
Abstract
Morphea, also known as localized scleroderma, encompasses a group of idiopathic sclerotic skin diseases. The spectrum ranges from relatively mild phenotypes, which generally cause few problems besides local discomfort and visible disfigurement, to subtypes with severe complications such as joint contractures and limb length discrepancies. Eosinophilic fasciitis (EF, Shulman syndrome) is often regarded as belonging to the severe end of the morphea spectrum. The exact driving mechanisms behind morphea and EF pathogenesis remain to be elucidated. However, extensive extracellular matrix formation and autoimmune dysfunction are thought to be key pathogenic processes. Likewise, these processes are considered essential in systemic sclerosis (SSc) pathogenesis. In addition, similarities in clinical presentation between morphea and SSc have led to many theories about their relatedness. Importantly, morphea may be differentiated from SSc based on absence of sclerodactyly, Raynaud’s phenomenon, and nailfold capillary changes. The diagnosis of morphea is often based on characteristic clinical findings. Histopathological evaluation of skin biopsies and laboratory tests are not necessary in the majority of morphea cases. However, full-thickness skin biopsies, containing fascia and muscle tissue, are required for the diagnosis of EF. Monitoring of disease activity and damage, especially of subcutaneous involvement, is one of the most challenging aspects of morphea care. Therefore, data harmonization is crucial for optimizing standard care and for comparability of study results. Recently, the localized scleroderma cutaneous assessment tool (LoSCAT) has been developed and validated for morphea. The LoSCAT is currently the most widely reported outcome measure for morphea. Care providers should take disease subtype, degree of activity, depth of involvement, and quality-of-life impairments into account when initiating treatment. In most patients with circumscribed superficial subtypes, treatment with topical therapies suffices. In more widespread disease, UVA1 phototherapy or systemic treatment with methotrexate (MTX), with or without a systemic corticosteroid combination, should be initiated. Disappointingly, few alternatives for MTX have been described and additional research is still needed to optimize treatment for these debilitating conditions. In this review, we present a state-of-the-art flow chart that guides care providers in the treatment of morphea and EF.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Progress has been made in elucidating the immunological pathways involved in morphea. |
Disease monitoring by reliable and sensitive outcome measures is improved by the localized scleroderma cutaneous assessment tool (LoSCAT); however, especially for deep involvement, additional validated outcome measures are required. |
This review provides two state-of-the-art algorithms that guide care providers with regard to (i) diagnostic work-up and disease monitoring, and (ii) treatment of morphea and eosinophilic fasciitis. |
1 Introduction
Morphea, also known as localized scleroderma, encompasses a group of idiopathic sclerotic skin diseases. Controversy exists with regard to nomenclature of the disease spectrum. In some countries, localized scleroderma is the preferred overarching term, because morphea is regarded as one of the subtypes of the wider disease spectrum. However, morphea is the preferred term in the United States (US), because localized scleroderma could lead to unwanted confusion with the term systemic sclerosis, a systemic disorder with a different plethora of clinical and pathological signs and symptoms [1]. For the purpose of this review, we will use the term morphea.
The spectrum of morphea consists of heterogeneous disease phenotypes. Solitary sclerotic lesions, which generally cause few problems besides local discomfort and visible disfigurement, reflect the mild side of the spectrum [2, 3]. Conversely, sclerosis can cause severe complications in the linear subtype; limb length discrepancies and joint contractures may occur [1, 4, 5].
This review encompasses a description of the clinical aspects of the morphea subtypes. Eosinophilic fasciitis (EF, also known as Shulman Syndrome), often regarded as part of the morphea spectrum, is included in this review. Additionally, we describe recent developments in understanding of disease pathogenesis and potential outcome measures. Lastly, we present two state-of-the-art flow charts, which guide care providers with regard to (i) diagnostic work-up and disease monitoring and (ii) treatment of morphea and EF.
2 Epidemiology, Classification and Presentation
2.1 Epidemiology
The rarity of morphea is reflected in the annual incidence rates which are reported to be between 3.4 and 27 cases per 1,000,000 [6–8]. Females are more frequently affected than males (ratio: 2.4–5.0 to 1) [1, 9–11]. The peak incidence is bimodal with peaks between 7 and 11 years for pediatric-onset disease [1, 10–13] and 44–47 years for adult-onset disease [10, 12]. The incidence and prevalence of EF is unknown. The disease predominantly affects patients in their fourth and fifth decade of life [14–16]. Only a few case reports describe childhood-onset disease [11, 14, 15, 17–21].
2.2 Classification
Multiple classification schemes have been proposed throughout the years. However, consensus with regard to one superior classification system is currently lacking. Classification by Laxer and Zulian [22] describes the following five subtypes: (i) circumscribed morphea (including a superficial and deep variant), (ii) linear morphea (including a limb/trunk variant and head variant), (iii) generalized morphea, (iv) the pansclerotic subtype, and (v) the mixed subtype (Table 1). Other classification systems include more uncommon subtypes such as guttate and bullous morphea [23, 24]. Secondly, these systems include diagnoses which are debated to be part of the morphea spectrum, such as atrophoderma of Pasini and Pierini, extragenital lichen sclerosis et atrophicus, and EF. In the authors’ opinion, EF belongs to the spectrum of morphea, based on the fact that the largest case series reported concomitant morphea in 29–40% of the patients [14–16].
2.3 Clinical Characteristics
2.3.1 General Findings
Early, progressive morphea is characterized by erythematous or violaceous cutaneous lesions. As the disease progresses, sclerotic plaques develop at the center of these lesions. This leads to the appearance of a yellow-white sclerotic plaque with an erythematous or violaceous border, the so-called ‘lilac ring’ (Fig. 1). In some patients, development of hyperpigmentation is the predominant feature and sclerosis can be limited or absent (Fig. 2). In the majority of patients, skin softens in months to years. Consecutively, signs of residual damage, such as pigment alterations and cutaneous and subcutaneous atrophy, may develop. In most patients, the disease is self-limiting within 3–5 years. However, in some patients morphea remains progressive for multiple years or flares of disease occur frequently [1, 10, 25].

Circumscribed superficial morphea (morphea en plaque). A yellow-white sclerotic plaque with an erythematous or violaceous border, the characteristic ‘lilac ring’

Generalized morphea. Example of a patient with absence of induration, but extensive hyperpigmentation
2.3.2 Circumscribed Superficial Morphea
In circumscribed superficial morphea, also known as ‘morphea en plaque’, involvement is limited to the dermis [9, 10, 13]. Solitary or few plaques predominantly affect the trunk, waist and submammary region. Circumscribed superficial morphea is the most common subtype in adults and generally causes few problems besides local discomfort and visible disfigurement.
2.3.3 Circumscribed Deep Morphea
In circumscribed deep morphea (morphea profunda), sclerosis reaching into the subcutis is present and may extend into the fascia and muscle. Deep morphea is a rare subtype in both the adult and pediatric populations (~5%) [1, 9, 10]. Lower extremities are often affected symmetrically, where sclerosis might cause contractures and lead to subcutaneous atrophy.
2.3.4 Linear Morphea
In linear morphea, sclerosis may be limited to the dermis, but deeper involvement is often present. The ‘band-like’ cutaneous sclerosis (Fig. 3) frequently causes contractures [1, 4]. Additionally, limb-length discrepancies may occur [1] (Fig. 4). In the majority of patients, the disease remains unilateral. Linear morphea is the most common subtype in childhood-onset morphea (~65%) [11], but disease onset might occur during adulthood as well [4, 10]. Linear morphea of the limbs and trunk is characterized by a chronic disease course, as the disease may remain active after many decennia [26, 27] and recurrences are reported in a large proportion of both adults and children [4, 10, 25, 28, 29].

Linear morphea of the upper extremity

Linear morphea of the lower extremity. Limb length discrepancy and subcutaneous atrophy in burned-out disease (left leg)
The ‘en coup de sabre’ (ECDS) subtype most frequently affects the paramedian forehead. Linear lesions may extent onto the scalp, where they cause alopecia (Fig. 5). This subtype might be accompanied by ocular [30, 31], neurological [1, 11, 32] and odontostomatologic [33–35] complications. Progressive hemifacial atrophy (Parry–Romberg syndrome) is characterized by diffuse unilateral subcutaneous atrophy of the face. This subtype is often regarded part of a spectrum with ECDS because 71% of the patients have overlaying cutaneous sclerosis [32, 36]. However, studies that investigate the connection between these two conditions are lacking.

Linear morphea on the paramedian forehead (en coup de sabre) crossing onto the scalp causing alopecia
2.3.5 Generalized Morphea
Generalized morphea is characterized by large superficial coalesced plaques on multiple body sites (Fig. 6). Generalized morphea is more frequently present in adults than children [9, 10, 13]. Sclerosis is usually present on the trunk, arms, and legs with sparing of the face, hands, and feet.

Severe case of generalized morphea on the trunk with atrophy of both breasts
2.3.6 Eosinophilic Fasciitis (EF)
The onset of EF is characterized by acute or subacute development of pitting edema and erythema. As the disease progresses, edema is gradually replaced by a ‘peau d’orange’ aspect as deep sclerosis starts to develop (Fig. 7). Extremities are affected symmetrically, with exclusion of hands and feet. Neck and truncal involvement is reported in widespread disease. Cutaneous involvement may be accompanied by general symptoms such as weight loss, myalgia and asthenia [14, 15, 21].

Peau d’orange aspect on upper extremity in a patient with eosinophilic fasciitis
3 Histopathology
3.1 Skin Biopsy in Morphea
Histopathological characteristics correlate with the clinical state of morphea. Evaluation of early active morphea, represented by a biopsy of the inflammatory border, reveals a perivascular infiltrate composed of lymphocytes and plasma cells, possibly accompanied by eosinophils and macrophages. Evaluation of sclerotic skin demonstrates thickened and homogenized collagen bundles at the papillary and reticular dermis. The abundant collagen bundles enclose eccrine glands and few dermal blood vessels with fibrotic walls and narrow lumina may be observed as the sclerosis progresses. Subcutaneous infiltration and sclerosis reflect deeper involvement.
We recommend a diagnostic skin biopsy only in case of an unclear clinical presentation of morphea. The clinical appearance of the biopsy site (inflammatory border versus sclerotic center) should be mentioned to the pathologists for optimal clinicopathological correlation.
3.2 Full-Thickness Biopsy in EF
A full-thickness biopsy, containing fascia and muscle, is the golden standard for the diagnosis of EF. Histology typically displays a thickened fascia infiltrated by lymphocytes accompanied by eosinophils, plasma cells, and macrophages [14, 15]. The presence of eosinophils is transient and may be absent if patients have prolonged disease or receive systemic corticosteroids (SCSs) or immunosuppressive drugs (ISDs). Adjacent myositis is frequently observed [37]. The presence of thickened dermal collagen fibers may reflect the presence of concomitant superficial morphea [14, 15]. More recently, magnetic resonance imaging (MRI) [38–40], ultrasound [41, 42], and positron emission tomography (PET) [43–45] have been reported to be helpful in establishing the diagnosis by decreasing the likelihood of sampling errors for deep biopsies or by visualizing the fasciitis.
4 Pathogenesis
4.1 Pathological Hallmarks of Morphea
The exact driving mechanisms behind morphea pathogenesis remain to be elucidated. However, extensive extracellular matrix (collagen) formation and autoimmune dysfunction are thought to be key pathogenic processes [46–48]. Likewise, these processes are considered essential in systemic sclerosis (SSc) pathogenesis. The similarities between the two diseases have led to many theories about their relatedness. Based on this possible connection, most theories for morphea pathogenesis are deduced from SSc. Endothelial dysfunction, immune dysregulation, and excessive collagen deposition are regarded as key hallmarks for SSc pathogenesis [49, 50]. In recent years, research in SSc has led to some insight into these hallmarks and their inter-relationship. The evidence and relevance for these hallmarks in morphea pathogenesis are described in the following sections.
4.1.1 Vascular Dysfunction
For SSc, endothelial dysfunction is clinically reflected in the presence of Raynaud’s phenomenon and visible changes on capillaroscopy (nailfold lesions) [51]. No such clinicopathological correlation exists for morphea. However, the propensity of pericyte hyperplasia in capillar and venular walls and increased capillary density in active morphea lesions have been reported [52].
4.1.2 Immune Dysregulation
4.1.2.1 Autoimmunity
Autoimmunity is likely involved in the pathogenesis because of the presence of autoantibodies in a proportion of the morphea patients [9, 11, 53–58]. Secondly, the presence of concomitant autoimmune diseases in morphea patients and their relatives supports involvement of autoimmunity [5, 11, 13, 59]. Recently, a study including 211 morphea patients and 726 matched controls investigated HLA class I and II typing. The strongest associations with morphea were found with the HLA Class II allele DRB1*04:04 and class I allele HLA–B*37. Comparison of the risk alleles for the morphea cohort versus other autoimmune diseases revealed only one allele in common between morphea and SSc. However, many more alleles showed similarity with other autoimmune diseases such as rheumatoid arthritis [60].
4.1.2.2 Cytokines and Chemokines
Increased serum levels of the adhesion molecules Vascular Cell Adhesion Molecule-1 (VCAM-1), Intracellular Cell Adhesion Molecule-1 (ICAM-1) and E-Selectin have been reported in morphea patients [61, 62]. These molecules might be involved in early recruitment of inflammatory cells such as T cells, monocytes, and other immune cells.
Initial studies demonstrated elevation of classic profibrotic T helper cell 2 (Th2) cytokines IL-4 and IL-13 (and IL-2, 6, and 8) in serum of 48 morphea patients. Likewise, IL-4 and IL-13 elevation has been demonstrated in circulation, skin, and lung tissue of SSc patients [63–65]. Recently, a cross-sectional study performed a 29-plex Luminex, which included Th1, Th2 and Th17 cytokines and chemokines on plasma of 69 pediatric morphea patients and 71 controls. Elevation of interferon-γ (IFN-γ)-induced protein 10 (IP-10, CXCL10), IL-12p70 and IFN-γ suggested a proinflammatory Th1 predominance, whereas IL-17a elevation signified a proinflammatory Th17 predominance. Elevation of these proinflammatory Th1 and Th17 cytokines and chemokines correlated with shorter disease duration. In contrast to the Th1/17 predominance, Th2 cytokines IL-4, IL-5, and IL-13 were not significantly elevated [66]. IL-17a gene upregulation has also been reported in peripheral blood mononuclear and skin samples from morphea patients. This study confirmed the correlation between IL-17a elevation and shorter disease duration [67]. Tumor necrosis factor-α (TNF-α), an effector cytokine affiliated with the Th1 and Th17 lineage was shown to be increased in active disease versus inactive disease [66]. The association of TNF-α levels and disease activity was also shown in a previous study [68].
In conclusion, it is unclear whether a Th1/17 or Th2 predominance is present in morphea. One postulated theory is Th1 and Th17 predominance in early, active disease and Th2 predominance in the fibrotic phase of the disease.
4.1.3 Excessive Extracellular Matrix Formation
Development of cutaneous or subcutaneous fibrosis is the key characteristic of morphea. Fibrosis is a result of excessive collagen synthesis and decreased degradation. Transforming growth factor-β1 (TGF-β1) has been shown to increase the expression of several collagen types and other extracellular matrix components in morphea and SSc [69–71]. Additionally, TGF-β1 has inhibitory effects on matrix degradation. Therefore, TGF-β is regarded a key player in morphea and SSc pathogenesis. However, the processes leading to excessive TGF-β synthesis in morphea remain poorly understood. Recent reports in SSc showed TGF-β-dependent fibrosis development via toll-like receptor (TLR) signaling [72–75]. Most interestingly, some of these reports demonstrated endogenous ligands (mitochondrial DNA [72], fibronectin EDA (extra domain-A) [73] and tenascin-C [75]) for these TLRs. These findings integrate innate immunity and fibrosis development. The role of the innate immune system, via TLR signaling, in fibrogenesis remains to be investigated in morphea. However, a previous report of tenascin upregulation in morphea skin samples might hint towards involvement of the innate immune system in morphea pathogenesis [76].
4.2 Epigenetics
Low concordance rates of autoimmune diseases in monozygotic twins suggest additional pathogenic mechanisms besides genetic factors [77]. The field of epigenetics investigates heritable changes that influence gene expression without altering the DNA sequence; these changes include DNA methylation, post-translational modification of histones and microRNAs (miRNAs) [78]. MiRNAs are short non-coding RNAs of 18–23 nucleotides that bind messenger RNAs (mRNAs) and thereby inhibit their translation or induce mRNA degradation. A study in 2013 was the first to report miRNA dysregulation in morphea. This study reported let-7a downregulation in morphea skin and circulation. Additionally, experimental let-7a overexpression, or inhibition, affected protein expression of type I collagen [79]. A second study reported miRNA-196a downregulation in serum and involved skin of morphea patients. Transfection of an miRNA-196a inhibitor into normal cultured fibroblasts upregulated type 1 collagen protein in vitro [80]. Lastly, a recent study reported upregulation of miRNA-155 in SSc and morphea skin. Most interestingly, this study showed the potential of miRNA as a therapeutic target by decreasing the dermal thickness, collagen deposition, and number of activated fibroblasts upon topical administration of a miRNA-155 blocking agent in a murine bleomycine-induced fibrosis model [81]. The potential of miRNA as a therapeutic option in humans is currently being investigated in a phase I trial with MRG-201, a molecule which mimics miRNA-29 activity (clinicaltrials.gov NCT02603224).
4.3 Environmental Factors
Multiple environmental factors have been proposed to be involved in morphea development. The role of friction, long-term pressure, or mechanical factors such as vaccination or other injections is repeatedly reported in 13–16% of morphea patients [1, 11, 82]. Secondly, morphea is an uncommon complication after radiation therapy. A recent literature review summarized 66 cases with radiation-induced morphea (RIM) [83]. Radiation may lead to secretion of Th2 cytokines (Il-4 and -5), which leads to TGF-β-mediated fibrogenesis.
For multiple decades, studies have reported inconsistent results with regard to the association of Borrelia burgdorferi (B. Burgdorferi) and morphea. Results vary from detection of B. burgdorferi in case reports and large proportions of patients in case series [84–87], to complete absence of detectable B. burgdorferi in other studies [88–97]. The different techniques being reported, in combination with different borrelia strains present in different geographical locations, makes it difficult to come to a conclusion for a large group of morphea patients. In the authors’ opinion, it is unlikely that B. burgdorferi is involved in morphea pathogenesis for a large proportion of patients.
Interestingly, recent case reports almost exclusively report TNF-α inhibitors in drug-induced morphea [98–101]. Conversely, two cases have described beneficial effects of infliximab in severe morphea cases [102, 103]. Moreover, elevation of TNF-α in circulation in active morphea endorses the role of this cytokine in the development of morphea [66, 68]. However, the exact role of TNF-α still remains to be elucidated. In addition to TNF-α inhibitors, multiple other drugs and injections have been implicated in cases of drug-induced morphea. A review including 15 cases of drug-induced morphea concluded that drug-induced morphea was extremely rare, drug withdrawal did not lead to remission in most patients, and some of the drugs could be directly linked to connective tissue metabolism [104].
4.4 Pathogenesis of EF
The pathogenesis of EF remains unknown. Numerous associations with potential etiological factors have been proposed in case reports. Most of these factors have also been reported in association with morphea (i.e., B. burgdorferi infection [105], radiation therapy [106], and insect bites [107]). The most commonly reported etiological factor for EF is strenuous exercise or trauma preceding disease onset. Two case series, consisting of 52 [15] and 63 patients [16], reported exercise to be suggestive for disease induction in 46 and 28% of the patients, respectively. However, the mechanisms leading to clinical phenotype remain unknown.
5 Laboratory Testing
5.1 Autoantibodies
To date, no morphea-specific autoantibodies have been discovered. This is in contrast to SSc, for which multiple specific autoantibodies (anticentromere, anti-topoisomerase 1 and anti-RNA polyisomerase III antibodies) have been reported and are routinely being screened in standard care [108].
Antinuclear antibodies (ANAs) have been reported in 5.9–68% of morphea patients [9, 11, 13, 54–56, 58]. Previous retrospective studies reported increased ANA frequencies in severe subtypes such as linear and generalized morphea [13, 54]. Recently, a prospective study with 187 morphea patients could not confirm differences in ANA prevalence between the different subtypes. This study reported ANAs to be present in 34% of the complete group, with ANA prevalences only varying between 33 and 36% in the different disease subtypes [53]. Additionally, this study showed single-stranded DNA antibodies (ssDNA abs) only to be present in 8% of the complete group and 13% in the linear subtype, versus 7% in controls. Retrospective studies reported ssDNA abs in 29–39% of the linear subtype [54, 55]. Antihistone antibodies (AHAs) are consistently reported to be increased in the linear subtype (18–42%) [53, 54, 56], whereas conflicting results exist with regard to AHA and the generalized subtype [53, 56]. Lastly, antibodies to cardiolipin [11, 109–111], phospholipid [110], U1 ribonucleoproteins [112], U3 ribonucleoproteins [113], MMP-1 [114], and antinucleosome [58] have been described in morphea.
For morphea, autoantibodies remain of limited clinical use; no specific autoantibodies have been identified and no associations have been reported between any of the previously reported antibodies or titers and clinical activity or severity measures in the complete group of morphea subtypes.
In conclusion, we do not recommend testing of autoantibodies for diagnostic and disease monitoring purposes.
5.2 Borrelia burgdorferi
Section 4.3 encompasses a more detailed description of the role of borrelia in morphea development. Based on evidence, serological testing may be considered in atypical presentation of morphea.
5.3 Other Laboratory Tests
Inflammatory serological markers are upregulated in a minority of morphea patients and do not correlate with disease activity [11]. Therefore, we do not recommend routine testing of inflammatory serological markers.
5.4 Laboratory Tests for EF
Peripheral eosinophilia and elevation of inflammatory markers may be present in the early stages of EF. However, these findings are transient and may be absent in later stages of the disease or when patients receive SCS or ISD treatment. We recommend routine testing of absolute eosinophil counts and inflammatory markers in the initial phase of the disease and for the detection of disease reactivation [14, 15]. Autoantibodies may be detected in 15–20% of patients, whereas specific antibodies are absent in the majority of patients. Autoantibody testing may be useful in the differentiation versus SSc [14, 15]. In addition, anti-neutrophil cytoplasmic antibody (ANCA) testing can be performed in cases where eosinophilic granulomatosis with polyangiitis (EGPA) is considered as part of the differential diagnosis [115].
6 Outcome Measures
Validated outcome measures for disease activity and damage are essential for optimal standard of care. Secondly, data harmonization is crucial for comparability of study results. Recently, a comprehensive systematic review described tools for determining disease activity in morphea [116]. In the following sections, we describe the most promising outcome measures.
6.1 Clinical Scores
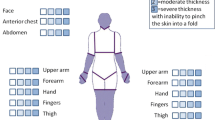
The localized scleroderma cutaneous assessment tool (LoSCAT) is a composite of the modified localized scleroderma severity index (mLoSSI) [117] and localized scleroderma skin damage index (LoSDI) [118]. The mLoSSI assesses signs of disease activity (expansion of disease, presence of erythema, and skin induration). The LoSDI scores cutaneous and subcutaneous atrophy, and pigment alterations; signs which reflect disease damage. In addition to the LoSCAT, physician global assessment of disease activity (PhysGA-A) and damage (PhysGA-D) have also been investigated in the development of the LoSCAT score [117, 118]. The Dyspigmentation, Induration, Erythema and Telangiectasia (DIET) score poorly discriminates between disease activity and disease damage; similar scores can reflect either active or inactive disease [119, 120]. Lastly, several one- or two-dimensional clinical scores, such as the modified Rodnan skin score [121–123] (mRSS), the modified skin score (mSS, by Zachariae) [124, 125] and the computerized skin score (CSS) [126] have been described. These scores share the downside of only capturing skin sclerosis as a sign of disease activity. Therefore, based on current literature, LoSCAT in combination with PhysGA-A and PhysGA-D are the recommended clinical outcome measures for superficial morphea assessment.
6.2 Miscellaneous Outcome Measures
Ultrasound is widely reported for the assessment of morphea activity [116]. However, the downside of ultrasound is the wide variability in equipment reported between studies. The Localized Scleroderma Clinical and Ultrasound Study Group (LOCUS) has developed a standardized ultrasound imaging protocol, which should lead to data harmonization [127]. Another technique commonly reported is infrared thermography (IRT). Multiple studies have demonstrated correlations between lesion temperature and clinical activity status of the morphea lesions [116]. Lastly, durometry is a tool used to measure material hardness. Originally, durometry was described as a reliable method for the assessment of morphea skin hardness [124]. Recently, a study with 23 pediatric patients demonstrated that durometry was able to discriminate between affected and unaffected skin and that it was sensitive to detect change in lesions [128]. The usefulness of durometry remains limited as lesions located at bony surfaces, such as scalp, and skin lesions are not suited to assessment with durometry [129, 130].
In conclusion, in superficial morphea, high frequency ultrasound and IRT are the most promising techniques for monitoring disease activity, in addition to the LoSCAT.
6.3 Outcome Measures for EF
To our knowledge, no outcome measures have been validated for EF. Therefore, we recommend the use of outcome measures known from SSc and morphea research. We recommend LoSCAT in combination with PhysGA-A and PhysGA-D, possibly accompanied by ultrasound and IRT, for the assessment of superficial involvement. Lastly, movement restrictions may be assessed by passive range of motion measurements.
7 Assessment of Musculoskeletal Involvement
Slow progression of musculoskeletal involvement may be notoriously difficult to measure between consecutive visits, but accumulated damage may be significant over a longer period of time. This emphasizes the necessity of additional imaging tools or biomarkers which reflect active deep disease.
7.1 Recommendations for Morphea
A retrospective study reported MRI findings in 43 morphea patients; MRI enabled confirmation of musculoskeletal abnormalities in all but one of the patients in whom musculoskeletal involvement was clinically suspected [131]. A recent retrospective study confirmed the potential of MRI in detection of musculoskeletal involvement [132]. More importantly, a longitudinal study reported MRI findings to be sensitive to changes in patients with deep morphea who were being treated with ISDs [133].
One study reported the additional value of electromyography (EMG) in detection of muscle dysfunction in patients with linear morphea [134]. Lastly, ultrasound has been reported in the monitoring of muscle involvement in deep morphea. However, the usefulness of this technique is highly operator-dependent [135]. In conclusion, the exact role of EMG and ultrasound requires further investigation. Based on the current evidence, we recommend MRI for monitoring of musculoskeletal involvement in morphea when indicated. However, we do not recommend routine MRI assessment.
7.2 Recommendations for EF
A retrospective study described serial MRI findings in six EF patients. Pretreatment MRI evaluation showed strong fascial enhancement, especially after intravenous (IV) administration of an extracellular gadolinium-based contrast agent. Post-therapy MRI evaluation during follow-up showed complete resolution of the characteristic MRI changes of the superficial and deep muscle fasciae in those patients who had complete clinical remission (n = 5) [40]. Another case series described MRI findings in six EF patients. Pretreatment MRI findings (n = 6) showed fascial thickening and hyperintense signal within the fascia in fluid-sensitive sequences. The post-therapy images (n = 3) showed marked improvement in two and mild but definite improvement in the other [136]. Case reports confirm the added value of MRI for both diagnostic purposes [137] and for the assessment of the fasciitis [39, 138]. In addition, ultrasound [41, 42] and positron emission tomography (PET) [43–45] have also been reported to be helpful for similar purposes. In conclusion, MRI may be considered for the assessment of EF.
8 Differential Diagnosis
8.1 Differentiation Diagnosis of Morphea
Evaluation of the differential diagnosis for the complete morphea spectrum is beyond the scope of this review and is described elsewhere [24]. Differentiation between morphea and SSc is commonly requested in an outpatient clinic. In the vast majority of patients, differentiation between the two diseases should be based on clinical findings and no additional testing is necessary in the absence of SSc suspicion.
Raynaud’s phenomenon or gastrointestinal problems are early signs of SSc and should therefore be checked in the patients’ history. Signs of SSc such as sclerodactyly, digital ulcers, pitting scars, puffy fingers, calcinosis cutis, telangiectasia, and diffuse facial sclerosis are rarely present in morphea and should be excluded by clinical examination.
Two studies investigated differentiating characteristics between morphea and SSc skin biopsies. These studies identified abundant cellular infiltrates in morphea compared with SSc, even in the sclerotic phase of morphea. However, most signs overlapped and differentiation remained difficult [139, 140]. Therefore, we do not recommend routine skin biopsies for the purpose of differentiation between morphea and SSc.
If SSc suspicion is present at any of the aforementioned steps, complete screening for SSc, including extensive laboratory testing, pulmonary imaging and functional testing, cardiac imaging, and nailfold capillaroscopy, is recommended [108].
8.2 Differential Diagnosis of EF
The initial presentation of EF is characterized by an acute or subacute onset of pitting edema, erythema, myalgia, and arthralgia, accompanied by elevated inflammatory markers [erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP)] and peripheral eosinophilia. After this initial inflammatory phase, sclerosis gradually starts to develop at affected body sites [15, 21]. The differential diagnosis of EF consists of morphea, SSc, scleroderma-like conditions, and miscellaneous diseases characterized by signs of inflammation and peripheral eosinophilia.
Differentiation between morphea and EF is mainly based on the characteristic distribution of cutaneous sclerosis in different morphea subtypes (i.e., linear morphea) [48]. In addition, histological demonstration of a fasciitis accompanied by eosinophils in the infiltrate support the diagnosis of EF [15]. However, histological differentiation with deep morphea, especially in the presence of widespread sclerosis, may be challenging. In such cases, clinical findings such as symmetrical distribution, a pronounced inflammatory phase, and detection of peripheral eosinophilia point towards EF.
Eosinophilic fasciitis and SSc are both characterized by initial inflammation, followed by cutaneous fibrosis. Facial and acral involvement are uncommon in EF and point toward SSc. In addition, presence of Raynaud’s phenomenon, abnormal nailfold capillaroscopy, specific autoantibodies, and internal organ involvement are absent in EF and frequently found in SSc. Lastly, histopathological demonstration of a fasciitis supports the diagnosis of EF [108].
Eosinophilia-myalgia syndrome (EMS) [141], caused by l-tryprophane ingestion, and toxic oil syndrome [142], are two examples of scleroderma-like syndromes which may be difficult to differentiate from EF, even by full-thickness biopsies. However, internal organ involvement differentiates the conditions from EF. Likewise, hypereosinophilic syndrome (HES) [143] and EGPA [144] are also characterized by organ involvement. In addition, the absence of cutaneous sclerosis in both HES and EGPA should aid differentiation with EF.
Lastly, nephrogenic systemic fibrosis should be suspected in patients with advanced renal failure, or a history of gadolinium administration. In addition to the patients’ history, involvement of the acra and absence of eosinophilia supports the diagnosis of nephrogenic systemic fibrosis.
9 Referral and Flow Chart
Figure 8 displays a flowchart which guides the care provider with regard to diagnoses and complication management.

Flow chart for considerations with regard to diagnosis and monitoring of morphea and eosinophilic fasciitis. EF eosinophilic fasciitis, EMG electromyography, MRI magnetic resonance imaging, SSc systemic sclerosis, US ultrasonography
9.1 Morphea ECDS and Progressive Facial Hemiatrophy (Parry–Romberg Syndrome)
Referral to an ophthalmologist should be considered to detect complications such as eyelid abnormalities, anterior uveitis, and episcleritis [30]. Odontostomatologic complications should be managed accordingly in cooperation with an oral maxillofacial surgeon and/or dentist [33–35]. Lastly, neurological manifestations consisting of seizures and headaches [145] may occur and referral to a neurologist could be considered to rule out central nervous system involvement [32, 146–150].
9.2 Eosinophilic Fasciitis
Eosinophilic fasciitis manifests secondary to malignancy in a minority of patients (5–10%). Association with hematological malignancy is more common than association with solid neoplasms [14, 15, 151]. Various screening modalities, such as laboratory testing, X-ray, CT or PET scan, ultrasound and endoscopy, may be employed, dependent on the patient risk profile and additional signs of malignancy.
9.3 Musculoskeletal Complications
Musculoskeletal complications (arthralgia, contractures, and arthritis) are reported in up to 40% of morphea and EF patients [1, 5, 15]. We recommend consultation of a rheumatologist in the presence of musculoskeletal complications.
10 Treatment
Care providers should take disease subtype, degree of activity, depth of involvement, and quality-of-life impairments into account when initiating treatment. In most patients with circumscribed superficial subtypes, treatment with topical therapies suffices. In widespread superficial disease, phototherapy or systemic treatment with methotrexate (MTX) should be initiated. For deep involvement, MTX is considered first-line treatment. The addition of SCS should be considered in case of severe disease, rapid progression, or (looming) contractures. Figure 9 shows an algorithm which guides care providers in treatment decisions for the different subtypes, including EF.

Flow chart for the management of morphea and eosinophilic fasciitis. * Topical Corticosteroids (TCS): moderately potent TCS once daily for 3 months. Highly potent TCS once daily for 1 month. ** Topical calcipotriol 0.005% ointment: once or twice daily, with or without occlusion. Possibly in combination with TCS. *** Topical tacrolimus 0.1% ointment: once or twice daily, with or without occlusion. Possibly in combination with TCS. † Phototherapy: preferably UVA1; suggested dose: 60 J/cm2 to a cumulative dose of 1460 J/cm2. If UVA1 is unavailable or impractical, alternative modalities are broadband UVA, PUVA or UVB.  Methotrexate (MTX): adult starting dose 15 mg/week, max dose 25 mg/week; pediatric starting dose 15 mg/m2, max dose 25 mg. Folic acid supplementation: 0.4–1 mg/day or 5–10 mg/week.
Methotrexate (MTX): adult starting dose 15 mg/week, max dose 25 mg/week; pediatric starting dose 15 mg/m2, max dose 25 mg. Folic acid supplementation: 0.4–1 mg/day or 5–10 mg/week.  Systemic corticosteroids (SCS): adult starting dose 0.5–1 mg/kg/day (max 60 mg) during a max of 3 months followed by tapering; pediatric dose: 1–2 mg/kg/day, max dose 60mg/day, followed by tapering.
Systemic corticosteroids (SCS): adult starting dose 0.5–1 mg/kg/day (max 60 mg) during a max of 3 months followed by tapering; pediatric dose: 1–2 mg/kg/day, max dose 60mg/day, followed by tapering.  Intravenous methylprednisolone (IVMP): adult dose 1000 mg/day for 3 days/month for 3–6 months, possibly followed by oral SCS. Pediatric dose 30 mg/kg/day for 3 days/month for 3 months, possibly followed by oral SCS. α Mycophenolate Mofetil (MMF, alternative to MTX): adult dose 1000 mg twice daily. Pediatric dose 600–1200 mg/m2/day twice daily. A Deep/linear subtypes: treatment with MTX monotherapy. Addition of SCS or IVMP in case of rapidly progressive disease or in the presence of (looming) contractures. B Eosinophilic Fasciitis: standard induction treatment with oral SCS or IVMP in combination with MTX . PUVA psoralen plus broadband UVA, UV ultraviolet
Intravenous methylprednisolone (IVMP): adult dose 1000 mg/day for 3 days/month for 3–6 months, possibly followed by oral SCS. Pediatric dose 30 mg/kg/day for 3 days/month for 3 months, possibly followed by oral SCS. α Mycophenolate Mofetil (MMF, alternative to MTX): adult dose 1000 mg twice daily. Pediatric dose 600–1200 mg/m2/day twice daily. A Deep/linear subtypes: treatment with MTX monotherapy. Addition of SCS or IVMP in case of rapidly progressive disease or in the presence of (looming) contractures. B Eosinophilic Fasciitis: standard induction treatment with oral SCS or IVMP in combination with MTX . PUVA psoralen plus broadband UVA, UV ultraviolet
10.1 Topical Therapy
10.1.1 Topical and Intralesional Corticosteroids
Topical corticosteroids (TCSs) are first-line therapy for superficial morphea. However, no studies have reported efficacy of TCSs in morphea. Based on expert opinion, we regard TCS first-line treatment for superficial circumscribed morphea. We recommend a highly potent TCS once a day for up to 4 weeks or a moderately potent TCS once a day for up to 3 months. Additionally, long-term TCS therapy should be in the form of interval therapy [24]. As with TCS, there are no studies reported for intralesional corticosteroids.
10.1.2 Topical Tacrolimus 0.1% Ointment
A double-blind, randomized controlled trial (RCT) with tacrolimus 0.1% ointment reported significant improvement in durometry and clinical scores [152]. A 3-month open-label study with topical tacrolimus 0.1% ointment twice daily under occlusion demonstrated complete resolution of early lesions and softening of late sclerotic lesions [153]. Another open-label study reported improvement in 9 out of 13 patients with topical tacrolimus 0.1% ointment, twice daily without occlusion [154]. We recommend topical tacrolimus 0.1% ointment, either with or without occlusion, twice daily as an alternative or additional topical therapy for superficial morphea. Additionally, based on a recent case report, we do not recommend topical tacrolimus in radiation-induced morphea [155].
10.1.3 Topical Calcipotriol 0.005% Ointment
Two uncontrolled studies investigated topical calcipotriene 0.005% ointment, either with [156] or without occlusion [157]. These studies included 31 patients and both studies reported beneficial effects in all patients. Lastly, an uncontrolled study with six patients reported efficacy of combination therapy with betamethasone dipropionate and calcipotriol 0.005% [158]. Based on the literature, we recommend topical calcipotriol 0.005% ointment, once or twice daily, with or without occlusion as an alternative topical therapy for superficial morphea. Additionally, topical calcipotriol 0.005% ointment may be prescribed combined with TCS therapy.
10.1.4 Miscellaneous Topical Therapies
A proof-of-concept study with topical imiquimod 5% cream reported effectiveness, measured by decrease of DIET and visual analog scale (VAS) scores in nine pediatric patients [159]. A second prospective vehicle-controlled study with 25 adult patients confirmed these results. Imiquimod 5% cream was superior to vehicle in reducing DIET scores at months 3, 6, 9, and 12 [120]. Both prospective studies reported no study withdrawals and adverse effects were minimal. Lastly, several case reports and series confirm the beneficial effects of imiquimod 5% [119, 160, 161]. Despite these positive study results, German guidelines do not recommend the use of imiquimod 5% cream, based on the authors’ experience [24].
Interestingly, an open phase II trial with 12 morphea patients reported significant reduction of mLoSSI and durometer scores at month 6 compared with baseline with 8% pirfenidone gel, three times daily. Pirfenidone gel was well tolerated and no side effects were reported [162].
10.2 Phototherapy
Several phototherapy modalities [ultraviolet (UV) B, psoralen plus broadband UVA (PUVA), broadband UVA and UVA1] have been investigated for morphea. Lower wave length phototherapy (UVA) has greater potency of tissue penetration than UVB and therefore has been studied more extensively.
10.2.1 UVA1
The treatment potential of UVA1 (wavelength of 340–400 mm) is superior to broadband UVA (320–400 mm), because of the lower risk for sunburn. This allows for higher dosage delivery with fewer side effects. Three different UVA1 dose regimens exist and have been investigated for morphea: low dose (LD, 10–20 J/cm2) [163–168], medium dose (MD, 30–50 J/cm2) [121, 122, 167–170], and high dose (HD, 60–130 J/cm2) [164]. One study demonstrated superior results for HD compared with LD-UVA1 [164]. Additionally, one randomized prospective study compared LD-UVA1, MD-UVA1, and UVB. This study showed superior efficacy of MD-UVA1 versus UVB but not versus LD-UVA1 [167]. Noteworthy, the first true randomized, blinded, and placebo-controlled trial with UVA1 is currently including patients (clinicaltrials.gov NCT01799174). The maximum dose investigated in a study consists of a cumulative dose of 3900 J/cm2. At our center, we treat patients with 60 J/cm2 3–5 times a week to a cumulative dose of 1460 J/cm2. In contrast to the excellent short-term results with UVA1, a study of 37 adults showed 2-year and 3-year recurrence rates of 44.5 and 48.4% after UVA1 treatment, respectively [29]. No data exists with regard to effectiveness of repeated treatment with UVA1.
10.2.2 Miscellaneous Phototherapy Modalities
One study investigated broadband UVA (320–400 mm) in three dose regimens (5, 10, and 20 J/cm2) for 20 sessions in 63 patients. All regimens were effective and none of them was superior to the other regimens [171]. Two prospective studies report beneficial effects of PUVA in 30 patients [172, 173]. Additionally, a retrospective single-center study confirmed these beneficial effects in 28 patients [174]. Lastly, one prospective study demonstrated equal effectiveness of UVB and low-dose UVA1 [167].
10.2.3 Summary and Recommendations
Based on the literature, we recommend UVA1 as first-choice phototherapy modality for adult patients. Although supported by less evidence, UVA1 phototherapy may also be used in a pediatric morphea. Despite the deeper penetration potential of UVA1, phototherapy should primarily be used for superficial subtypes of morphea. Additionally, it may be used as add-on therapy to systemic treatment in patients with both superficial and deep manifestations.
10.3 Systemic Treatments
10.3.1 Systemic Corticosteroids (SCSs)
One open study investigated an oral SCS (prednisone, starting dose 0.5–1 mg/kg/day followed by tapering) in 17 patients with severe morphea. Patients were treated for 5 to 70 months. This study reported a rapid response. However, in six patients (35%), disease relapse was observed after treatment discontinuation [175]. Additionally, a retrospective study with 28 adult patients reported a favorable response in 24 patients (86%) with an oral SCS (prednisone, starting dose 0.3–1.0 mg/kg/day). Patients were treated for 3–39 months. Similar to the first study, recurrence rate was 45% after treatment discontinuation [176]. High recurrence rates combined with an unfavorable long-term side-effect profile leads to the recommendation that oral SCS monotherapy is no viable long-term treatment option for morphea. Intravenous methylprednisolone (IVMP) has only been investigated in combination with MTX and will be discussed in the following section.
10.3.2 Methotrexate
MTX, with or without the combination of IVMP and/or oral SCS, is the most reported systemic treatment for morphea. Best evidence results from a double-blind RCT with 70 pediatric morphea patients, which investigated oral MTX (15 mg/m2/week, max 20 mg) versus placebo for 12 months. Both arms received an oral SCS (prednisone, starting dose 1 mg/kg/day, max 50 mg, followed by tapering) for the first 3 months. Reported outcome measures consisted of a computerized skin score rate and IRT. Improvements in outcome measures at month 12 could only be observed in the MTX treatment arm, whereas the placebo arm showed worsening [28].
Two prospective non-controlled studies investigated MTX in combination with IVMP [177] or oral SCS [178] in children. One study included ten pediatric patients who received subcutaneous MTX 0.3–0.6 mg/kg/week (max 20 mg/week), of whom nine patients simultaneously received IVMP (30 mg/kg for 3 days, monthly for 3 months). At the last follow-up visit, all patients who continued (n = 9) had inactive skin lesions [177]. The second study investigated subcutaneous MTX (1 mg/kg/week, max 25 mg) in combination with an oral SCS (prednisone 2 mg/kg/day, maximum 60 mg/day followed by tapering) in 36 pediatric patients. All patients demonstrated significant improvement in mLoSSI and PhysGA-A at a median of 1.77 months [178].
Two additional prospective studies investigated adult patients. One study investigated oral MTX (15 mg/week) in combination with IVMP (1000 mg for 3 days, monthly) for at least 6 months in 15 patients with severe morphea. In the majority of patients (n = 14), a significant decrease in mSS was observed, supported by histologic and ultrasound assessments [179]. Another uncontrolled prospective study investigated oral MTX (15 mg/week) monotherapy for 24 weeks in nine adult morphea patients [180]. Significant improvements in MSS and VAS for tightness were observed at week 24.
In addition to prospective trials, we identified 11 retrospective studies which report MTX with or without IVMP and/or oral SCS [1, 4, 25, 27, 32, 109, 181–185]. A total of 353 unique patients were identified (124 adults and 229 pediatric patients). Response rates to MTX with or without IVMP and/or oral SCS ranged between 80–94%. In contrast to the high short-term response rates, (long-term) disease recurrences are reported in 28–44% of the patients after MTX discontinuation [1, 25, 182, 184]. A prospective long-term follow-up study described that MTX treatment duration was a predictor for relapse, suggesting that longer treatment duration prevents disease relapse [186]. Lastly, some of the retrospective studies report superior response rates for combination therapy with MTX plus SCS compared with MTX monotherapy [4, 185]. However, these studies are prone to bias because of confounding by indication as a result of the retrospective design of these studies.
In conclusion, MTX is an effective treatment for morphea. However, no comparative studies between MTX monotherapy and MTX plus SCS have been reported. Therefore, no recommendations can be given as to whether MTX should be applied with or without SCS. However, either oral SCS or IVMP should be added to MTX in the case of severe disease, rapidly progressive disease, or in the presence of (looming) contractures. Recommended dosages are displayed in the legend of the flowchart (Fig. 9). Optimal timing of systemic treatment discontinuation remains a difficult aspect in therapeutic management.
10.3.2.1 Folic/Folinic Acid Supplementation
A retrospective analysis of MTX treatment in 107 adult patients showed that folic acid (5–10 mg once a week) protected against MTX discontinuation due to adverse events [181]. Additionally, folic acid (0.4–1 mg/day) or folinic acid (5 mg/week) is recommended by Childhood Arthritis and Rheumatology Research Alliance (CARRA) [187].
10.3.3 Mycophenolate Mofetil (MMF)
Two studies reported MMF in severe refractory morphea. The first study retrospectively described ten MTX- and corticosteroid-refractory pediatric morphea patients who were treated with MMF (600–1200 mg/m2/day). Six out of ten patients received MMF combined with MTX treatment. Arrest of disease progression was reported in eight patients (80%) a reduction of erythema in seven (70%), skin softening in nine (90%), and extracutaneous manifestations in four patients (40%). MMF was well tolerated in all patients [188]. The second study retrospectively described MMF in seven morphea patients (three adults and four children). Doses ranged from 500 to 2500 mg. This study reported disease remission in four patients (57%) and maintenance of disease remission in one patient (14%). In the remaining two patients (29%), MMF treatment was discontinued due to disease progression or side effects [189]. Despite the limited evidence for MMF in morphea, two studies showed extensive experience with MMF in large proportions of involved care providers [187, 190]. Based on current evidence, we recommend MMF (adults 1000 mg twice daily; children 600–1200 mg/m2/day twice daily) as an alternative to MTX.
10.3.4 Miscellaneous Systemic Treatment Options
Recent case reports describe imatinib [191–194], infliximab [102, 103], rituximab [98], abatacept [195], the mTOR inhibitors tacrolimus [196] and everolimus [197], and mesenchymal stem-cell therapy [198] as alternative treatment options in severe morphea. The authors recommend restraint from the use of TNF-α inhibitors in the treatment of morphea as multiple reports show morphea can be provoked by these agents [98–101].
10.4 Recommendations for EF
Historically, HD SCS monotherapy (prednisone 0.5–2.0 mg/kg/day) was regarded as first-line treatment for EF [14, 15, 199]. However, multiple recent retrospective studies have shown superior response rates in patients who are treated with a combination of HD SCS and an ISD, especially for weekly MTX (15–25 mg/week) [14, 16, 200]. These studies are retrospective and thus prone to confounding by indication. Future prospective studies should investigate the additional (long-term) effect of MTX as was done for pediatric morphea [28].
Case series reported non- or partial responses to conventional treatment in a proportion of patients [14–16]. Until recently, only case reports or small case series described alternative treatment options [21]. However, a recent study prospectively investigated HD pulse IV MTX (4 mg/kg/month; median monthly MTX dose 288 mg) as an alternative treatment option in 12 patients with EF. The median mSS improved significantly after six pulses (p = 0.001). Additionally, the range of motion of affected joints and patient-reported outcomes showed significant increases. Treatment was well tolerated and adverse events could be managed accordingly. In this study, one patient had to be withdrawn due to an adverse event [201]. Another recent prospective study reported superior clinical improvement in severe EF patients treated with D-penicillamine (D-pen) plus oral SCS (n = 10) versus SCS monotherapy (n = 6). Disappointingly, four out of ten patients in the D-pen-arm had to discontinue treatment due to adverse events [202]. Lastly, the following alternative treatments have been reported in case reports and series: infliximab [203], azathioprine [204, 205], sulfasalazine [206], cyclosporine [207], cyclophosphamide [208], rituximab [209], tocilizumab [210], sirolimus [211], PUVA [212], IV immunoglobulins [213], and bone marrow transplantation [214, 215].
We recommend a combination of SCS and MTX as first-line treatment for EF. Induction therapy may consist of HD oral SCS (prednisone 1 mg/kg/day, followed by tapering) or IVMP (1000 mg for 3 days monthly, followed by oral SCS) started simultaneously with weekly MTX (15–25 mg/week) maintenance therapy.
10.5 Treatment of Disease Damage
Multiple studies report effective strategies for correction of disease damage. However, timing of these correctional procedures is crucial, as disease flare has frequently been reported even after many years of disease quiescence [10, 26]. A recent study reviews the surgical treatment options for Parry–Romberg syndrome and ECDS and describes the debate with regard to timing of these procedures [36]. Autologous fat grafting in ECDS is reported in multiple case reports and series [216–221]. Lastly, one case report describes correction for limb-length discrepancy in linear morphea of a lower extremity [222]. In the authors’ opinion, these procedures should only be performed in long-term quiescent disease by a care provider with (extensive) experience in this field. Lastly, although evidence is lacking, physical therapy should be considered in case of decreased range of motion of an affected joint or when postural deformities are present as a result of limb-length discrepancies.
11 Conclusions
Morphea and EF pathogenesis remains to be elucidated as well as the exact immunological relationship with SSc. This may lead to insight into the disease mechanisms behind these debilitating conditions and the identification of new therapeutic targets.
Progression has been made in standardization of outcome measures for morphea. Currently, LoSCAT is the most promising and frequently used outcome measure. This should lead to data harmonization and the potential to compare future studies.
Monitoring of deep involvement of morphea is one of the most difficult aspects of the disease. The lack of validated outcome measures for deep involvement emphasizes the need for new biomarkers.
References
Christen-Zaech S, Hakim MD, Afsar FS, Paller AS. Pediatric morphea (localized scleroderma): review of 136 patients. J Am Acad Dermatol. 2008;59(3):385–96. doi:10.1016/j.jaad.2008.05.005.
Nelson AM. Localized scleroderma including morphea, linear scleroderma, and eosinophilic fasciitis. Curr Probl Pediatr. 1996;26(9):318–24.
Klimas NK, Shedd AD, Bernstein IH, Jacobe H. Health-related quality of life in morphoea. Br J Dermatol. 2015;172(5):1329–37. doi:10.1111/bjd.13572.
Mazori DR, Wright NA, Patel M, Liu SW, Ramachandran SM, Franks AG Jr, et al. Characteristics and treatment of adult-onset linear morphea: a retrospective cohort study of 61 patients at 3 tertiary care centers. J Am Acad Dermatol. 2016;74(3):577–9. doi:10.1016/j.jaad.2015.09.069.
Zulian F, Vallongo C, Woo P, Russo R, Ruperto N, Harper J, et al. Localized scleroderma in childhood is not just a skin disease. Arthritis Rheum. 2005;52(9):2873–81. doi:10.1002/art.21264.
Peterson LS, Nelson AM, Su WP, Mason T, O’Fallon WM, Gabriel SE. The epidemiology of morphea (localized scleroderma) in Olmsted County 1960–1993. J Rheumatol. 1997;24(1):73–80.
Silman A, Jannini S, Symmons D, Bacon P. An epidemiological study of scleroderma in the West Midlands. Br J Rheumatol. 1988;27(4):286–90.
Herrick AL, Ennis H, Bhushan M, Silman AJ, Baildam EM. Incidence of childhood linear scleroderma and systemic sclerosis in the UK and Ireland. Arthritis Care Res. 2010;62(2):213–8. doi:10.1002/acr.20070.
Kreuter A, Wischnewski J, Terras S, Altmeyer P, Stucker M, Gambichler T. Coexistence of lichen sclerosus and morphea: a retrospective analysis of 472 patients with localized scleroderma from a German tertiary referral center. J Am Acad Dermatol. 2012;67(6):1157–62. doi:10.1016/j.jaad.2012.04.003.
Mertens JS, Seyger MM, Kievit W, Hoppenreijs EP, Jansen TL, van de Kerkhof PC, et al. Disease recurrence in localized scleroderma: a retrospective analysis of 344 patients with paediatric- or adult-onset disease. Br J Dermatol. 2015;172(3):722–8. doi:10.1111/bjd.13514.
Zulian F, Athreya BH, Laxer R, Nelson AM, Feitosa de Oliveira SK, Punaro MG, et al. Juvenile localized scleroderma: clinical and epidemiological features in 750 children. An international study. Rheumatology (Oxford, England) 2006;45(5):614–20. doi:10.1093/rheumatology/kei251.
Condie D, Grabell D, Jacobe H. Comparison of outcomes in adults with pediatric-onset morphea and those with adult-onset morphea: a cross-sectional study from the morphea in adults and children cohort. Arthritis Rheumatol (Hoboken, NJ) 2014;66(12):3496–504. doi:10.1002/art.38853.
Leitenberger JJ, Cayce RL, Haley RW, Adams-Huet B, Bergstresser PR, Jacobe HT. Distinct autoimmune syndromes in morphea: a review of 245 adult and pediatric cases. Arch Dermatol. 2009;145(5):545–50. doi:10.1001/archdermatol.2009.79.
Lebeaux D, Frances C, Barete S, Wechsler B, Dubourg O, Renoux J, et al. Eosinophilic fasciitis (Shulman disease): new insights into the therapeutic management from a series of 34 patients. Rheumatology (Oxford, England). 2012;51(3):557–61. doi:10.1093/rheumatology/ker366.
Lakhanpal S, Ginsburg WW, Michet CJ, Doyle JA, Moore SB. Eosinophilic fasciitis: clinical spectrum and therapeutic response in 52 cases. Semin Arthritis Rheum. 1988;17(4):221–31.
Wright NA, Mazori DR, Patel M, Merola JF, Femia AN, Vleugels RA. Epidemiology and treatment of eosinophilic fasciitis: an analysis of 63 patients from 3 tertiary care centers. JAMA Dermatol. 2016;152(1):97–9. doi:10.1001/jamadermatol.2015.3648.
Papa R, Nozza P, Granata C, Caorsi R, Gattorno M, Martini A, et al. Juvenile eosinophilic fasciitis: three case reports with review of the literature. Clin Exp Rheumatol. 2016;34(3):527–30.
Ortega-Loayza AG, Merritt BG, Groben PA, Morrell DS. Eosinophilic fasciitis in a female child. J Am Acad Dermatol. 2008;58(5 Suppl 1):S72–4. doi:10.1016/j.jaad.2007.05.014.
Ching DW, Petrie JP. Childhood eosinophilic fasciitis presenting as inflammatory polyarthritis and associated with selective IgA deficiency. Ann Rheum Dis. 1991;50(9):647–8.
Williams HJ, Ziter FA, Banta CA. Childhood eosinophilic fasciitis—progression to linear scleroderma. J Rheumatol. 1986;13(5):961–2.
Lebeaux D, Sene D. Eosinophilic fasciitis (Shulman disease). Best Pract Res Clin Rheumatol. 2012;26(4):449–58. doi:10.1016/j.berh.2012.08.001.
Laxer RM, Zulian F. Localized scleroderma. Curr Opin Rheumatol. 2006;18(6):606–13. doi:10.1097/01.bor.0000245727.40630.c3.
Peterson LS, Nelson AM, Su WP. Classification of morphea (localized scleroderma). Mayo Clin Proc. 1995;70(11):1068–76. doi:10.1016/s0025-6196(11)64442-x.
Kreuter A, Krieg T, Worm M, Wenzel J, Moinzadeh P, Kuhn A, et al. German guidelines for the diagnosis and therapy of localized scleroderma. J German Soc Dermatol JDDG. 2016;14(2):199–216. doi:10.1111/ddg.12724.
Mirsky L, Chakkittakandiyil A, Laxer RM, O’Brien C, Pope E. Relapse after systemic treatment in paediatric morphoea. Br J Dermatol. 2012;166(2):443–5. doi:10.1111/j.1365-2133.2011.10535.x.
Saxton-Daniels S, Jacobe HT. An evaluation of long-term outcomes in adults with pediatric-onset morphea. Arch Dermatol. 2010;146(9):1044–5. doi:10.1001/archdermatol.2010.239.
Piram M, McCuaig CC, Saint-Cyr C, Marcoux D, Hatami A, Haddad E, et al. Short- and long-term outcome of linear morphoea in children. Br J Dermatol. 2013;169(6):1265–71. doi:10.1111/bjd.12606.
Zulian F, Martini G, Vallongo C, Vittadello F, Falcini F, Patrizi A, et al. Methotrexate treatment in juvenile localized scleroderma: a randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2011;63(7):1998–2006. doi:10.1002/art.30264.
Vasquez R, Jabbar A, Khan F, Buethe D, Ahn C, Jacobe H. Recurrence of morphea after successful ultraviolet al phototherapy: a cohort study. J Am Acad Dermatol. 2014;70(3):481–8. doi:10.1016/j.jaad.2013.10.018.
Zannin ME, Martini G, Athreya BH, Russo R, Higgins G, Vittadello F, et al. Ocular involvement in children with localised scleroderma: a multi-centre study. Br J Ophthalmol. 2007;91(10):1311–4. doi:10.1136/bjo.2007.116038.
Miller MT, Sloane H, Goldberg MF, Grisolano J, Frenkel M, Mafee MF. Progressive hemifacial atrophy (Parry–Romberg disease). J Pediatr Ophthalmol Strabismus. 1987;24(1):27–36.
Tollefson MM, Witman PM. En coup de sabre morphea and Parry–Romberg syndrome: a retrospective review of 54 patients. J Am Acad Dermatol. 2007;56(2):257–63. doi:10.1016/j.jaad.2006.10.959.
Trainito S, Favero L, Martini G, Pedersen TK, Favero V, Herlin T, et al. Odontostomatologic involvement in juvenile localised scleroderma of the face. J Paediatr Child Health. 2012;48(7):572–6. doi:10.1111/j.1440-1754.2012.02435.x.
Talacko AA, Reade PC. Hemifacial atrophy and temporomandibular joint pain-dysfunction syndrome. Int J Oral Maxillofac Surg. 1988;17(4):224–6.
O’Flynn S, Kinirons M. Parry–Romberg syndrome: a report of the dental findings in a child followed up for 9 years. Int J Paediatr Dent. 2006;16(4):297–301. doi:10.1111/j.1365-263X.2006.00730.x.
Tolkachjov SN, Patel NG, Tollefson MM. Progressive hemifacial atrophy: a review. Orphanet J Rare Dis. 2015;10:39. doi:10.1186/s13023-015-0250-9.
Huang KW, Chen XH. Pathology of eosinophilic fasciitis and its relation to polymyositis. Can J Neurol Sci. 1987;14(4):632–7.
Kirchgesner T, Dallaudiere B, Omoumi P, Malghem J, Vande Berg B, Lecouvet F, et al. Eosinophilic fasciitis: typical abnormalities, variants and differential diagnosis of fasciae abnormalities using MR imaging. Diagn Intervent Imaging. 2015;96(4):341–8. doi:10.1016/j.diii.2014.06.018.
Agnew KL, Blunt D, Francis ND, Bunker CB. Magnetic resonance imaging in eosinophilic fasciitis. Clin Exp Dermatol. 2005;30(4):435–6. doi:10.1111/j.1365-2230.2005.01753.x.
Baumann F, Bruhlmann P, Andreisek G, Michel BA, Marincek B, Weishaupt D. MRI for diagnosis and monitoring of patients with eosinophilic fasciitis. AJR Am J Roentgenol. 2005;184(1):169–74. doi:10.2214/ajr.184.1.01840169.
Dybowski F, Neuen-Jacob E, Braun J. Eosinophilic fasciitis and myositis: use of imaging modalities for diagnosis and monitoring. Ann Rheum Dis. 2008;67(4):572–4. doi:10.1136/ard.2007.076844.
Mondal S, Goswami RP, Sinha D, Ghosh A. Ultrasound is a useful adjunct in diagnosis of eosinophilic fasciitis. Rheumatology (Oxford, England) 2015;54(11):2041. doi:10.1093/rheumatology/kev290.
Marie I, Sauvetre G. Fluorodeoxyglucose positron emission tomography in eosinophilic fasciitis. Jt Bone Spine Revue Rhum. 2014;81(6):541. doi:10.1016/j.jbspin.2014.07.001.
Kim HJ, Lee SW, Kim GJ, Lee JH. Usefulness of FDG PET/CT in the diagnosis of eosinophilic fasciitis. Clin Nucl Med. 2014;39(9):801–2. doi:10.1097/rlu.0000000000000260.
Cheriet S, Chastan M, Levesque H, Marie I. Positron emission tomography in the diagnosis of eosinophilic fasciitis. QJM. 2011;104(11):987–8. doi:10.1093/qjmed/hcq218.
Takehara K, Sato S. Localized scleroderma is an autoimmune disorder. Rheumatology (Oxford, England) 2005;44(3):274–9. doi:10.1093/rheumatology/keh487.
Zulian F, Cuffaro G, Sperotto F. Scleroderma in children: an update. Curr Opin Rheumatol. 2013;25(5):643–50. doi:10.1097/BOR.0b013e3283641f61.
Fett N, Werth VP. Update on morphea: Part I. Epidemiology, clinical presentation, and pathogenesis. J Am Acad Dermatol. 2011;64(2):217–28. doi:10.1016/j.jaad.2010.05.045. (quiz 29–30).
Gabrielli A, Avvedimento EV, Krieg T. Scleroderma. N Engl J Med. 2009;360(19):1989–2003. doi:10.1056/NEJMra0806188.
Stern EP, Denton CP. The pathogenesis of systemic sclerosis. Rheum Dis Clin N Am. 2015;41(3):367–82. doi:10.1016/j.rdc.2015.04.002.
Cutolo M, Pizzorni C, Sulli A, Smith V. Early diagnostic and predictive value of capillaroscopy in systemic sclerosis. Curr Rheumatol Rev. 2013;9(4):249–53.
Helmbold P, Fiedler E, Fischer M, Marsch W. Hyperplasia of dermal microvascular pericytes in scleroderma. J Cutan Pathol. 2004;31(6):431–40. doi:10.1111/j.0303-6987.2004.00203.x.
Dharamsi JW, Victor S, Aguwa N, Ahn C, Arnett F, Mayes MD, et al. Morphea in adults and children cohort III: nested case–control study—the clinical significance of autoantibodies in morphea. JAMA Dermatol. 2013;149(10):1159–65. doi:10.1001/jamadermatol.2013.4207.
Arkachaisri T, Fertig N, Pino S, Medsger TA Jr. Serum autoantibodies and their clinical associations in patients with childhood- and adult-onset linear scleroderma. A single-center study. J Rheumatol. 2008;35(12):2439–44. doi:10.3899/jrheum.080098.
Falanga V, Medsger TA Jr, Reichlin M. Antinuclear and anti-single-stranded DNA antibodies in morphea and generalized morphea. Arch Dermatol. 1987;123(3):350–3.
Sato S, Fujimoto M, Ihn H, Kikuchi K, Takehara K. Clinical characteristics associated with antihistone antibodies in patients with localized scleroderma. J Am Acad Dermatol. 1994;31(4):567–71.
Falanga V, Medsger TA, Reichlin M. High titers of antibodies to single-stranded DNA in linear scleroderma. Arch Dermatol. 1985;121(3):345–7.
Sato S, Kodera M, Hasegawa M, Fujimoto M, Takehara K. Antinucleosome antibody is a major autoantibody in localized scleroderma. Br J Dermatol. 2004;151(6):1182–8. doi:10.1111/j.1365-2133.2004.06256.x.
Harrington CI, Dunsmore IR. An investigation into the incidence of auto-immune disorders in patients with localized morphoea. Br J Dermatol. 1989;120(5):645–8.
Jacobe H, Ahn C, Arnett FC, Reveille JD. Major histocompatibility complex class I and class II alleles may confer susceptibility to or protection against morphea: findings from the morphea in adults and children cohort. Arthritis Rheumatol (Hoboken, NJ). 2014;66(11):3170–7. doi:10.1002/art.38814.
Yamane K, Ihn H, Kubo M, Yazawa N, Kikuchi K, Soma Y, et al. Increased serum levels of soluble vascular cell adhesion molecule 1 and E-selectin in patients with localized scleroderma. J Am Acad Dermatol. 2000;42(1 Pt 1):64–9.
Ihn H, Fujimoto M, Sato S, Kikuchi K, Igarashi A, Soma Y, et al. Increased levels of circulating intercellular adhesion molecule-1 in patients with localized scleroderma. J Am Acad Dermatol. 1994;31(4):591–5.
Needleman BW, Wigley FM, Stair RW. Interleukin-1, interleukin-2, interleukin-4, interleukin-6, tumor necrosis factor alpha, and interferon-gamma levels in sera from patients with scleroderma. Arthritis Rheum. 1992;35(1):67–72.
Hasegawa M, Fujimoto M, Kikuchi K, Takehara K. Elevated serum levels of interleukin 4 (IL-4), IL-10, and IL-13 in patients with systemic sclerosis. J Rheumatol. 1997;24(2):328–32.
Salmon-Ehr V, Serpier H, Nawrocki B, Gillery P, Clavel C, Kalis B, et al. Expression of interleukin-4 in scleroderma skin specimens and scleroderma fibroblast cultures. Potential role in fibrosis. Arch Dermatol. 1996;132(7):802–6.
Torok KS, Kurzinski K, Kelsey C, Yabes J, Magee K, Vallejo AN, et al. Peripheral blood cytokine and chemokine profiles in juvenile localized scleroderma: T-helper cell-associated cytokine profiles. Semin Arthritis Rheum. 2015;45(3):284–93. doi:10.1016/j.semarthrit.2015.06.006.
Danczak-Pazdrowska A, Kowalczyk M, Szramka-Pawlak B, Gornowicz-Porowska J, Szewczyk A, Silny W, et al. Interleukin-17A and interleukin-23 in morphea. Arch Med Sci. 2012;8(6):1089–95. doi:10.5114/aoms.2012.32421.
Hasegawa M, Sato S, Nagaoka T, Fujimoto M, Takehara K. Serum levels of tumor necrosis factor and interleukin-13 are elevated in patients with localized scleroderma. Dermatology (Basel, Switzerland) 2003;207(2):141–7.
Lafyatis R. Transforming growth factor beta—at the centre of systemic sclerosis. Nat Rev Rheumatol. 2014;10(12):706–19. doi:10.1038/nrrheum.2014.137.
Higley H, Persichitte K, Chu S, Waegell W, Vancheeswaran R, Black C. Immunocytochemical localization and serologic detection of transforming growth factor beta 1. Association with type I procollagen and inflammatory cell markers in diffuse and limited systemic sclerosis, morphea, and Raynaud’s phenomenon. Arthritis Rheum. 1994;37(2):278–88.
Kubo M, Ihn H, Yamane K, Tamaki K. Up-regulated expression of transforming growth factor beta receptors in dermal fibroblasts in skin sections from patients with localized scleroderma. Arthritis Rheum. 2001;44(3):731–4. doi:10.1002/1529-0131(200103)44:3<731:aid-anr124>3.0.co;2-u.
Fang F, Goncalves Marangoni R, Zhou X, Yang Y, Ye B, Shangguang A, et al. Toll-like receptor 9 signaling is augmented in systemic sclerosis and elicits transforming growth factor beta-dependent fibroblast activation. Arthritis Rheumatol (Hoboken, NJ) 2016;68(8):1989–2002. doi:10.1002/art.39655.
Bhattacharyya S, Tamaki Z, Wang W, Hinchcliff M, Hoover P, Getsios S, et al. Fibronectin EDA promotes chronic cutaneous fibrosis through toll-like receptor signaling. Sci Transl Med. 2014;6(232):232ra50. doi:10.1126/scitranslmed.3008264.
van Bon L, Affandi AJ, Broen J, Christmann RB, Marijnissen RJ, Stawski L, et al. Proteome-wide analysis and CXCL4 as a biomarker in systemic sclerosis. N Engl J Med. 2014;370(5):433–43. doi:10.1056/NEJMoa1114576.
Bhattacharyya S, Wang W, Morales-Nebreda L, Feng G, Wu M, Zhou X, et al. Tenascin-C drives persistence of organ fibrosis. Nat Commun. 2016;7:11703. doi:10.1038/ncomms11703.
Seyger MM, van den Hoogen FH, van Vlijmen-Willems IM, van de Kerkhof PC, de Jong EM. Localized and systemic scleroderma show different histological responses to methotrexate therapy. J Pathol. 2001;193(4):511–6. doi:10.1002/1096-9896(2000)9999:9999<:aid-path779>3.0.co;2-8.
Feghali-Bostwick C, Medsger TA Jr, Wright TM. Analysis of systemic sclerosis in twins reveals low concordance for disease and high concordance for the presence of antinuclear antibodies. Arthritis Rheum. 2003;48(7):1956–63. doi:10.1002/art.11173.
Broen JC, Radstake TR, Rossato M. The role of genetics and epigenetics in the pathogenesis of systemic sclerosis. Nat Rev Rheumatol. 2014;10(11):671–81. doi:10.1038/nrrheum.2014.128.
Makino K, Jinnin M, Hirano A, Yamane K, Eto M, Kusano T, et al. The downregulation of microRNA let-7a contributes to the excessive expression of type I collagen in systemic and localized scleroderma. J Immunol. (Baltimore, MD, 1950) 2013;190(8):3905–15. doi:10.4049/jimmunol.1200822.
Makino T, Jinnin M, Etoh M, Yamane K, Kajihara I, Makino K, et al. Down-regulation of microRNA-196a in the sera and involved skin of localized scleroderma patients. Eur J Dermatol. 2014;24(4):470–6. doi:10.1684/ejd.2014.2384.
Yan Q, Chen J, Li W, Bao C, Fu Q. Targeting miR-155 to treat experimental scleroderma. Sci Rep. 2016;6:20314. doi:10.1038/srep20314.
Grabell D, Hsieh C, Andrew R, Martires K, Kim A, Vasquez R, et al. The role of skin trauma in the distribution of morphea lesions: a cross-sectional survey of the morphea in adults and children cohort IV. J Am Acad Dermatol. 2014;71(3):493–8. doi:10.1016/j.jaad.2014.04.009.
Spalek M, Jonska-Gmyrek J, Galecki J. Radiation-induced morphea—a literature review. J Eur Acad Dermatol Venereol JEADV. 2015;29(2):197–202. doi:10.1111/jdv.12704.
Eisendle K, Grabner T, Zelger B. Morphoea: a manifestation of infection with Borrelia species? Br J Dermatol. 2007;157(6):1189–98. doi:10.1111/j.1365-2133.2007.08235.x.
Prinz JC, Kutasi Z, Weisenseel P, Poto L, Battyani Z, Ruzicka T. “Borrelia-associated early-onset morphea”: a particular type of scleroderma in childhood and adolescence with high titer antinuclear antibodies? Results of a cohort analysis and presentation of three cases. J Am Acad Dermatol. 2009;60(2):248–55. doi:10.1016/j.jaad.2008.09.023.
Verberkt RM, Janssen M, Wesseling J. A boy with a tight skin: Borrelia-associated early-onset morphea. Clin Exp Rheumatol. 2014;32(1):121–2.
Miglino B, Viana M, Zavattaro E, Bonin S, Valente G, Colombo E. Localized scleroderma unius lateri and Borrelia burgdoferi infection. Indian J Dermatol Venereol Leprol. 2012;78(3):383–5. doi:10.4103/0378-6323.95460.
Espinoza-Leon F, Hassanhi-Hassanhi M, Arocha-Sandoval F, Urbina-Lopez M. Absence of Borrelia burgdorferi antibodies in the sera of Venezuelan patients with localized scleroderma (morphea). Invest Clin. 2006;47(3):283–8.
Weide B, Schittek B, Klyscz T, Schuz K, Stark M, Rassner G, et al. Morphoea is neither associated with features of Borrelia burgdorferi infection, nor is this agent detectable in lesional skin by polymerase chain reaction. Br J Dermatol. 2000;143(4):780–5.
Colome-Grimmer MI, Payne DA, Tyring SK, Sanchez RL. Borrelia burgdorferi DNA and Borrelia hermsii DNA are not associated with morphea or lichen sclerosus et atrophicus in the southwestern United States. Arch Dermatol. 1997;133(9):1174.
Alonso-Llamazares J, Persing DH, Anda P, Gibson LE, Rutledge BJ, Iglesias L. No evidence for Borrelia burgdorferi infection in lesions of morphea and lichen sclerosus et atrophicus in Spain. A prospective study and literature review. Acta Dermato Venereol. 1997;77(4):299–304.
Akimoto S, Ishikawa O, Miyachi Y. The absence of antibodies against Borrelia burgdorferi in the sera of Japanese patients with localized scleroderma. J Rheumatol. 1996;23(3):573–4.
De Vito JR, Merogi AJ, Vo T, Boh EE, Fung HK, Freeman SM, et al. Role of Borrelia burgdorferi in the pathogenesis of morphea/scleroderma and lichen sclerosus et atrophicus: a PCR study of thirty-five cases. J Cutan Pathol. 1996;23(4):350–8.
Fan W, Leonardi CL, Penneys NS. Absence of Borrelia burgdorferi in patients with localized scleroderma (morphea). J Am Acad Dermatol. 1995;33(4):682–4.
Dillon WI, Saed GM, Fivenson DP. Borrelia burgdorferi DNA is undetectable by polymerase chain reaction in skin lesions of morphea, scleroderma, or lichen sclerosus et atrophicus of patients from North America. J Am Acad Dermatol. 1995;33(4):617–20.
Wienecke R, Schlupen EM, Zochling N, Neubert U, Meurer M, Volkenandt M. No evidence for Borrelia burgdorferi-specific DNA in lesions of localized scleroderma. J Invest Dermatol. 1995;104(1):23–6.
Gutierrez-Gomez C, Godinez-Hana AL, Garcia-Hernandez M, Suarez-Roa Mde L, Toussaint-Caire S, Vega-Memije E, et al. Lack of IgG antibody seropositivity to Borrelia burgdorferi in patients with Parry–Romberg syndrome and linear morphea en coup de sabre in Mexico. Int J Dermatol. 2014;53(8):947–51. doi:10.1111/ijd.12105.
Chimenti MS, Teoli M, Di Stefani A, Giunta A, Esposito M, Perricone R. Resolution with rituximab of localized scleroderma occurring during etanercept treatment in a patient with rheumatoid arthritis. Eur J Dermatol EJD. 2013;23(2):273–4. doi:10.1684/ejd.2013.1929.
Stewart FA, Gavino AC, Elewski BE. New side effect of TNF-alpha inhibitors: morphea. Skinmed. 2013;11(1):59–60.
Ramirez J, Hernandez MV, Galve J, Canete JD, Sanmarti R. Morphea associated with the use of adalimumab: a case report and review of the literature. Mod Rheumatol/Jpn RheumAssoc. 2012;22(4):602–4. doi:10.1007/s10165-011-0550-4.
Mattozzi C, Richetta AG, Cantisani C, Giancristoforo S, D’Epiro S, Gonzalez Serva A, et al. Morphea, an unusual side effect of anti-TNF-alpha treatment. Eur J Dermatol EJD. 2010;20(3):400–1. doi:10.1684/ejd.2010.0946.
Diab M, Coloe JR, Magro C, Bechtel MA. Treatment of recalcitrant generalized morphea with infliximab. Arch Dermatol. 2010;146(6):601–4. doi:10.1001/archdermatol.2010.120.
Ferguson ID, Weiser P, Torok KS. A case report of successful treatment of recalcitrant childhood localized scleroderma with infliximab and leflunomide. Open Rheumatol J. 2015;9:30–5. doi:10.2174/18743129014090100030.
Peroni A, Zini A, Braga V, Colato C, Adami S, Girolomoni G. Drug-induced morphea: report of a case induced by balicatib and review of the literature. J Am Acad Dermatol. 2008;59(1):125–9. doi:10.1016/j.jaad.2008.03.009.
Granter SR, Barnhill RL, Duray PH. Borrelial fasciitis: diffuse fasciitis and peripheral eosinophilia associated with Borrelia infection. Am J Dermatopathol. 1996;18(5):465–73.
Sherber NS, Wigley FM, Paget SA. Diffuse fasciitis with eosinophilia developing after local irradiation for breast cancer. Clin Rheumatol. 2009;28(6):729–32. doi:10.1007/s10067-009-1122-2.
Mallepalli JR, Quinet RJ, Sus R. Eosinophilic fasciitis induced by fire ant bites. Ochsner J. 2008;8(3):114–8.
van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65(11):2737–47. doi:10.1002/art.38098.
Beltramelli M, Vercellesi P, Frasin A, Gelmetti C, Corona F. Localized severe scleroderma: a retrospective study of 26 pediatric patients. Pediatr Dermatol. 2010;27(5):476–80. doi:10.1111/j.1525-1470.2010.01258.x.
Lis-Swiety A, Brzezinska-Wcislo L, Arasiewicz H, Bergler-Czop B. Antiphospholipid antibodies in localized scleroderma: the potential role of screening tests for the detection of antiphospholipid syndrome. Postepy dermatol Alergol. 2014;31(2):65–70. doi:10.5114/pdia.2014.40978.
Sato S, Fujimoto M, Hasegawa M, Takehara K. Antiphospholipid antibody in localised scleroderma. Ann Rheum Dis. 2003;62(8):771–4.
Yamane K, Ihn H, Kubo M, Kuwana M, Asano Y, Yazawa N, et al. Anti-U1RNP antibodies in patients with localized scieroderma. Arch Dermatol Res. 2001;293(9):455–9.
Yimane K, Ihn H, Kubo M, Asano Y, Yazawa N, Tamaki K. Anti-U3 snRNP antibodies in localised scleroderma. Ann Rheum Dis. 2001;60(12):1157–8.
Tomimura S, Ogawa F, Iwata Y, Komura K, Hara T, Muroi E, et al. Autoantibodies against matrix metalloproteinase-1 in patients with localized scleroderma. J Dermatol Sci. 2008;52(1):47–54. doi:10.1016/j.jdermsci.2008.04.013.
Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg–Strauss syndrome. Arthritis Rheum. 2005;52(9):2926–35. doi:10.1002/art.21250.
Lis-Swiety A, Janicka I, Skrzypek-Salamon A, Brzezinska-Wcislo L. A systematic review of tools for determining activity of localized scleroderma in paediatric and adult patients. J Eur Acad Dermatol Venereol JEADV. 2016;. doi:10.1111/jdv.13790.
Arkachaisri T, Vilaiyuk S, Li S, O’Neil KM, Pope E, Higgins GC, et al. The localized scleroderma skin severity index and physician global assessment of disease activity: a work in progress toward development of localized scleroderma outcome measures. J Rheumatol. 2009;36(12):2819–29. doi:10.3899/jrheum.081284.
Arkachaisri T, Vilaiyuk S, Torok KS, Medsger Jr TA. Development and initial validation of the localized scleroderma skin damage index and physician global assessment of disease damage: a proof-of-concept study. Rheumatology (Oxford, England) 2010;49(2):373–81. doi:10.1093/rheumatology/kep361.
Dytoc M, Ting PT, Man J, Sawyer D, Fiorillo L. First case series on the use of imiquimod for morphoea. Br J Dermatol. 2005;153(4):815–20. doi:10.1111/j.1365-2133.2005.06776.x.
Dytoc M, Wat H, Cheung-Lee M, Sawyer D, Ackerman T, Fiorillo L. Evaluation of the efficacy and safety of topical imiquimod 5% for plaque-type morphea: a multicenter, prospective, vehicle-controlled trial. J Cutan Med Surg. 2015;19(2):132–9. doi:10.2310/7750.2014.14072.
de Rie MA, Enomoto DN, de Vries HJ, Bos JD. Evaluation of medium-dose UVA1 phototherapy in localized scleroderma with the Cutometer and fast Fourier transform method. Dermatology (Basel, Switzerland) 2003;207(3):298–301.
Andres C, Kollmar A, Mempel M, Hein R, Ring J, Eberlein B. Successful ultraviolet al phototherapy in the treatment of localized scleroderma: a retrospective and prospective study. Br J Dermatol. 2010;162(2):445–7. doi:10.1111/j.1365-2133.2009.09438.x.
Porta F, Kaloudi O, Garzitto A, Prignano F, Nacci F, Falcini F, et al. High frequency ultrasound can detect improvement of lesions in juvenile localized scleroderma. Mod Rheumatol/Jpn Rheum Assoc. 2014;24(5):869–73. doi:10.3109/14397595.2013.844301.
Seyger MM, van den Hoogen FH, de Boo T, de Jong EM. Reliability of two methods to assess morphea: skin scoring and the use of a durometer. J Am Acad Dermatol. 1997;37(5 Pt 1):793–6.
Zachariae H, Bjerring P, Halkier-Sorensen L, Heickendorff L, Sondergaard K. Skin scoring in systemic sclerosis: a modification–relations to subtypes and the aminoterminal propeptide of type III procollagen (PIIINP). Acta Dermato Venereol. 1994;74(6):444–6.
Zulian F, Meneghesso D, Grisan E, Vittadello F, Belloni Fortina A, Pigozzi B, et al. A new computerized method for the assessment of skin lesions in localized scleroderma. Rheumatology (Oxford, England) 2007;46(5):856–60. doi:10.1093/rheumatology/kel446.
Li SC, Liebling MS, Haines KA, Weiss JE, Prann A. Initial evaluation of an ultrasound measure for assessing the activity of skin lesions in juvenile localized scleroderma. Arthritis Care Res. 2011;63(5):735–42. doi:10.1002/acr.20407.
Poff S, Li SC, Kelsey CE, Foeldvari I, Torok KS. Durometry as an outcome measure in juvenile localized scleroderma. Br J Dermatol. 2016;174(1):228–30. doi:10.1111/bjd.14103.
Moon KW, Song R, Kim JH, Lee EY, Lee EB, Song YW. The correlation between durometer score and modified Rodnan skin score in systemic sclerosis. Rheumatol Int. 2012;32(8):2465–70. doi:10.1007/s00296-011-1993-9.
Falanga V, Bucalo B. Use of a durometer to assess skin hardness. J Am Acad Dermatol. 1993;29(1):47–51.
Schanz S, Fierlbeck G, Ulmer A, Schmalzing M, Kummerle-Deschner J, Claussen CD, et al. Localized scleroderma: MR findings and clinical features. Radiology. 2011;260(3):817–24. doi:10.1148/radiol.11102136.
Wang FD, Wang HW, Wu ZH, Hou B, Jiang B, Zhang Y, et al. Localized scleroderma of lower extremities: clinical and magnetic resonance imaging features. Zhongguo Yi Xue Ke Xue Yuan Xue Bao Acta Acad Med Sin. 2015;37(4):392–7. doi:10.3881/j.issn.1000-503X.2015.04.004.
Schanz S, Henes J, Ulmer A, Kotter I, Fierlbeck G, Claussen CD, et al. Response evaluation of musculoskeletal involvement in patients with deep morphea treated with methotrexate and prednisolone: a combined MRI and clinical approach. AJR Am J Roentgenol. 2013;200(4):W376–82. doi:10.2214/ajr.12.9335.
Saad Magalhaes C, de Albuquerque Pedrosa Fernandes T, Dias Fernandes T, de Lima Resende LA. A cross-sectional electromyography assessment in linear scleroderma patients. Pediatr Rheumatol Online J. 2014;12:27. doi:10.1186/1546-0096-12-27.
Voermans NC, Pillen S, de Jong EM, Creemers MC, Lammens M, van Alfen N. Morphea profunda presenting as a neuromuscular mimic. J Clin Neuromuscul Dis. 2008;9(4):407–14. doi:10.1097/CND.0b013e318175c495.
Moulton SJ, Kransdorf MJ, Ginsburg WW, Abril A, Persellin S. Eosinophilic fasciitis: spectrum of MRI findings. AJR Am J Roentgenol. 2005;184(3):975–8. doi:10.2214/ajr.184.3.01840975.
Ronneberger M, Janka R, Schett G, Manger B. Can MRI substitute for biopsy in eosinophilic fasciitis? Ann Rheum Dis. 2009;68(10):1651–2. doi:10.1136/ard.2008.103903.
Desvignes-Engelbert A, Sauliere N, Loeuille D, Blum A, Chary-Valckenaere I. From diagnosis to remission: place of MRI in eosinophilic fasciitis. Clin Rheumatol. 2010;29(12):1461–4. doi:10.1007/s10067-010-1508-1.
Succaria F, Kurban M, Kibbi AG, Abbas O. Clinicopathological study of 81 cases of localized and systemic scleroderma. J Eur Acad Dermatol Venereol JEADV. 2013;27(2):e191–6. doi:10.1111/j.1468-3083.2012.04581.x.
Torres JE, Sanchez JL. Histopathologic differentiation between localized and systemic scleroderma. Am J Dermatopathol. 1998;20(3):242–5.
Allen JA, Peterson A, Sufit R, Hinchcliff ME, Mahoney JM, Wood TA, et al. Post-epidemic eosinophilia-myalgia syndrome associated with l-tryptophan. Arthritis Rheum. 2011;63(11):3633–9. doi:10.1002/art.30514.
Kaufman LD, Krupp LB. Eosinophilia-myalgia syndrome, toxic-oil syndrome, and diffuse fasciitis with eosinophilia. Curr Opin Rheumatol. 1995;7(6):560–7.
Roufosse F. Hypereosinophilic syndrome variants: diagnostic and therapeutic considerations. Haematologica. 2009;94(9):1188–93. doi:10.3324/haematol.2009.010421.
Noth I, Strek ME, Leff AR. Churg–Strauss syndrome. Lancet (London, England) 2003;361(9357):587–94. doi:10.1016/S0140-6736(03)12518-4.
Polcari I, Moon A, Mathes EF, Gilmore ES, Paller AS. Headaches as a presenting symptom of linear morphea en coup de sabre. Pediatrics. 2014;134(6):e1715–9. doi:10.1542/peds.2014-0019.
Chiu YE, Vora S, Kwon EK, Maheshwari M. A significant proportion of children with morphea en coup de sabre and Parry–Romberg syndrome have neuroimaging findings. Pediatr Dermatol. 2012;29(6):738–48. doi:10.1111/pde.12001.
Blaszczyk M, Krolicki L, Krasu M, Glinska O, Jablonska S. Progressive facial hemiatrophy: central nervous system involvement and relationship with scleroderma en coup de sabre. J Rheumatol. 2003;30(9):1997–2004.
Appenzeller S, Montenegro MA, Dertkigil SS, Sampaio-Barros PD, Marques-Neto JF, Samara AM, et al. Neuroimaging findings in scleroderma en coup de sabre. Neurology. 2004;62(9):1585–9.
Amaral TN, Marques Neto JF, Lapa AT, Peres FA, Guirau CR, Appenzeller S. Neurologic involvement in scleroderma en coup de sabre. Autoimmune Dis. 2012;2012:719685. doi:10.1155/2012/719685.
Doolittle DA, Lehman VT, Schwartz KM, Wong-Kisiel LC, Lehman JS, Tollefson MM. CNS imaging findings associated with Parry–Romberg syndrome and en coup de sabre: correlation to dermatologic and neurologic abnormalities. Neuroradiology. 2015;57(1):21–34. doi:10.1007/s00234-014-1448-6.
Bischoff L, Derk CT. Eosinophilic fasciitis: demographics, disease pattern and response to treatment: report of 12 cases and review of the literature. Int J Dermatol. 2008;47(1):29–35. doi:10.1111/j.1365-4632.2007.03544.x.
Kroft EB, Groeneveld TJ, Seyger MM, de Jong EM. Efficacy of topical tacrolimus 0.1% in active plaque morphea: randomized, double-blind, emollient-controlled pilot study. Am J Clin Dermatol. 2009;10(3):181–7. doi:10.2165/00128071-200910030-00004.
Mancuso G, Berdondini RM. Localized scleroderma: response to occlusive treatment with tacrolimus ointment. Br J Dermatol. 2005;152(1):180–2. doi:10.1111/j.1365-2133.2004.06318.x.
Stefanaki C, Stefanaki K, Kontochristopoulos G, Antoniou C, Stratigos A, Nicolaidou E, et al. Topical tacrolimus 0.1% ointment in the treatment of localized scleroderma. An open label clinical and histological study. J Dermatol. 2008;35(11):712–8. doi:10.1111/j.1346-8138.2008.00552.x.
Chu CH, Cheng YP, Liang CW, Chiu HC, Jee SH, Lisa Chan JY, et al. Radiation recall dermatitis induced by topical tacrolimus for post-irradiation morphea. J Eur Acad Dermatol Venereol JEADV. 2016;. doi:10.1111/jdv.13739.
Cunningham BB, Landells ID, Langman C, Sailer DE, Paller AS. Topical calcipotriene for morphea/linear scleroderma. J Am Acad Dermatol. 1998;39(2 Pt 1):211–5.
Kreuter A, Gambichler T, Avermaete A, Jansen T, Hoffmann M, Hoffmann K, et al. Combined treatment with calcipotriol ointment and low-dose ultraviolet al phototherapy in childhood morphea. Pediatr Dermatol. 2001;18(3):241–5.
Dytoc MT, Kossintseva I, Ting PT. First case series on the use of calcipotriol-betamethasone dipropionate for morphoea. Br J Dermatol. 2007;157(3):615–8. doi:10.1111/j.1365-2133.2007.07971.x.
Pope E, Doria AS, Theriault M, Mohanta A, Laxer RM. Topical imiquimod 5% cream for pediatric plaque morphea: a prospective, multiple-baseline, open-label pilot study. Dermatology (Basel, Switzerland) 2011;223(4):363–9. doi:10.1159/000335560.
Man J, Dytoc MT. Use of imiquimod cream 5% in the treatment of localized morphea. J Cutan Med Surg. 2004;8(3):166–9. doi:10.1007/s10227-003-0112-2.
Campione E, Paterno EJ, Diluvio L, Orlandi A, Bianchi L, Chimenti S. Localized morphea treated with imiquimod 5% and dermoscopic assessment of effectiveness. J Dermatol Treat. 2009;20(1):10–3. doi:10.1080/09546630802132668.
Rodriguez-Castellanos M, Tlacuilo-Parra A, Sanchez-Enriquez S, Velez-Gomez E, Guevara-Gutierrez E. Pirfenidone gel in patients with localized scleroderma: a phase II study. Arthritis Res Ther. 2014;16(6):510. doi:10.1186/s13075-014-0510-4.
Kreuter A, Gambichler T, Avermaete A, Happe M, Bacharach-Buhles M, Hoffmann K, et al. Low-dose ultraviolet al phototherapy for extragenital lichen sclerosus: results of a preliminary study. J Am Acad Dermatol. 2002;46(2):251–5.
Stege H, Berneburg M, Humke S, Klammer M, Grewe M, Grether-Beck S, et al. High-dose UVA1 radiation therapy for localized scleroderma. J Am Acad Dermatol. 1997;36(6 Pt 1):938–44.
Gruss CJ, Von Kobyletzki G, Behrens-Williams SC, Lininger J, Reuther T, Kerscher M, et al. Effects of low dose ultraviolet A-1 phototherapy on morphea. Photodermatol Photoimmunol Photomed. 2001;17(4):149–55.
Kerscher M, Volkenandt M, Gruss C, Reuther T, von Kobyletzki G, Freitag M, et al. Low-dose UVA phototherapy for treatment of localized scleroderma. J Am Acad Dermatol. 1998;38(1):21–6.
Kreuter A, Hyun J, Stucker M, Sommer A, Altmeyer P, Gambichler T. A randomized controlled study of low-dose UVA1, medium-dose UVA1, and narrowband UVB phototherapy in the treatment of localized scleroderma. J Am Acad Dermatol. 2006;54(3):440–7. doi:10.1016/j.jaad.2005.11.1063.
Sator PG, Radakovic S, Schulmeister K, Honigsmann H, Tanew A. Medium-dose is more effective than low-dose ultraviolet al phototherapy for localized scleroderma as shown by 20-MHz ultrasound assessment. J Am Acad Dermatol. 2009;60(5):786–91. doi:10.1016/j.jaad.2008.12.013.
Camacho NR, Sanchez JE, Martin RF, Gonzalez JR, Sanchez JL. Medium-dose UVA1 phototherapy in localized scleroderma and its effect in CD34-positive dendritic cells. J Am Acad Dermatol. 2001;45(5):697–9. doi:10.1067/mjd.2001.117735.
Su O, Onsun N, Onay HK, Erdemoglu Y, Ozkaya DB, Cebeci F, et al. Effectiveness of medium-dose ultraviolet al phototherapy in localized scleroderma. Int J Dermatol. 2011;50(8):1006–13. doi:10.1111/j.1365-4632.2010.04843.x.
El-Mofty M, Mostafa W, El-Darouty M, Bosseila M, Nada H, Yousef R, et al. Different low doses of broad-band UVA in the treatment of morphea and systemic sclerosis. Photodermatol Photoimmunol Photomed. 2004;20(3):148–56. doi:10.1111/j.1600-0781.2004.00081.x.
Kerscher M, Meurer M, Sander C, Volkenandt M, Lehmann P, Plewig G, et al. PUVA bath photochemotherapy for localized scleroderma. Evaluation of 17 consecutive patients. Arch Dermatol. 1996;132(11):1280–2.
Usmani N, Murphy A, Veale D, Goulden V, Goodfield M. Photochemotherapy for localized morphoea: effect on clinical and molecular markers. Clin Exp Dermatol. 2008;33(6):698–704. doi:10.1111/j.1365-2230.2008.02890.x.
Pavlotsky F, Sakka N, Lozinski A, Barzilai A. Bath psoralen-UVA photochemotherapy for localized scleroderma: experience from a single institute. Photodermatol Photoimmunol Photomed. 2013;29(5):247–52. doi:10.1111/phpp.12063.
Joly P, Bamberger N, Crickx B, Belaich S. Treatment of severe forms of localized scleroderma with oral corticosteroids: follow-up study on 17 patients. Arch Dermatol. 1994;130(5):663–4.
Amy de la Breteque M, Rybojad M, Cordoliani F, Petit A, Juillard C, Flageul B, et al. Relapse of severe forms of adult morphea after oral corticosteroid treatment. J Eur Acad Dermatol Venereol JEADV. 2013;27(9):1190–1. doi:10.1111/jdv.12039.
Uziel Y, Feldman BM, Krafchik BR, Yeung RS, Laxer RM. Methotrexate and corticosteroid therapy for pediatric localized scleroderma. J Pediatr. 2000;136(1):91–5.
Torok KS, Arkachaisri T. Methotrexate and corticosteroids in the treatment of localized scleroderma: a standardized prospective longitudinal single-center study. J Rheumatol. 2012;39(2):286–94. doi:10.3899/jrheum.110210.
Kreuter A, Gambichler T, Breuckmann F, Rotterdam S, Freitag M, Stuecker M, et al. Pulsed high-dose corticosteroids combined with low-dose methotrexate in severe localized scleroderma. Arch Dermatol. 2005;141(7):847–52. doi:10.1001/archderm.141.7.847.
Seyger MM, van den Hoogen FH, de Boo T, de Jong EM. Low-dose methotrexate in the treatment of widespread morphea. J Am Acad Dermatol. 1998;39(2 Pt 1):220–5.
Mertens JS, van den Reek JM, Kievit W, van de Kerkhof PC, Thurlings RM, Radstake TR, et al. Drug survival and predictors of drug survival for methotrexate treatment in a retrospective cohort of adult patients with localized scleroderma. Acta Dermato Venereol. 2016;. doi:10.2340/00015555-2411.
Weibel L, Sampaio MC, Visentin MT, Howell KJ, Woo P, Harper JI. Evaluation of methotrexate and corticosteroids for the treatment of localized scleroderma (morphoea) in children. Br J Dermatol. 2006;155(5):1013–20. doi:10.1111/j.1365-2133.2006.07497.x.
Fitch PG, Rettig P, Burnham JM, Finkel TH, Yan AC, Akin E, et al. Treatment of pediatric localized scleroderma with methotrexate. J Rheumatol. 2006;33(3):609–14.
Cox D, Or G, Collins S, Byrne A, Irvine A, Watson R. Juvenile localised scleroderma: a retrospective review of response to systemic treatment. Ir J Med Sci. 2008;177(4):343–6. doi:10.1007/s11845-008-0217-0.
Kroft EB, Creemers MC, van den Hoogen FH, Boezeman JB, de Jong EM. Effectiveness, side-effects and period of remission after treatment with methotrexate in localized scleroderma and related sclerotic skin diseases: an inception cohort study. Br J Dermatol. 2009;160(5):1075–82. doi:10.1111/j.1365-2133.2008.09017.x.
Zulian F, Vallongo C, Patrizi A, Belloni-Fortina A, Cutrone M, Alessio M, et al. A long-term follow-up study of methotrexate in juvenile localized scleroderma (morphea). J Am Acad Dermatol. 2012;67(6):1151–6. doi:10.1016/j.jaad.2012.03.036.
Li SC, Torok KS, Pope E, Dedeoglu F, Hong S, Jacobe HT, et al. Development of consensus treatment plans for juvenile localized scleroderma: a roadmap toward comparative effectiveness studies in juvenile localized scleroderma. Arthritis Care Res. 2012;64(8):1175–85. doi:10.1002/acr.21687.
Martini G, Ramanan AV, Falcini F, Girschick H, Goldsmith DP, Zulian F. Successful treatment of severe or methotrexate-resistant juvenile localized scleroderma with mycophenolate mofetil. Rheumatology (Oxford, England) 2009;48(11):1410–3. doi:10.1093/rheumatology/kep244.
Mertens JS, Marsman D, van de Kerkhof PC, Hoppenreijs EP, Knaapen HK, Radstake TR, et al. Use of mycophenolate mofetil in patients with severe localized scleroderma resistant or intolerant to methotrexate. Acta Dermato Venereol. 2016;96(4):510–3. doi:10.2340/00015555-2297.
Hawley DP, Pain CE, Baildam EM, Murphy R, Taylor AE, Foster HE. United Kingdom survey of current management of juvenile localized scleroderma. Rheumatology (Oxford, England) 2014;53(10):1849–54. doi:10.1093/rheumatology/keu212.
Alcantara-Reifs CM, Garnacho-Saucedo GM, Salido-Vallejo R, de la Corte-Sanchez S, Garcia-Nieto AV. Imatinib treatment of therapy resistant generalized deep morphea. Dermatol Ther. 2015;28(5):271–3. doi:10.1111/dth.12248.
Coelho-Macias V, Mendes-Bastos P, Assis-Pacheco F, Cardoso J. Imatinib: a novel treatment approach for generalized morphea. Int J Dermatol. 2014;53(10):1299–302. doi:10.1111/ijd.12387.
Inamo Y, Ochiai T. Successful combination treatment of a patient with progressive juvenile localized scleroderma (morphea) using imatinib, corticosteroids, and methotrexate. Pediatr Dermatol. 2013;30(6):e191–3. doi:10.1111/j.1525-1470.2012.01882.x.
Moinzadeh P, Krieg T, Hunzelmann N. Imatinib treatment of generalized localized scleroderma (morphea). J Am Acad Dermatol. 2010;63(5):e102–4. doi:10.1016/j.jaad.2010.02.030.
Stausbol-Gron B, Olesen AB, Deleuran B, Deleuran MS. Abatacept is a promising treatment for patients with disseminated morphea profunda: presentation of two cases. Acta Dermato Venereol. 2011;91(6):686–8. doi:10.2340/00015555-1136.
Hirohata A, Hanafusa T, Igawa K, Inoue-Nishimoto T, Mabuchi-Kiyohara E, Nishide M, et al. Oral tacrolimus for the treatment of generalized morphea. Eur J Dermatol EJD. 2016;26(1):112–3. doi:10.1684/ejd.2015.2689.
Frumholtz L, Roux J, Bagot M, Rybojad M, Bouaziz JD. Treatment of generalized deep morphea with everolimus. JAMA Dermatol. 2016;. doi:10.1001/jamadermatol.2016.2338.
Scuderi N, Ceccarelli S, Onesti MG, Fioramonti P, Guidi C, Romano F, et al. Human adipose-derived stromal cells for cell-based therapies in the treatment of systemic sclerosis. Cell Transplant. 2013;22(5):779–95. doi:10.3727/096368912X639017.
Michet CJ Jr, Doyle JA, Ginsburg WW. Eosinophilic fasciitis: report of 15 cases. Mayo Clin Proc. 1981;56(1):27–34.
Berianu F, Cohen MD, Abril A, Ginsburg WW. Eosinophilic fasciitis: clinical characteristics and response to methotrexate. Int J Rheum Dis. 2015;18(1):91–8. doi:10.1111/1756-185x.12499.
Mertens JS, Zweers MC, Kievit W, Knaapen HK, Gerritsen M, Radstake TR, et al. High-dose intravenous pulse methotrexate in patients with eosinophilic fasciitis. JAMA Dermatol. 2016;. doi:10.1001/jamadermatol.2016.2873.
Mendoza FA, Bai R, Kebede AG, Jimenez SA. Severe eosinophilic fasciitis: comparison of treatment with d-penicillamine plus corticosteroids vs. corticosteroids alone. Scand J Rheumatol. 2016;45(2):129–34. doi:10.3109/03009742.2015.1067713.
Khanna D, Agrawal H, Clements PJ. Infliximab may be effective in the treatment of steroid-resistant eosinophilic fasciitis: report of three cases. Rheumatology (Oxford, England) 2010;49(6):1184–8. doi:10.1093/rheumatology/keq062.
Nassonova VA, Ivanova MM, Akhnazarova VD, Oskilko TG, Bjelle A, Hofer PA, et al. Eosinophilic fasciitis. Review and report of six cases. Scand J Rheumatol. 1979;8(4):225–33.
Alonso-Castro L, de las Heras E, Moreno C, Fleta-Asin B, Munoz-Zato E, Carrillo R et al. Eosinophilic fasciitis/generalized morphea overlap successfully treated with azathioprine. Int J Dermatol. 2014;53(11):1386–8. doi:10.1111/j.1365-4632.2012.05741.x.
Jones AC, Doherty M. Eosinophilic fasciitis with late onset arthritis responsive to sulfasalazine. J Rheumatol. 1993;20(4):750–1.
Bukiej A, Dropinski J, Dyduch G, Szczeklik A. Eosinophilic fasciitis successfully treated with cyclosporine. Clin Rheumatol. 2005;24(6):634–6. doi:10.1007/s10067-005-1099-4.
De Jonge-Bok JM, Steven MM, Eulderink F, Cats A. Diffuse (eosinophilic) fasciitis. A series of six cases. Clin Rheumatol. 1984;3(3):365–73.
Scheinberg M, Hamerschlak N, Kutner JM, Ribeiro AA, Ferreira E, Goldenberg J, et al. Rituximab in refractory autoimmune diseases: Brazilian experience with 29 patients (2002–2004). Clin Exp Rheumatol. 2006;24(1):65–9.
Espinoza F, Jorgensen C, Pers YM. Efficacy of tocilizumab in the treatment of eosinophilic fasciitis: report of one case. Jt Bone Spine Revue Rhum. 2015;82(6):460–1. doi:10.1016/j.jbspin.2015.02.008.
Oza VS, Walsh R, North J, Berger TG, Murase JE. Treatment of eosinophilic fasciitis with sirolimus. JAMA Dermatol. 2016;152(4):488–90. doi:10.1001/jamadermatol.2016.0048.
Schiener R, Behrens-Williams SC, Gottlober P, Pillekamp H, Peter RU, Kerscher M. Eosinophilic fasciitis treated with psoralen-ultraviolet A bath photochemotherapy. Br J Dermatol. 2000;142(4):804–7.
Pimenta S, Bernardes M, Bernardo A, Brito I, Castro L, Simoes-Ventura F. Intravenous immune globulins to treat eosinophilic fasciitis: a case report. Jt Bone Spine Revue Rhum. 2009;76(5):572–4. doi:10.1016/j.jbspin.2009.06.001.
Cetkovsky P, Koza V, Cetkovska P, Svojgrova M. Successful treatment of severe Shulman’s syndrome by allogeneic bone marrow transplantation. Bone Marrow Transplant. 1998;21(6):637–9. doi:10.1038/sj.bmt.1701137.
Kim SW, Rice L, Champlin R, Udden MM. Aplastic anemia in eosinophilic fasciitis: responses to immunosuppression and marrow transplantation. Haematologia. 1997;28(3):131–7.
Bang RL. Case report: use of skin grafting to keep a scleroderma patient ambulant. Plast Reconstr Surg. 1979;63(5):732–4.
Consorti G, Tieghi R, Clauser LC. Frontal linear scleroderma: long-term result in volumetric restoration of the fronto-orbital area by structural fat grafting. J Craniofac Surg. 2012;23(3):e263–5. doi:10.1097/SCS.0b013e31824ef7e8.
Ibler KS, Gramkow C, Siemssen PA. Autologous fat transplantation for the treatment of linear scleroderma en coup de sabre. Skinmed. 2015;13(1):74–6.
Karaaltin MV, Akpinar AC, Baghaki S, Akpinar F. Treatment of “en coup de sabre” deformity with adipose-derived regenerative cell-enriched fat graft. J Craniofac Surg. 2012;23(2):e103–5. doi:10.1097/SCS.0b013e3182418ce8.
Zanelato TP, Marquesini G, Colpas PT, Magalhaes RF, Moraes AM. Implantation of autologous fat globules in localized scleroderma and idiopathic lipoatrophy—report of five patients. Anais Bras Dermatol. 2013;88(6 Suppl 1):120–3. doi:10.1590/abd1806-4841.20132115.
Deshmukh SP, Dogra BB, Sharma YK, Deo KS. Autologus fat transfer for restoration of facial contour in “progressive facial hemiatrophy”. Indian J dermatol Venereol Leprol. 2012;78(6):775. doi:10.4103/0378-6323.102398.
Handler MZ, Wulkan AJ, Stricker SJ, Schachner LA. Linear morphea and leg length discrepancy: treatment with a leg-lengthening procedure. Pediatr Dermatol. 2013;30(5):616–8. doi:10.1111/pde.12169.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this review.
Conflict of interest
Jorre S. Mertens, Marieke M.B. Seyger, Rogier M. Thurlings, Timothy R.D.J. Radstake, and Elke M.G.J. de Jong have no conflicts of interest.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Mertens, J.S., Seyger, M.M.B., Thurlings, R.M. et al. Morphea and Eosinophilic Fasciitis: An Update. Am J Clin Dermatol 18, 491–512 (2017). https://doi.org/10.1007/s40257-017-0269-x
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40257-017-0269-x