multiorgan syndrome: a reappraisal of paraneoplastic pemphigus. Arch Dermatol. 2001;137(2):193–206.
Hashimoto T, Ohzono A, Ishii N. “Reply to: clinical and immunological findings in 104 cases of paraneoplastic pemphigus”: reply from the authors. Br J Dermatol. 2016;174:461–2.
Siu A, Landon K, Ramos DM. Differential diagnosis and management of oral ulcers. Semin Cutan Med Surg. 2015;34:171–7.
Kardaun SH, Jonkman MF. Stevens-Johnson syndrome/toxic epidermal necrolysis and erythema exsudativum multiforme. Autoimmune bullous diseases. London: Springer; 2016. p. 183–93.
Nikolskiy P. Trois nouveaux cas de pemphigus foliacé étudiés au point de vue de la symptomatologie. Ann Derm Syph. 1898;13:1026–31.
van Gijn J, Gijselhart JP. Nikolsky and his sign. Ned Tijdschr Geneeskd. 2011;155:A2846.
Uzun S, Durdu M. The specificity and sensitivity of Nikolskiy sign in the diagnosis of pemphigus. J Am Acad Dermatol. 2006;54:411–5.
Mignogna MD, Fortuna G, Leuci S, Ruoppo E, Marasca F, Matarasso S. Nikolsky’s sign on the gingival mucosa: a clinical tool for oral health practitioners. J Periodontol. 2008;79:2241–6.
Fortuna G, Marinkovich MP. Linear immunoglobulin A bullous dermatosis. Clin Dermatol. 2012;30:38–50.
Rose C, Weyers W, Denisjuk N, Hillen U, Zillikens D, Shimanovich I. Histopathology of anti-p200 pemphigoid. Am J Dermatopathol. 2007;29:119–24.
Hrabovska Z, Jautova J, Hrabovsky V. A study of clinical, histopathological and direct immunofluorescence diagnosis in pemphigus group: utility of direct immunofluorescence. Bratisl Lek Listy. 2017;118:243–9.
Horn TD, Anhalt GJ. Histologic features of paraneoplastic pemphigus. Arch Dermatol. 1992;128(8):1091–5.
Jordon RE, Beutner EH, Witebsky E, Blumental G, Hale WL, Lever WF. Basement zone antibodies in bullous pemphigoid. JAMA. 1967;200:751–6.
Giurdanella F, Diercks GFH, Jonkman MF, Pas HH. Laboratory diagnosis of pemphigus: direct immunofluorescence remains the gold standard. Br J Dermatol. 2016;175(1):185–6.
Meijer JM, Diercks GFH, de Lang EWG, Pas HH, Jonkman MF. Assessment of diagnostic strategy for early recognition of bullous and nonbullous variants of pemphigoid. JAMA Dermatol. 2019. https://doi.org/10.1001/jamadermatol.2018.4390.
Diercks GF, Pas HH, Jonkman MF. Immunofluorescence of autoimmune bullous diseases. Surg Pathol Clin. 2017;10(2):505–12.
Jonkman MF, Diercks GFH. How to take a biopsy. Autoimmune bullous diseases. London: Springer; 2016. p. 33–7.
Kamaguchi M, Iwata H, Ujiie I, Ujiie H, Sato J, Kitagawa Y, et al. Direct immunofluorescence using non-lesional buccal mucosa in mucous membrane pemphigoid. Front Med (Lausanne). 2018;5:20.
Carey B, Joshi S, Abdelghani A, Mee J, Andiappan M, Setterfield J. The optimal oral biopsy site for diagnosis of mucous membrane pemphigoid and pemphigus vulgaris. Br J Dermatol. 2019. https://doi.org/10.1111/bjd.18032.
Vodegel RM, de Jong MCJM, Meijer HJ, Weytingh MB, Pas HH, Jonkman MF. Enhanced diagnostic immunofluorescence using biopsies transported in saline. BMC Dermatol. 2004;4:10.
Meijer JM, Atefi I, Diercks GFH, Vorobyev A, Zuiderveen J, Meijer HJ, et al. Serration pattern analysis for differentiating epidermolysis bullosa acquisita from other pemphigoid diseases. J Am Acad Dermatol. 2018;78(754–9):e6.
Thorne JE, Anhalt GJ, Jabs DA. Mucous membrane pemphigoid and pseudopemphigoid. Ophthalmology. 2004;111:45–52.
Shimanovich I, Nitz JM, Zillikens D. Multiple and repeated sampling increases the sensitivity of direct immunofluorescence testing for the diagnosis of mucous membrane pemphigoid. J Am Acad Dermatol. 2017;77(700–5):e3.
Bean SF. Cicatricial pemphigoid: immunofluorescent studies. Arch Dermatol. 1974;110:552–5.
Rogers RS, Perry HO, Bean SF, Jordon RE. Immunopathology of cicatricial pemphigoid: studies of complement deposition. J Investig Dermatol. 1977;68:39–43.
Rogers RS, Van Hale HM. Immunopathologic diagnosis of oral mucosal inflammatory diseases. Australas J Dermatol. 1986;27:51–7.
Terra JB, Jonkman MF, Diercks GFH, Pas HH. Low sensitivity of type VII collagen enzyme-linked immunosorbent assay in epidermolysis bullosa acquisita: serration pattern analysis on skin biopsy is required for diagnosis. Br J Dermatol. 2013;169:164–7.
Vodegel RM, De Jong MCJM, Pas HH, Jonkman MF. IgA-mediated epidermolysis bullosa acquisita: two cases and review of the literature. J Am Acad Dermatol. 2002;47:919–25.
Joly P, Richard C, Gilbert D, Chosidow O, Roujeau JC, et al. Sensitivity and specificity of clinical, histologic, and immunologic features in the diagnosis of paraneoplastic pemphigus. J Am Acad Dermatol. 2000;43(4):619–26.
Poot AM, Siland J, Jonkman MF, Pas HH, Diercks GFH. Direct and indirect immunofluorescence staining patterns in the diagnosis of paraneoplastic pemphigus. Br J Dermatol. 2016;174:912–5.
Feibelman C, Stolzner G, Provost TT. Pemphigus vulgaris: superior sensitivity of monkey esophagus in the determination of pemphigus antibody. Arch Dermatol. 1981;117:561–2.
Pas H. Immunoassays. In: Jonkman M, editor. Autoimmune bullous diseases. London: Springer; 2016. p. 57–62.
Pas HH. Immunoblot assay in differential diagnosis of autoimmune blistering skin diseases. Clin Dermatol. 2001;19:622–30.
Izumi K, Nishie W, Mai Y, Ujiie H, Iwata H, Natsuga K, et al. Detection of mucous membrane pemphigoid autoantibodies by full-length BP180 enzyme-linked immunosorbent assay. J Dermatol Sci. 2017;88:247–8.
Charneux J, Lorin J, Vitry F, Antonicelli F, Reguiai Z, Barbe C, et al. Usefulness of BP230 and BP180-NC16a enzyme-linked immunosorbent assays in the initial diagnosis of bullous pemphigoid: a retrospective study of 138 patients. Arch Dermatol. 2011;147:286–91.
Schmidt E, Dahnrich C, Rosemann A, Probst C, Komorowski L, Saschenbrecker S, et al. Novel ELISA systems for antibodies to desmoglein 1 and 3: correlation of disease activity with serum autoantibody levels in individual pemphigus patients. Exp Dermatol. 2010;19:458–63.
Abasq C, Mouquet H, Gilbert D, Tron F, Grassi V, Musette P, et al. ELISA testing of anti-desmoglein 1 and 3 antibodies in the management of pemphigus. Arch Dermatol. 2009;145:529–35.
Ishii K, Harada R, Matsuo I, Shirakata Y, Hashimoto K, Amagai M. In vitro keratinocyte dissociation assay for evaluation of the pathogenicity of anti-desmoglein 3 IgG autoantibodies in pemphigus vulgaris. J Investig Dermatol. 2005;124:939–46.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
Barbara Horvath has received grants from AbbVIe, Janssen-Cilag, Solenne B.V., and Celgene; consulting fees from AbbVie, Janssen-Cilag, Novartis, UCB Pharma, Akari Pharmaceutics, Philips, and Roche and support for travel from AbbVie, Janssen-Cilag, and Novartis. Hanan Rashid, Aniek Lamberts, Gilles F.H. Diercks, Hendri H. Pas, Joost M. Meijer, and Maria C. Bollin have no conflicts of interest that are directly relevant to the content of this article.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rashid, H., Lamberts, A., Diercks, G.F.H. et al. Oral Lesions in Autoimmune Bullous Diseases: An Overview of Clinical Characteristics and Diagnostic Algorithm. Am J Clin Dermatol 20, 847–861 (2019). https://doi.org/10.1007/s40257-019-00461-7
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-019-00461-7