
Abstract
Despite treatment with statins, patients with elevated low-density lipoprotein cholesterol (LDL-C) and triglycerides remain at increased risk for adverse cardiovascular events. Consequently, novel pharmaceutical drugs have been developed to control and modify the composition of blood lipids to ultimately prevent fatal cardiovascular events in patients with dyslipidaemia. This article reviews established and emerging lipid-lowering drugs regarding their mechanism of action, development stage, ongoing clinical trials, side effects, effect on blood lipids and reduction in cardiovascular morbidity and mortality. We conducted a keyword search to identify studies on established and emerging lipid modifying drugs. Results were summarized in a narrative overview. Established pharmaceutical treatment options include the Niemann-Pick-C1 like-1 protein (NPC1L1) inhibitor ezetimibe, the protein convertase subtilisin-kexin type 9 (PCSK9) inhibitors alirocumab and evolocumab, fibrates as peroxisome proliferator receptor alpha (PPAR-α) activators, and the omega-3 fatty acid icosapent ethyl. Statins are recommended as the first-line therapy for primary and secondary cardiovascular prevention in patients with hypercholesterinaemia and hypertriglyceridemia. For secondary prevention in hypercholesterinaemia, second-line options such as statin add-on or statin-intolerant treatments are ezetimibe, alirocumab and evolocumab. For secondary prevention in hypertriglyceridemia, second-line options such as statin add-on or statin-intolerant treatments are icosapent ethyl and fenofibrate. Robust data for these add-on therapeutics in primary cardiovascular prevention remains scarce. Recent biotechnological advances have led to the development of innovative small molecules (bempedoic acid, lomitapide, pemafibrate, docosapentaenoic and eicosapentaenoic acid), antibodies (evinacumab), antisense oligonucleotides (mipomersen, volanesorsen, pelcarsen, olezarsen), small interfering RNA (inclisiran, olpasiran), and gene therapies for patients with dyslipidemia. These molecules specifically target new cellular pathways, such as the adenosine triphosphate-citrate lyase (bempedoic acid), PCSK9 (inclisiran), angiopoietin-like 3 (ANGPTL3: evinacumab), microsomal triglyceride transfer protein (MTP: lomitapide), apolipoprotein B-100 (ApoB-100: mipomersen), apolipoprotein C-III (ApoC-III: volanesorsen, olezarsen), and lipoprotein (a) (Lp(a): pelcarsen, olpasiran). The authors are hopeful that the development of new treatment modalities alongside new therapeutic targets will further reduce patients’ risk of adverse cardiovascular events. Apart from statins, data on new drugs’ use in primary cardiovascular prevention remain scarce. For their swift adoption into clinical routine, these treatments must demonstrate safety and efficacy as well as cost-effectiveness in randomized cardiovascular outcome trials.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Statins remain the cornerstone for the primary and secondary prevention and cardiovascular events in patients with hypercholesterinaemia and hypertriglyceridemia. |
Ezetimibe, alirocumab, evolocumab, icosapent ethyl and fenofibrate are second-line treatment alternatives as add-on treatment to statin or as monotherapy for statin-intolerant patients with dyslipidaemia. |
Notable emerging lipid-lowering treatments include bempedoic acid, the siRNA PCSK9 inhibitor inclisiran, the ANGPTL3 inhibitor evinacumab, the MTP inhibitor lomitapide, the ApoB-100 inhibitor mipomersen and the ApoC-III degradation molecules volanesorsen and olezarsen, as well as the Lp(a) inhibitors pelcarsen and olpasiran. |
1 Introduction
Cardiovascular diseases (CVD) are the leading cause of death worldwide [1]. In 2019, out of a total of 56.5 million deaths, 32.8% (18.6 million) were attributable to CVD—compared with 17.8% (10.1 million) deaths from neoplasms [2]. Among CVD, most deaths are attributable to ischemic heart diseases (16.2%, 9.1 million), strokes (11.6%, 6.6 million) and hypertensive heart diseases (2.0%, 1.2 million) [2].
Metabolic, behavioural, environmental and occupational risk factors adversely affect the incidence and progression of CVD [1, 3,4,5,6]. High systolic blood pressure, dietary risks and elevated low-density lipoprotein cholesterol (LDL-C) levels count among the top three cardiovascular risk factors; each attributable for 25.0%, 17.2%, and 11.0% of CVD deaths, respectively [2]. Despite the widespread availability of low-cost statins, the majority of patients with dyslipidaemia do not attain adequate LDL-C levels by existing lifestyle modifications and pharmacological treatment strategies [7,8,9,10,11]. The DA VINCI study found that out of 5888 patients with dyslipidaemia who were enrolled in 18 European countries, only 33% achieved their risk-based LDL-C goal recommended in the European Society of Cardiology (ESC) 2019 guidelines [12]. These results were consistent across European countries [13,14,15]. Accordingly, an Australian study of 61,407 patients reported that only 36% achieved their recommended LDL-C levels [16]. Similarly, Klimchak et al. use claims data from 44 million US inhabitants to estimate that only 36% of high-risk patients with atherosclerotic CVD attain the recommended LDL-C level of < 70 mg/dL [17]. There are several reasons why the majority of patients do not sufficiently reach LDL-C levels. First, ESC and AHA/ACC/multi-society guidelines have recently pursued more string LDL-C reductions according to the principle ‘the lower, the better’ [18,19,20] For example, the recommended LDL-C level for very high-risk patients has been lowered from < 70 to < 55 mg/mL. Second, patients may be reluctant to pursue lower LDL-C levels with lifestyle modifications and regular pharmaceutical therapy in absence of short-term consequences. Third, high prices for new treatments, such as PCSK9 inhibitors, have been identified as a barrier to swift coverage and reimbursement by insurers [21,22,23,24]. Finally, there may simply not be a sufficient number of add-on and alternative therapies to statins given that approximately 9.1% of patients are intolerant to statins [25].
As a consequence, multiple authors have concluded that there is a significant unmet medical need for patients with dyslipidaemia to reduce blood lipid levels to prevent the incidence of adverse cardiovascular events [26,27,28,29,30]. Therefore, in this article, we describe and review established and emerging lipid-lowering drugs regarding their molecular mechanism of action, effects on blood lipids, reduction of cardiovascular morbidity and mortality in primary and secondary prevention, and side effects.
2 Established Pharmaceutical Therapies
Available guidelines (ESC 2019 and AHA/ACC/multi-society 2018) entail a list of pharmaceutical interventions that may be used in patients with dyslipidaemia to lower blood lipids and ultimately the risk of adverse cardiovascular events [19, 20]. An overview of these drugs is provided in Fig. 1.

Established lipid-lowering therapies for cardiovascular prevention. LDL-C, TG and MACE reductions for ezetimibe, evolocumab, alirocumab, fibrates and icosapent ethyl are presented for combination therapy with statins. Data sources as referenced in the accompanying text passages. aThe presented MACE reduction for high-intensity statins refers to the incremental benefit relative to low-/medium-intensity statins. HMG-CoA 3-hydroxy-3-methylglutaryl coenzyme A, IV intravenous, LDL-C low-density lipoprotein cholesterol, MACE major adverse cardiovascular events, MoA mechanism of action, NPC1L1 Niemann-Pick C1-like 1 protein, PCSK9 proprotein convertase subtilisin-kexin type 9, PO perioral, PPAR-α peroxisome proliferator receptor alpha, RoA route of administration, SC subcutaneous, TG triglyceride
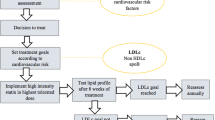
For hypercholesterinaemia (Fig. 2), the ESC and AHA/ACC/multi-society guidelines recommend a risk-stratified treatment with statins, ezetimibe and PCSK9 inhibitors. Both guidelines recommend statins as the first-line therapy in primary and secondary prevention for patients with hypercholesterinaemia. Ezetimibe is recommended as the second-line treatment in patients who do not attain their LDL-C goal on maximum tolerated statin therapy by both guidelines for primary and secondary prevention. PCSK9 inhibitors are recommended by both guidelines for secondary cardiovascular prevention in very high-risk patients who do not reach their LDL-C goal with maximum tolerated statin therapy and ezetimibe. ESC guidelines further state that PCSK9 inhibitors may be considered for primary cardiovascular prevention in very high-risk patients who do not achieve their LDL-C goal with maximum tolerated statin therapy and ezetimibe. AHA/ACC/multi-society guidelines specify that treatment with PSCK9 inhibitors should include a physician–patient discussion about the net benefit and cost, given that PCSK9 inhibitors were found to be of low value. ESC guidelines further specify that ezetimibe and/or PSCK9 inhibitors should be considered in patients with hypercholesterinaemia who are intolerant to statins.

ESC and AHA/ACC guidelines for cholesterol-lowering treatments. We extracted recommendations for statins, ezetimibe and PCSK9 inhibitors in the treatment of patients with elevated plasma cholesterol from ESC 2019 and AHA/ACC/multi-society 2018 guidelines. For AHA/ACC/multi-society guidelines, the COR is categorized as class I (strong), class IIa (moderate), class IIb (weak), class III: no benefit (moderate) and class III: harm (strong). For ESC guidelines, the COR is categorized as class I (recommended) class IIa (should be considered), class IIb (may be considered) and class III (not recommended). For US guidelines, the LOE is categorized as level A (high-quality randomized evidence), level B-R (moderate-quality randomized evidence), level B-NR (moderate-quality non-randomized evidence), level C-LD (limited data) and level C-EO (expert opinion). For EU guidelines, the LOE is categorized as level A (data from multiple RCT or meta-analyses), level B (data from one RCT or large non-randomized trial) and level C (consensus expert opinion and/or small studies, retrospective studies, registries). ACC American College of Cardiology, AHA American Heart Association, ASCVD atherosclerotic cardiovascular disease, COR class (strength) of recommendation, CVD cardiovascular disease, ESC European Society of Cardiology, FH familial hypercholesterolemia, LDL-C low-density lipoprotein cholesterol, LOE level (quality) of evidence, PCSK9 proprotein convertase subtilisin-kexin type 9, RCT randomized controlled trial
For hypertriglyceridemia (Fig. 3), both guidelines recommend a risk-stratified treatment with statins, icosapent ethyl and/or fibrates. Statins are the first-line treatment for hypertriglyceridemia in both guidelines. Icosapent ethyl and fibrate are second-line treatments that should/may be considered in combination with a statin. ESC guidelines refer to triglyceride levels > 2.3 mmol/L (> 200 mg/dL) for icosapent ethyl and/or fibrates, whilst AHA/ACC/multi-society guidelines refer to triglyceride levels beyond > 5.6 mmol/L (> 500 mg/dL).

ESC and AHA/ACC guidelines for hypertriglyceridemia treatments. We extracted recommendations for statins, icosapent ethyl and fibrates in the treatment of hypertriglyceridemia from ESC 2019 and AHA/ACC/multi-society 2018 guidelines. For AHA/ACC/multi-society guidelines, the COR is categorized as class I (strong), class IIa (moderate), class IIb (weak), class III: no benefit (moderate) and class III: harm (strong). For ESC guidelines, the COR is categorized as class I (recommended) class IIa (should be considered), class IIb (may be considered) and class III (not recommended). For US guidelines, the LOE is categorized as level A (high-quality randomized evidence), level B-R (moderate-quality randomized evidence), level B-NR (moderate-quality non-randomized evidence), level C-LD (limited data) and level C-EO (expert opinion). For EU guidelines, the LOE is categorized as level A (data from multiple RCT or meta-analyses), level B (data from one RCT or large non-randomized trial) and level C (consensus expert opinion and/or small studies, retrospective studies, registries). ACC American College of Cardiology, AHA American Heart Association, ASCVD atherosclerotic cardiovascular disease, COR class (strength) of recommendation, CVD cardiovascular disease, ESC European Society of Cardiology, LDL-C low-density lipoprotein cholesterol, LOE level (quality) of evidence, RCT randomized-controlled trial, TG triglyceride
In this section, we review the clinical trial evidence that supports these recommendations. Furthermore, we briefly summarize these lipid-lowering drugs’ mechanism of action, side effects and effects on blood lipids.
2.1 Statins
Statins reduce the internal synthesis of cholesterol by a competitive inhibition of the rate-limiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase [19]. The reduced internal synthesis of cholesterol results in an increased surface expression of LDL-C receptors, which then increases the uptake of LDL-C. Ultimately, this results in lower serum LDL-C, ApoB-100 and triglyceride levels. Given the competitive inhibition of HMG-CoA, statins’ dose-dependent efficacy is used in clinical practice to escalate treatment from low-/medium-intensity to high-intensity statin treatment for at-risk patients. Low-/medium-intensity statins reduce LDL-C by − 30% to − 50% and triglycerides by − 20%, whilst high-intensity statins reduce LDL-C by more than − 50% and triglycerides by up to − 40% [19, 31]. Statins also elevate high-density lipoprotein cholesterol (HDL-C) in a dose-dependent manner by up to + 10% [32]. Studies reported no or only a minor increase in lipoprotein (a) (Lp(a)) levels among patients treated with statins [33, 34]. Beyond the modification of blood lipid levels, in vitro and in vivo studies showed that statins exert beneficial cardioprotective pleiotropic effects [35, 36]. These ‘include improvement of endothelial dysfunction, increased nitric oxide bioavailability, antioxidant properties, inhibition of inflammatory responses, and stabilization of atherosclerotic plaques’ [35]. However, the significance of these pleiotropic effects in clinical practice remains debated [37].
Statins’ relevance in cardiovascular prevention is supported by multiple meta-analyses across several patient subgroups [38,39,40,41,42,43,44,45,46,47,48,49,50]. According to an analysis of 170,000 patients, a 38.67 mg/dl (1 mmol/l) reduction in LDL-C was associated with a − 22% (95% CI − 20 to − 24, p < 0.0001) decreased risk in major adverse cardiovascular events (MACE) [38], thereby confirming LDL-C as a valid surrogate parameter for the clinical endpoints MACE and cardiovascular death. Overall, low-/moderate-intensive statin therapy reduces the risk of MACE by − 22% (95% CI − 19 to − 24, p < 0.0001) [38]. Further intensifying statin therapy provides an additional MACE risk reduction of − 15% (95% CI − 11 to − 18, p < 0.0001) [38]. Results were consistent across primary and secondary prevention cohorts [40, 41, 48, 49, 51]. In patients with low risk of CVD (< 10% risk), statin therapy reduced the risk of major vascular events (RR 0.79, 95% CI 0.77–0.81 per 1.0 mmol/L reduction in LDL-C) irrespective of previous vascular diseases [40]. Statin therapy reduced the risk of all-cause mortality by 9% per 1.0 mmol/L LDL-C reduction.
The most clinically significant adverse effects of statin therapy are muscle-associated symptoms including myopathy (11 per 100,000 patient years) up to rhabdomyolysis (3 per 100,000 patient years) [52], an elevation of liver enzymes, hyperglycaemia [53], new onset of diabetes mellitus type 2 [54] and proteinuria. Evidence for an increased risk of haemorrhagic stroke remains inconclusive [38, 54, 55]. As statins are metabolized through the hepatic pathway, inhibitors and inducers of the cytochromes P450 (CYP) system, for example, anti-infectives, calcium antagonists, cyclosporine and grapefruit juice, may cause drug–drug interactions which impact their bioavailability; ultimately leading to the aggravation of side effects or a limited therapeutic efficacy. A recent meta-analysis of 176 studies with a total of 4.1 million patients found that 9.1% of patients are intolerant to statins [25]. Age, female gender, Asian and Black race, obesity, diabetes mellitus, hypothyroidism, chronic liver disease, renal failure, antiarrhythmic drugs, calcium channel blockers, alcohol consumption and higher statin dose were associated with a greater risk of statin intolerance [25].
Albeit statins are the first-line therapeutic option to lower blood lipids in patients with dyslipidaemia, for example, those with hypercholesterinaemia or hypertriglyceridemia, many patients’ blood lipids remain above the risk-stratified target levels defined by ESC guidelines. To reduce the risk of adverse cardiovascular events among these at-risk patients, new pharmacological treatment options have been developed over the past two decades. New treatments may be prescribed adjuvant to moderate-/high-intensity statins or as monotherapy for patients who are intolerant to statins.
2.2 Ezetimibe
Ezetimibe selectively inhibits the absorption of cholesterol in the small intestine [by interfering with the Niemann-Pick C1-like 1 protein (NPC1L1)] without modifying uptake of other nutrients and vitamins [56, 57]. A genetic sequencing study involving more than 90,000 patients found significantly lower LDL-C levels and a lower risk of coronary heart diseases among patients with NPC1L1 mutations [58] Compared with statin monotherapy, a combination of ezetimibe and statins reduces LDL-C by − 24%, ApoB-100 by − 14%, triglycerides by − 12% and high-sensitivity C-reactive protein (hsCRP) by − 13%, whilst maintaining HDL-C levels [59,60,61]. The randomized controlled double-blind Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT) entailing 18,144 patients after acute coronary syndrome with a follow-up of 6 years found that ezetimibe added to simvastatin compared with simvastatin monotherapy significantly reduced the risk of MACE by − 6% (95% CI − 1 to − 11, p = 0.016). Although the earlier ENHANCE trial could not confirm that an addition of ezetimibe to statins significantly reduces intima-media thickness in patients with familial hypercholesterolemia, the SHARP and SEAS trials highlight ezetimibe’s role in the prevention of ischaemic cardiovascular events [62,63,64,65]. Ezetimibe’s safety and efficacy has been confirmed in multiple meta-analyses [61, 66,67,68,69,70].
The Japanese EWTOPIA 75 trial investigated ezetimibe as monotherapy for primary cardiovascular prevention in patients aged 75 years or older. A total of 3796 patients were randomly assigned to ezetimibe (10 mg daily) versus usual care. After a median follow-up of 4.1 years, ezetimibe reduced the incidence of MACE by − 34% (HR 0.66, 95% CI 0.50–0.86, p = 0.002) [71].
Compared with statin monotherapy, adjuvant ezetimibe was not found to increase the occurrence of side effects or treatment discontinuation.
2.3 PCSK9 Inhibitors
Evolocumab and alirocumab are monoclonal antibodies which inhibit the proprotein convertase subtilisin-kexin type 9 (PCSK9). These antibodies reduce the concentration of PCSK9 plasma levels, which results in an increased expression of LDL-C receptors and in turn reduced LDL-C levels. Consequently, genetic studies found significantly lower LDL-C levels and fewer cardiovascular events in patients with PCSK9 loss-of-function mutations compared with control [72,73,74]. Treatment with PCSK9 inhibitors reduces LDL-C by approximately − 60% [75,76,77,78,79]. PCSK9 inhibitors also lower triglycerides and Lp(a), whilst increasing ApoA-I. Coherent with the LDL-C reduction, the Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk (FOURIER) and the Evaluation of Cardiovascular Outcomes After an Acute Coronary Syndrome During Treatment With Alirocumab (ODYSSEY) trials found that PCSK9 inhibitors significantly reduce the risk of MACE by − 15% in patients with prior acute cardiovascular syndrome. These double-blinded trials randomized 27,564 and 18,924 patients, respectively, to receive evolcoumab/alirocumab or placebo in addition to statins with a follow-up period of 2.2 and 2.8 years, respectively. The cardioprotective effects of PCSK9 inhibitors were subsequently confirmed in other clinical trials and meta-analyses [80,81,82,83,84,85].
Schmidt et al.’s meta-analysis of PCSK9 inhibitors includes trials with primary and secondary prevention cohorts [80]. Trials either include patients with established CVD or at high CVD risk. However, outcome data are infrequently reported for the primary and secondary prevention cohorts, separately. Data from the GLAGOV, ODYSSEY COMBO I and ODYSSEY LONG TERM trials show greater efficacy of PCSK9 inhibitors in patients with prior CVD (or myocardial infarction) in terms of LDL-C and percent atheroma volume [86,87,88].
Subcutaneous application of the monoclonal antibodies results in local injection site reactions. PCSK9 inhibitors were also discussed to induce the expression of auto-antibodies and increase the risk of diabetes mellitus [89, 90].
2.4 Fibrates
Fibrates, such as fenofibrate, bezafibrat and gemfibrozil, activate the peroxisome proliferator receptor alpha (PPAR-α) and thereby reduce blood lipid levels. Fibrates reduce concentrations of triglycerides by approximately − 50% and LDL-C by − 20% whilst increasing HDL-C by up to + 20% depending on the fibrate class [19]. Treatment with fibrates is associated with an increased risk of gastrointestinal pain, skin rashes, myopathy and liver enzyme elevation [19]. Evidence supporting the prescription of fibrates for cardiovascular prevention remains debated. The ACCORD, LEADER and VA-COOP trials could not confirm that fibrates reduce the incidence of MACE at a 5% significance level, whilst a significant MACE reduction was observed in the FIELD and VA-HIT trials [91,92,93,94]. A meta-analysis pooling outcomes from these trials found that fibrates significantly reduce the risk of MACE by −10% (95% CI − 0 to − 18, p = 0.048) [94, 95]. The same analysis also finds that fibrates reduce the incidence of coronary events, but not death. Consequently, fibrates’ role in cardiovascular prevention remains debated. ESC and AHA/ACC/multi-society guidelines recommend fibrates for patients with dyslipidaemia with elevated triglycerides despite statin treatment. However, their role in cardiovascular prevention and triglyceride reduction is projected to diminish following icosapent ethyl’s approval.
2.5 Omega-3 Fatty Acids
Since observational studies suggested high omega-3 fatty acid levels are associated with a lower risk of cardiovascular events and death, researchers evaluated these acids in randomized controlled trials (RCTs). Particular interest surrounded the ‘fish oils’ docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA) and most recently docosapentaenoic acid (DPA). Although several large-scale RCTs evaluated these omega-3 fatty acids, the public and scientific community remains puzzled about their differential outcomes. The STRENGTH, VITAL, ASCEND, ORIGIN, OMEGA, ALPHA-OMEGA, GISSI-HF and GISSI-P trials randomized patients to receive either a combination of EPA and DHA or placebo; yet only the GISSI trials noted a statistically significant effect on cardiovascular events and mortality [96,97,98,99,100,101,102,103]. In contrast, patients receiving only high-dose EPA in the REDUCE-IT and JELIS trials were at − 25% (95% CI − 17 to − 32, p < 0.001) and −19% (95% CI − 5 to − 31, p = 0.011) lower risk for MACE, respectively [104, 105].
These differential treatment effects could be subject to the administered omega-3 fatty acid, dosing regimen, comparator and studied patient population. In vitro and in vivo experimental studies highlight EPA’s and DHA’s distinct effects on inflammation, cellular membranes, platelets and triglycerides, which could result in the observed differential MACE outcomes [106, 107]. Albeit the exact MoA is unknown, scientists hypothesize that EPA exerts pleiotropic cardiovascular effects beyond lowering triglycerides: anti-inflammatory, anti-thrombotic, membrane stabilizing, plaque stabilizing, anti-arrhythmic and anti-hypertensive [105, 106, 108, 109]. Systematic reviews of the aforementioned studies with meta-analyses and meta-regressions show a dose-dependent association between omega-3 fatty acids’ titration and cardiovascular events [104, 106, 109,110,111]. The REDUCE-IT and JELIS trials randomized patients to receive 4 g and 1.8 g of highly purified EPA, whilst the EPA dosage was significantly lower in the other trials. Although there has been significant public debate surrounding the comparator used in the REDUCE-IT trial (mineral oil) [112, 113], experts in both regulatory agencies in the USA and the European Union (EU)—from the US Food and Drug Administration (FDA) and the European Medicines Agency (EMA)—approved highly purified EPA, icosapent ethyl, for cardiovascular prevention in patients with dyslipidaemia. It is estimated that the inappropriate use of mineral oil as a comparator may have overestimated the MACE outcome by up to 3% [114]. The resulting net MACE reduction of − 22% then closely resembles the − 19% MACE reduction observed in the JELIS trial.
Notably, the REDUCE-IT trial enrolled 29% of patients without CVD (primary prevention) and 71% of patients with established CVD (secondary prevention) [105]. Subgroup analyses showed a non-significantly greater MACE reduction in the secondary prevention (HR 0.73, 95% CI 0.65–0.81) than the primary prevention cohort (HR 0.88, 95% CI 0.70–1.10, p = 0.14). A post hoc analysis of the REDUCE-IT trial confirmed icosapent ethyl’s efficacy among patients with prior myocardial infarction (MACE HR 0.74, 95% CI 0.65–0.85, p < 0.001) [115]. Similarly, the JELIS trial, which predominantly included patients without established CVD, demonstrated a consistent efficacy of EPA in the primary (HR 0.82, 95% CI 0.63–1.06, p = 0.132) and secondary prevention cohorts (HR 0.81, 95% CI 0.657–0.998, p = 0.048) [104]. Consequently, recent economic analyses found icosapent ethyl could be a cost-effective treatment, particularly for secondary cardiovascular prevention, in the USA, Germany, UK, Canada, Australia, and Japan [22, 23, 116,117,118,119,120,121].
Treatment with EPA was observed to be associated with a higher incidence of arterial fibrillation, serious bleeding events (e.g. haemorrhagic strokes) and peripheral oedema, but with a lower incidence of diarrhoea, gastrointestinal pain and anaemia than monotherapy with statins [105, 106]
2.6 Other Drugs
Several other pharmacological treatments are/were used to treat patients with dyslipidaemia, such as bile acid sequestrants and niacin. Bile acid sequestrants, such as cholestyramine, colesevelam and colestipol, bind to intestinal cholesterol and thereby inhibit its absorption in the small intestine, effectively removing it from the enterohepatic cholesterol circulation [19]. Although bile acid sequestrants are associated with a significant reduction in LDL-C and MACE, clinical trials supporting their efficacy were conducted before the introduction of statins, ezetimibe, PCSK9 inhibitors and icosapent ethyl [122]. In the EU, treatment with niacin was stopped after the AIM-HIGH and HPS2-THRIVE trials showed no improvement in MACE and an increased occurrence of undesirable side effects [123, 124].
3 Emerging Pharmaceutical Therapies
Recently, several novel pharmaceutical therapies with innovative MoA have been approved by the FDA and/or EMA to treat patients with dyslipidaemia [125], many of which are currently being investigated in cardiovascular outcome trials (CVOT). An even greater number of therapeutics are currently under clinical development. If successful, these novel pharmaceutical therapies could soon be approved and could transform the management of patients with dyslipidaemia. This section reviews these emerging pharmaceutical therapies regarding their MoA, first safety and efficacy results, and ongoing clinical trials (Fig. 4).

Emerging pharmaceutical therapies for the treatment of dyslipidemia. Drug types were categorized and illustrated as small molecules (capsule), antibodies (syringe) and gene therapeutics (double-stranded DNA helix). Data sources as referenced in the accompanying text passages. aThe drug class could not be identified for the compound NNC0385-0434 A. ANGPTL3 angiopoietin-like 3, ApoB-100 apolipoprotein B-100, ApoC-III apolipoprotein C-III, ATP adenosine triphosphate, CVOT cardiovascular outcome trial, FCS familial chylomicronemia syndrome, FDA US Food and Drug Administration approval, HeFH heterozygous familial hypercholesterolemia, HoFH homozygous familial hypercholesterolemia, HTG hypertriglyceridemia, IV intravenous, Lp(a) lipoprotein(a), MoA mechanism of action, MTG microsomal triglyceride transfer protein, P0 pre-clinic, P1 phase 1, P2 phase 2, P3 phase 3, PCSK9 proprotein convertase subtilisin-kexin type 9, PPAR-α peroxisome proliferator receptor alpha, PO perioral, RoA route of administration, SC subcutaneous, sHTG severe hypertriglyceridemia, siRNA small interfering ribonucleic acid
3.1 Bempedoic Acid
Bempedoic acid inhibits adenosine triphosphate (ATP)-citrate lyase and thereby cholesterol biosynthesis [126]. The ATP-citrate lyase enzyme specifically catalyses the synthesis of Acetyl-CoA, which is an underlying substrate to synthesize HMG-CoA. Although both statins and bempedoic acid inhibit the cholesterol biosynthesis, bempedoic acid inhibits an enzyme that is located upstream of the HMG-CoA-reductase. Furthermore, bempedoic acid is a prodrug that is activated by the SLC27A2 enzyme which is not present in muscle cells. In contrast to statins, a lower incidence of myopathy, rhabdomyolysis or other muscle-related side effects were observed in patients receiving bempedoic acid [127]. The most common side effects are increased uric acid levels and gout [128]. Therefore, bempedoic acid has been an eagerly awaited pharmacological alternative to reduce LDL-C levels among statin-intolerant patients.
Bempedoic acid’s safety and efficacy in statin-intolerant patients has been evaluated across the family of phase 3 CLEAR trials: [127, 129,130,131]
-
The CLEAR Tranquillity trial evaluated bempedoic acid compared with placebo in 269 patients receiving ezetimibe as background therapy [132]. Relative to placebo, bempedoic acid reduced LDL-C by − 29%, non-HDL-C by − 24%, total cholesterol by − 18%, ApoB-100 by −19% and hsCRP by − 31%.
-
The CLEAR Serenity trial randomized 345 statin-intolerant patients with hypercholesterolemia to receive bempedoic acid or placebo [127]. Bempedoic acid lowered LDL-C by − 21%, non-HDL-C by − 18%, total cholesterol by − 15%, ApoB-100 by − 15% and hsCRP by − 24%.
-
The CLEAR Wisdom trial randomized 779 patients with atherosclerosis or heterozygous familial hypercholesterolemia (HeFH) to receive bempedoic acid or placebo [129]. Bempedoic acid reduced LDL-C by − 17%, non-HDL by − 13%, total cholesterol by − 11%, ApoB-100 by −13% and hsCRP by − 9% compared with placebo.
-
The CLEAR Harmony trial assessed bempedoic acid’s safety and efficacy in 2230 statin-treated patients with atherosclerosis or HeFH compared with placebo [130]. Relative to placebo, bempedoic acid reduced LDL-C by − 16%, non-HDL-C by − 13%, total cholesterol by − 11%, ApoB-100 by − 12% and hsCRP by − 22%. The incidence of adverse events was similar across both interventional groups, yet a higher percentage discounted treatment with bempedoic acid than placebo (10.9% versus 7.1%). Gout was more frequently observed among patients treated with bempedoic acid (1.2% versus 0.3%).
-
The CLEAR Outcomes trial randomized 13,970 patients with established or high risk for CVD, intolerance to statins, and LDL-C level ≥ 100 mg/dL to receive bempedoic acid (180 mg daily) or placebo [131]. Bempedoic acid reduced the risk of MACE by − 13% (HR 0.87, 95% CI 0.79–0.96, p = 0.004) and resulted in a − 21.1% greater reduction in LDL-C levels compared with placebo [133]. A higher frequency of gout (3.1% versus 2.1%) and cholelithiasis (2.2% versus 1.2%) was observed with bempedoic acid than with placebo [133].
On the basis of these clinical trials, bempedoic acid has been approved to lower LDL-C in patients with HeFH or established atherosclerotic cardiovascular disease by the FDA and for primary hypercholesterolemia or mixed dyslipidaemia by the EMA. Even though data for primary CVD prevention is missing, the International Lipid Expert Panel concludes that bempedoic acid’s ‘favourable effects on plasma glucose and inflammatory markers make this drug a rational choice in the patient-centred care of specific groups of primary prevention’ [134].
3.2 Inclisiran
Inclisiran is a small interfering ribonucleic acid (siRNA) inhibitor of the PCSK9 biosynthesis. Although the underlying MoA is similar to evolocumab and alirocumab, the novel siRNA technology permits an infrequent subcutaneous administration (initially 284 mg for two doses 3 months apart and 284 mg every 6 months thereafter) with potentially fewer injection site reactions and improved patient adherence. Inclisiran’s efficacy was and is evaluated across the family of ORION trials in patients receiving maximum tolerable statins and other lipid-lowering agents: [135,136,137,138,139]
-
The phase 3 ORION-10 and ORION-11 trials evaluated inclisiran compared with placebo across a total of 1561 and 1617 patients with atherosclerosis or an atherosclerotic CVD risk-equivalent, respectively [137]. Compared with placebo, inclisiran reduced LDL-C levels by − 52% and − 50%, respectively.
-
These results were confirmed in the phase 3 ORION-9 trial which showed a − 48% LDL-C reduction of inclisiran relative to placebo among 482 patients with HeFH [138].
-
The ORION-2 pilot trial showed inclisiran could also be effective for patients with homozygous familial hypercholesterinaemia (HoFH) [139]. On the basis of these results a phase 3 study involving 56 patients with HoFH has been initiated (ORION-5) [135].
-
Results of the ORION-9, ORION-10, and ORION-11 trials were consistent in pooled patient- and trial-level meta-analyses [140, 141]. Across all three trials, LDL-C reductions amounted to −51%. Except for injection site rejections, patients were not at an increased risk for any adverse events. In contrast to the other PCSK9 inhibitors, inclisiran does not induce the expression of auto-antibodies according to results from the ORION-1 trial [142].
Inclisiran has been approved to lower LDL-C in patients with HeFH or clinical atherosclerotic cardiovascular disease by the FDA and in patients with primary hypercholesterolemia or mixed dyslipidaemia by the EMA. The CVOT, enrolling a total of 15,000 participants (ORION-4), is expected to be completed by July 2026 [136].
3.3 Evinacumab
Evinacumab is a monoclonal antibody inhibiting angiopoietin-like 3 (ANGPTL3). ANGPTL3 inhibits the lipoprotein lipase, which results in increased blood lipid levels [143,144,145]. Consequently, ANGPTL3 loss-of-function mutations result in lower triglycerides, LDL-C and a − 41% decreased risk of coronary heart diseases, despite reduced HDL-C levels [146, 147]. In contrast to other pharmacological treatments, evinacumab’s MoA is therefore independent of the LDL-C receptor [143, 148], providing new options for more synergistic combination treatments.
After evinacumab’s proof of concept was established in a single-group phase 2 trial [149], the phase 3 ELIPSE trial was conducted [150]. In total, 65 patients with HoFH were randomized to evinacumab (15 mg per kilogram body weight intravenous) or placebo every 4 weeks. Evinacumab lowered LDL-C by − 49% compared with placebo without a significant increase in adverse events. Evinacumab also significantly reduced ApoB-100 by − 37%, non-HDL by − 52%, total cholesterol by − 48%, triglycerides by − 50%, and ApoC-III by − 90%. Another phase 2 trial studied evinacumab in 272 patients with refractory hypercholesterinaemia (either HeFH or established atherosclerosis). At a dose of 450 mg per week, evinacumab reduced LDL-C levels by − 56% compared with baseline [151].
Evinacumab has been approved by the FDA and EMA to lower LDL-C in patients aged 12 years and older with HoFH. Ongoing trials are evaluating evinacumab for paediatric patients with HoFH [152] and for patients with severe hypertriglyceridemia at high risk of pancreatitis [153], as well as its long-term safety and efficacy profile [154].
3.4 Lomitapide
Lomitapide lowers cholesterol by inhibiting the microsomal triglyceride transfer protein (MTG) [155, 156]. MTP facilitates the transfer and loading of triglycerides and phospholipids onto ApoB-100 in hepatic cells’ endoplasmic reticulum. Thereby, the assembly of very-low-density lipoprotein (VLDL) particles, which turn into LDL-C after release into the blood serum, is inhibited. After conducting a successful dose-escalation study [157], a single-arm, open-label, phase 3 study of 29 patients with HoFH showed that lomitapide lowers LDL-C by − 50% and reduces the frequency of lipid apheresis [158]. Due to the accumulation of lipids in hepatocytes, increased aminotransferase levels alongside gastrointestinal symptoms, which can be well managed by dose reductions or treatment suspensions, were noted in the clinical trial.
Lomitapide is approved for the treatment of HoFH adjunct to a low-fat diet and therapy with other lipid-lowering therapies by the FDA and the EMA. Lomitapide’s effect on CV outcomes has not been established. Lomitapide is currently evaluated in paediatric patients with HoFH, in pregnant patients, and a real-world evidence study [159,160,161].
3.5 Mipomersen
Mipomersen is an antisense oligonucleotide that is administered via a weekly subcutaneous injection, inhibiting the messenger ribonucleic acid (mRNA) of ApoB-100 [19, 162]. By inhibiting the synthesis of ApoB-100, mipomersen also inhibits the assembly and synthesis of VLDL, which in turn results in lower LDL-C serum concentrations. A systematic review and meta-analysis of 13 trials with a total of 1053 patients found mipomersen significantly reduces LDL-C, total cholesterol, non-HDL-C, ApoB-100, Lp(a), triglycerides, VLDL and ApoA-I without effecting HDL-C [163]. Treatment with mipomersen was frequently associated with injection site reactions, hepatic steatosis, elevated liver enzymes and flu-like symptoms [163].
Mipomersen is approved for HoFH adjunct to lipid-lowering therapies and diet by the FDA. However, the EMA refused to grant marketing authorization due to the frequent gastrointestinal and cardiovascular side effects observed in clinical trials that could outweigh mipomersen’s potential beneficial cardiovascular benefits. Mipomersen’s long-term safety and efficacy is currently under investigation [19].
3.6 Volanesorsen
Volanesorsen is a second-generation hepatocyte-directed antisense oligonucleotide that reduces mRNA levels of ApoC-III. ApoC-III serves as an independent risk marker for CVD due to its central role in lipid metabolism [164, 165]. High ApoC-III levels were shown to increase total triglyceride levels by an accumulation of VLDL and chylomicrons [166,167,168,169] In contrary, loss-of-function mutations of the ApoC-III are associated with a lower risk of CVD [170,171,172].
After a first proof-of-concept study [173], volanesorsen’s dose-dependent efficacy has been established in a phase 1 trial of three patients with familial chylomicronemia syndrome (FCS) and elevated triglycerides [174]. In a randomized, placebo-controlled, double-blind phase 2 trial of 57 patients with elevated triglycerides, volanesorsen reduced ApoC-III by up to − 80% and triglycerides by − 71% [175]. A similar trial design enrolling 114 patients with established CVD and elevated triglycerides demonstrated an up to − 60% reduction in triglyceride levels [176]. Consistently, volanesorsen reduced triglycerides by − 71% in a trial of 114 patients with severe hypertriglyceridemia or FCS [177]. However, between 24% and 61% of patients treated with volanesorsen suffered from injection site reactions. Further side effects include serious bleeding and thrombocytopenia. Volanesorsen was approved by the EMA but not the FDA due to safety concerns for the treatment of patients with FCS.
3.7 Pemafibrate
Pemafibrate is a fibrate that modulates PPAR-α [178]. In contrast to fenofibrate, pemafibrate is selective for PPAR-α and exerts a higher potency. On the basis of promising results from multiple phase 1, 2 and 3 trials [178], the double-blind, randomized, placebo-controlled trial PROMINENT investigated pemafibrate’s effect on MACE in 10,497 patients with diabetes mellitus type 2 and hypertriglyceridemia [179, 180]. At a median follow-up of 3.4 years, pemafibrate lowered triglycerides by − 26%, VLDL by − 26%, remnant cholesterol by − 26% and ApoC-III by − 28% compared with placebo. However, pemafibrate did not significantly reduce the risk of MACE (HR 1.03, 95% CI 0.91–1.15).
3.8 Pelacarsen
Pelacarsen is a second-generation hepatocyte-directed antisense oligonucleotide that lowers Lp(a) levels [181]. Lp(a) has been identified as an independent risk marker for CVD as it exerts pro-atherogenic, pro-inflammatory, and pro-thrombotic effects which may play a critical role in the pathogenesis of atherosclerosis [182]. Patients’ serum Lp(a) levels are primarily (> 90%) genetically determined by the apo(a) gene [183]. Consequently, genetic studies found a causal link between elevated Lp(a) levels and a higher risk for CVD diseases,[184,185,186,187,188,189] and vice versa [190]. However, the risk increase was of lower magnitude than the one observed with LDL-C [186, 191, 192]. In the FOURIER trial, greater MACE reductions were seen among patients whose highly elevated Lp(a) levels were reduced by evolocumab [193]. Two studies reported a significantly lower cardiovascular event rate in patients treated with lipid apheresis for Lp(a) [194, 195]. However, substantial reductions in Lp(a) levels are necessary to provide a clinically meaningful effect on cardiovascular outcomes [186, 196].
Pre-clinical studies confirmed the concept (proof of concept) that antisense oligonucleotides specifically lower Lp(a) levels [197, 198]. Subsequently, phase 1 and 2 trials enrolling a total of 103 patients with elevated Lp(a) and CVD showed that treatment with antisense oligonucleotides reduced Lp(a) levels by up to − 80% [199, 200]. Consistently, in a randomized, double-blind, placebo-controlled phase 2 trial of 286 patients with elevated Lp(a) levels and established CVD, pelacarsen reduced Lp(a) in a dose-dependent manner by up to − 80% from baseline [182]. At the same time, pelacarsen was well tolerated. Only injection site reactions were more frequently observed in the treatment group. HORIZON, the multi-centre CVOT investigating pelacarsen’s effect on MACE in 7680 patients, is currently ongoing and expected to be completed by 29 May 2025 [201].
3.9 DPA and EPA
A combination therapy of the omega-3 fatty acids EPA, DHA and DPA is currently in clinical development. According to the manufacturer Matinas Biopharma, their product MAT9001 contains a significant amount of EPA and DPA mixed with a small dose of DHA. After promising experimental studies and a clinical pharmacokinetic trial [202, 203], MAT9001 (4 g per day) was compared with icosapent ethyl (4 g per day) in a phase 2 head-to-head crossover trial enrolling 42 patients with hypertriglyceridemia [204] MAT9001 achieved significantly greater reductions in triglycerides (− 33% versus −11%, p < 0.001), VLDL-C (− 33% versus − 8%, p < 0.001), non-HDL-C (− 9% versus − 5%, p = 0.027), total cholesterol (− 9% versus − 6%, p = 0.0013), ApoC-III (− 26% versus − 5%, p = 0.006), ApoA-I (− 15% versus − 10%, p = 0.003) and PCSK9 (−12% versus +9%, p < 0.001), yet not ApoB-100 (− 4% versus − 1%, p = 0.058), HDL-C (− 11% versus − 11%, p = 0.337) and LDL-C (− 2% versus − 4%, p = 0.116). However, the ENHANCE-IT trial could not reproduce MAT9001’s superior efficacy to icosapent ethyl in patients undergoing a therapeutic lifestyle change diet [205]. No difference in triglyceride, LDL-C and other lipoprotein levels could be observed between the two interventional groups except for hsCRP (− 6% versus − 9%, p = 0.034). Although Matinas Biopharma planned to investigate MAT9001’s efficacy in a CVOT, active clinical development has been suspended in light of the ENHANCE-IT trial.
3.10 Olezarsen
Olezarsen is a next-generation ApoC-III hepatocyte-directed antisense oligonucleotide, which is conjugated with n-acetylgalactosamine. This novel molecular design resulted in an improved efficacy and safety profile compared with volanesorsen. In a phase 1/2a trial enrolling 40 healthy volunteers, olezarsen reduced ApoC-III by up to − 92% and triglycerides by − 77%, whilst only 1 participant (2.5%) suffered from an injection site reaction [206] Olezarsen is currently being investigated in five phase 2 and 3 trials for patients with (severe) hypertriglyceridemia and FCS [207,208,209,210,211].
3.11 NNC0385-0434 A
NNC0385-0434 A is the first oral PCSK9 inhibitor that is currently under phase 2 development in a trial of 255 patients with established CVD [212]. This novel route of administration is expected to improve adherence compared with PCSK9 inhibitors that are currently injected subcutaneously every 2–4 weeks.
3.12 Olpasiran
Olpasiran is a siRNA that reduces Lp(a) levels. In pre-clinical studies olpasiran successfully reduced Lp(a) levels in transgenic mice and cynomolgus monkeys [213]. In a phase 1 trial of 64 patients, olpasiran reduced Lp(a) by − 71% to − 97% [213]. A phase 2 dose-finding study of 281 patients with established CVD, OCEAN(a)-DOSE, is currently ongoing [214]. Topline results ‘demonstrated a significant reduction from baseline in Lp(a) of up to or greater than 90 percent at week 36 (primary endpoint) and week 48 (end of treatment period) for the majority of doses’ [215].
3.13 Other therapeutics
Beyond the presented clinical development programs, there are a variety of innovative small molecules, monoclonal antibodies, vaccines and gene therapies currently in pre-clinical development [216]
4 Conclusion
In this article, we reviewed the mechanism of action, targets, efficacy and safety of established and emerging lipid-lowering drugs. Emerging drugs could offer patients’ benefit in multiple regards. Bempedoic acid presents a viable alternative for statin-intolerant patients. Inclisiran could improve patient adherence to PCSK9 inhibitor therapy due to its semi-annual administration. Pelacarsen and olpasiran could be the first treatments that reduce the independent cardiovascular risk factor Lp(a). Evinacumab, lomitapide, mipomersen volanesorsen and olezarsen are new treatments that primarily affect aspects of the lipid metabolism other than LDL-C synthesis or triglycerides, and thereby present a first step towards patient-tailored treatment approaches in cardiovascular prevention.
Furthermore, this extensive review highlights that many new treatments are first developed by pharmaceutical companies to treat patients with rare genetic diseases, for example, FeFH, FoFH and FCS. Thereafter, these treatments are typically tested for patients with established cardiovascular diseases (secondary prevention), and then for patients with high-risk for cardiovascular diseases (primary prevention). Ultimately, new drugs must not only demonstrate efficacy in large, randomized, cardiovascular outcomes trials to improve patients’ morbidity and mortality, but also demonstrate cost-effectiveness to be swiftly adopted into clinical routines. The journey for emerging to established pharmaceutical treatments entails a lengthy and costly process of small and large clinical trials that is plagued by attrition. To overcome the unmet needs for patients with dyslipidaemia, we must not only find more targets and developed better treatments, but also improve clinical trial design and execution.
The authors are hopeful that the presented ‘new wave’ of lipid-modifying drugs with innovative treatment modalities will further reduce patients’ risk of adverse cardiovascular events. With this arsenal of new drugs, we will eventually be able to personalize lipid-lowering treatment according to patient-specific needs.
References
Roth GA, Mensah GA, Johnson CO, et al. Global burden of cardiovascular diseases and risk factors, 1990–2019: update from the GBD 2019 Study. J Am Coll Cardiol. 2020;76:2982–3021. https://doi.org/10.1016/j.jacc.2020.11.010.
Global Burden of Disease Collaborative Network. Global burden of disease study 2019 (GBD 2019) Reference Life Table. 2021.https://doi.org/10.6069/1D4Y-YQ37 (accessed 24 Sep 2022).
Timmis A, Townsend N, Gale CP, et al. European Society of Cardiology: cardiovascular disease statistics 2019. Eur Heart J. 2020;41:12–85. https://doi.org/10.1093/eurheartj/ehz859.
Timmis A, Vardas P, Townsend N, et al. European Society of Cardiology: cardiovascular disease statistics 2021. Eur Heart J. 2022;43:716–99. https://doi.org/10.1093/eurheartj/ehab892.
Yusuf S, Reddy S, Ôunpuu S, et al. Global burden of cardiovascular diseases. Circulation. 2001;104:2746–53. https://doi.org/10.1161/hc4601.099487.
Yusuf S, Reddy S, Ôunpuu S, et al. Global burden of cardiovascular diseases. Circulation. 2001;104:2855–64. https://doi.org/10.1161/hc4701.099488.
März W, Dippel F-W, Theobald K, et al. Utilization of lipid-modifying therapy and low-density lipoprotein cholesterol goal attainment in patients at high and very-high cardiovascular risk: real-world evidence from Germany. Atherosclerosis. 2018;268:99–107. https://doi.org/10.1016/j.atherosclerosis.2017.11.020.
Santos RD, Waters DD, Tarasenko L, et al. A comparison of non-HDL and LDL cholesterol goal attainment in a large, multinational patient population: the Lipid Treatment Assessment Project 2. Atherosclerosis. 2012;224:150–3. https://doi.org/10.1016/j.atherosclerosis.2012.06.052.
Kuiper JG, Sanchez RJ, Houben E, et al. Use of lipid-modifying therapy and LDL-C goal attainment in a high-cardiovascular-risk population in the Netherlands. Clin Ther. 2017;39:819-827.e1. https://doi.org/10.1016/j.clinthera.2017.03.001.
Chi MD, Vansomphone SS, Liu I-LA, et al. Adherence to statins and LDL-cholesterol goal attainment. Am J Manag Care. 2014;20:e105-112.
Parris ES, Lawrence DB, Mohn LA, et al. Adherence to statin therapy and LDL cholesterol goal attainment by patients with diabetes and dyslipidemia. Diabetes Care. 2005;28:595–9. https://doi.org/10.2337/diacare.28.3.595.
Ray KK, Molemans B, Schoonen WM, et al. EU-wide cross-sectional observational study of lipid-modifying therapy use in secondary and primary care: the DA VINCI study. Eur J Prev Cardiol. 2021;28:1279–89. https://doi.org/10.1093/eurjpc/zwaa047.
Vrablik M, Seifert B, Parkhomenko A, et al. Lipid-lowering therapy use in primary and secondary care in Central and Eastern Europe: DA VINCI observational study. Atherosclerosis. 2021;334:66–75. https://doi.org/10.1016/j.atherosclerosis.2021.08.035.
Gouni-Berthold I, Schaper F, Schatz U, et al. Low-density lipoprotein cholesterol goal attainment in Germany: results from the DA VINCI study. Atheroscler Plus. 2022;50:10–6. https://doi.org/10.1016/j.athplu.2022.07.024.
van de Borne P, Peeters A, Janssens L, et al. Lipid-lowering therapy and risk-based LDL-C goal attainment in Belgium: DA VINCI observational study. Acta Cardiol. 2022. https://doi.org/10.1080/00015385.2022.2030568.
Talic S, Marquina C, Zomer E, et al. Attainment of low-density lipoprotein cholesterol goals in statin treated patients: Real-world evidence from Australia. Curr Probl Cardiol. 2022;47:101068. https://doi.org/10.1016/j.cpcardiol.2021.101068.
Klimchak AC, Patel MY, Iorga ŞR, et al. Lipid treatment and goal attainment characteristics among persons with atherosclerotic cardiovascular disease in the United States. Am J Prev Cardiol. 2020;1:100010. https://doi.org/10.1016/j.ajpc.2020.100010.
Wang N, Fulcher J, Abeysuriya N, et al. Intensive LDL cholesterol-lowering treatment beyond current recommendations for the prevention of major vascular events: a systematic review and meta-analysis of randomised trials including 327 037 participants. Lancet Diabetes Endocrinol. 2020;8:36–49. https://doi.org/10.1016/S2213-8587(19)30388-2.
Mach F, Baigent C, Catapano AL, et al. 2019 ESC/EAS Guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and European Atherosclerosis Society (EAS). Eur Heart J. 2020;41:111–88. https://doi.org/10.1093/eurheartj/ehz455.
Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139:e1082–143. https://doi.org/10.1161/CIR.0000000000000625.
Marquina C, Zomer E, Vargas-Torres S, et al. Novel treatment strategies for secondary prevention of cardiovascular disease: a systematic review of cost-effectiveness. Pharmacoeconomics. 2020;38:1095–113. https://doi.org/10.1007/s40273-020-00936-0.
Michaeli DT, Michaeli JC, Boch T, et al. Cost-effectiveness of lipid-lowering therapies for cardiovascular prevention in Germany. Cardiovasc Drugs Ther. 2022. https://doi.org/10.1007/s10557-021-07310-y.
Michaeli DT, Michaeli JC, Boch T, et al. Cost-effectiveness of icosapent ethyl, evolocumab, alirocumab, ezetimibe, or fenofibrate in combination with statins compared to statin monotherapy. Clin Drug Investig. 2022;42:643–56. https://doi.org/10.1007/s40261-022-01173-3.
Michaeli DT, Michaeli JC, Boch T, et al. Cost-effectiveness of cholesterol-lowering drugs for secondary cardiovascular prevention in the UK: ezetimibe, evolocumab, and alirocumab. Eur Heart J. 2022;43:ehac544.2367. https://doi.org/10.1093/eurheartj/ehac544.2367.
Bytyçi I, Penson PE, Mikhailidis DP, et al. Prevalence of statin intolerance: a meta-analysis. Eur Heart J. 2022;43:3213–23. https://doi.org/10.1093/eurheartj/ehac015.
Ganda OP, Bhatt DL, Mason RP, et al. Unmet need for adjunctive dyslipidemia therapy in hypertriglyceridemia management. J Am Coll Cardiol. 2018;72:330–43. https://doi.org/10.1016/j.jacc.2018.04.061.
Krähenbühl S, Pavik-Mezzour I, von Eckardstein A. Unmet needs in LDL-C lowering: when statins won’t do! Drugs. 2016;76:1175–90. https://doi.org/10.1007/s40265-016-0613-0.
Schleyer T, Hui S, Wang J, et al. Quantifying unmet need in statin-treated hyperlipidemia patients and the potential benefit of further LDL-C reduction through an EHR-based retrospective cohort study. J Manag Care Spec Pharm. 2019;25:544–54. https://doi.org/10.18553/jmcp.2019.25.5.544.
Böhler S, Scharnagl H, Freisinger F, et al. Unmet needs in the diagnosis and treatment of dyslipidemia in the primary care setting in Germany. Atherosclerosis. 2007;190:397–407. https://doi.org/10.1016/j.atherosclerosis.2006.02.025.
Mitchell S, Roso S, Samuel M, et al. Unmet need in the hyperlipidaemia population with high risk of cardiovascular disease: a targeted literature review of observational studies. BMC Cardiovasc Disord. 2016;16:74. https://doi.org/10.1186/s12872-016-0241-3.
Reiner Z. Managing the residual cardiovascular disease risk associated with HDL-cholesterol and triglycerides in statin-treated patients: a clinical update. Nutr Metab Cardiovasc Dis NMCD. 2013;23:799–807. https://doi.org/10.1016/j.numecd.2013.05.002.
Barter PJ, Brandrup-Wognsen G, Palmer MK, et al. Effect of statins on HDL-C: a complex process unrelated to changes in LDL-C: analysis of the VOYAGER Database. J Lipid Res. 2010;51:1546–53. https://doi.org/10.1194/jlr.P002816.
Tsimikas S, Witztum JL, Miller ER, et al. High-dose atorvastatin reduces total plasma levels of oxidized phospholipids and immune complexes present on apolipoprotein B-100 in patients with acute coronary syndromes in the MIRACL trial. Circulation. 2004;110:1406–12. https://doi.org/10.1161/01.CIR.0000141728.23033.B5.
Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin). Circulation. 2014;129:635–42. https://doi.org/10.1161/CIRCULATIONAHA.113.004406.
Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109:III39-43. https://doi.org/10.1161/01.CIR.0000131517.20177.5a.
Oesterle A, Laufs U, Liao JK. Pleiotropic Effects of Statins on the Cardiovascular System. Circ Res. 2017;120:229–43. https://doi.org/10.1161/CIRCRESAHA.116.308537.
Pedersen TR. Pleiotropic effects of statins: evidence against benefits beyond LDL-cholesterol lowering. Am J Cardiovasc Drugs Drugs Devices Interv. 2010;10(Suppl 1):10–7. https://doi.org/10.2165/1158822-S0-000000000-00000.
Baigent C, Blackwell L, Cholesterol Treatment Trialists’ (CTT) Collaboration, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet Lond Engl. 2010;376:1670–81. https://doi.org/10.1016/S0140-6736(10)61350-5.
Fulcher J, O’Connell R, Cholesterol Treatment Trialists’ (CTT) Collaboration, et al. Efficacy and safety of LDL-lowering therapy among men and women: meta-analysis of individual data from 174,000 participants in 27 randomised trials. Lancet Lond Engl. 2015;385:1397–405. https://doi.org/10.1016/S0140-6736(14)61368-4.
Mihaylova B, Emberson J, Cholesterol Treatment Trialists’ (CTT) Collaborators, et al. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet Lond Engl. 2012;380:581–90. https://doi.org/10.1016/S0140-6736(12)60367-5.
Mills EJ, Rachlis B, Wu P, et al. Primary prevention of cardiovascular mortality and events with statin treatments: a network meta-analysis involving more than 65,000 patients. J Am Coll Cardiol. 2008;52:1769–81. https://doi.org/10.1016/j.jacc.2008.08.039.
Genser B, März W. Low density lipoprotein cholesterol, statins and cardiovascular events: a meta-analysis. Clin Res Cardiol Off J Ger Card Soc. 2006;95:393–404. https://doi.org/10.1007/s00392-006-0403-x.
Gould AL, Rossouw JE, Santanello NC, et al. Cholesterol reduction yields clinical benefit. A new look at old data. Circulation. 1995;91:2274–82. https://doi.org/10.1161/01.cir.91.8.2274.
Gould AL, Rossouw JE, Santanello NC, et al. Cholesterol reduction yields clinical benefit: impact of statin trials. Circulation. 1998;97:946–52. https://doi.org/10.1161/01.cir.97.10.946.
LaRosa JC, He J, Vupputuri S. Effect of statins on risk of coronary disease: a meta-analysis of randomized controlled trials. JAMA. 1999;282:2340–6. https://doi.org/10.1001/jama.282.24.2340.
Mills EJ, Wu P, Chong G, et al. Efficacy and safety of statin treatment for cardiovascular disease: a network meta-analysis of 170,255 patients from 76 randomized trials. QJM Mon J Assoc Physicians. 2011;104:109–24. https://doi.org/10.1093/qjmed/hcq165.
Ray KK, Seshasai SRK, Erqou S, et al. Statins and all-cause mortality in high-risk primary prevention: a meta-analysis of 11 randomized controlled trials involving 65,229 participants. Arch Intern Med. 2010;170:1024–31. https://doi.org/10.1001/archinternmed.2010.182.
Taylor F, Huffman MD, Macedo AF, et al. Statins for the primary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2013. https://doi.org/10.1002/14651858.CD004816.pub5.
Herrington W, Emberson J, Cholesterol Treatment Trialists’ (CTT) Collaboration, et al. Impact of renal function on the effects of LDL cholesterol lowering with statin-based regimens: a meta-analysis of individual participant data from 28 randomised trials. Lancet Diabetes Endocrinol. 2016;4:829–39. https://doi.org/10.1016/S2213-8587(16)30156-5.
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet Lond Engl. 2005;366:1267–78. https://doi.org/10.1016/S0140-6736(05)67394-1.
Naci H, Brugts JJ, Fleurence R, et al. Comparative benefits of statins in the primary and secondary prevention of major coronary events and all-cause mortality: a network meta-analysis of placebo-controlled and active-comparator trials. Eur J Prev Cardiol. 2013;20:641–57. https://doi.org/10.1177/2047487313480435.
Law M, Rudnicka AR. Statin safety: a systematic review. Am J Cardiol. 2006;97:52C-60C. https://doi.org/10.1016/j.amjcard.2005.12.010.
Sukhija R, Prayaga S, Marashdeh M, et al. Effect of statins on fasting plasma glucose in diabetic and nondiabetic patients. J Investig Med Off Publ Am Fed Clin Res. 2009;57:495–9. https://doi.org/10.2310/JIM.0b013e318197ec8b.
Mach F, Ray KK, Wiklund O, et al. Adverse effects of statin therapy: perception vs. the evidence—focus on glucose homeostasis, cognitive, renal and hepatic function, haemorrhagic stroke and cataract. Eur Heart J. 2018;39:2526–39. https://doi.org/10.1093/eurheartj/ehy182.
McKinney JS, Kostis WJ. Statin therapy and the risk of intracerebral hemorrhage: a meta-analysis of 31 randomized controlled trials. Stroke. 2012;43:2149–56. https://doi.org/10.1161/STROKEAHA.112.655894.
Kosoglou T, Meyer I, Veltri EP, et al. Pharmacodynamic interaction between the new selective cholesterol absorption inhibitor ezetimibe and simvastatin. Br J Clin Pharmacol. 2002;54:309–19. https://doi.org/10.1046/j.1365-2125.2002.01633.x.
Sudhop T, Lütjohann D, Kodal A, et al. Inhibition of intestinal cholesterol absorption by ezetimibe in humans. Circulation. 2002;106:1943–8. https://doi.org/10.1161/01.cir.0000034044.95911.dc.
Stitziel NO, Won H-H, Myocardial Infarction Genetics Consortium Investigators, et al. Inactivating mutations in NPC1L1 and protection from coronary heart disease. N Engl J Med. 2014;371:2072–82. https://doi.org/10.1056/NEJMoa1405386.
Cannon CP, Blazing MA, Giugliano RP, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–97. https://doi.org/10.1056/NEJMoa1410489.
Ballantyne CM, Blazing MA, King TR, et al. Efficacy and safety of ezetimibe co-administered with simvastatin compared with atorvastatin in adults with hypercholesterolemia. Am J Cardiol. 2004;93:1487–94. https://doi.org/10.1016/j.amjcard.2004.02.060.
Morrone D, Weintraub WS, Toth PP, et al. Lipid-altering efficacy of ezetimibe plus statin and statin monotherapy and identification of factors associated with treatment response: a pooled analysis of over 21,000 subjects from 27 clinical trials. Atherosclerosis. 2012;223:251–61. https://doi.org/10.1016/j.atherosclerosis.2012.02.016.
Sharp Collaborative Group null. Study of Heart and Renal Protection (SHARP): randomized trial to assess the effects of lowering low-density lipoprotein cholesterol among 9,438 patients with chronic kidney disease. Am Heart J. 2010;160:785-794.e10. https://doi.org/10.1016/j.ahj.2010.08.012.
Rossebø AB, Pedersen TR, Boman K, et al. Intensive lipid lowering with simvastatin and ezetimibe in aortic stenosis. N Engl J Med. 2008;359:1343–56. https://doi.org/10.1056/NEJMoa0804602.
Baigent C, Landray MJ, Reith C, et al. The effects of lowering LDL cholesterol with simvastatin plus ezetimibe in patients with chronic kidney disease (Study of Heart and Renal Protection): a randomised placebo-controlled trial. Lancet Lond Engl. 2011;377:2181–92. https://doi.org/10.1016/S0140-6736(11)60739-3.
Kastelein JJP, Akdim F, Stroes ESG, et al. Simvastatin with or without ezetimibe in familial hypercholesterolemia. N Engl J Med. 2008;358:1431–43. https://doi.org/10.1056/NEJMoa0800742.
Pandor A, Ara RM, Tumur I, et al. Ezetimibe monotherapy for cholesterol lowering in 2,722 people: systematic review and meta-analysis of randomized controlled trials. J Intern Med. 2009;265:568–80. https://doi.org/10.1111/j.1365-2796.2008.02062.x.
Savarese G, De Ferrari GM, Rosano GMC, et al. Safety and efficacy of ezetimibe: a meta-analysis. Int J Cardiol. 2015;201:247–52. https://doi.org/10.1016/j.ijcard.2015.08.103.
Zhu Y, Hu H, Yang J, et al. The efficacy and safety of statin in combination with ezetimibe compared with double-dose statin in patients with high cardiovascular risk: a meta-analysis. Bosn J Basic Med Sci. 2020;20:169–82. https://doi.org/10.17305/bjbms.2019.4437.
Yu M, Liang C, Kong Q, et al. Efficacy of combination therapy with ezetimibe and statins versus a double dose of statin monotherapy in participants with hypercholesterolemia: a meta-analysis of literature. Lipids Health Dis. 2020;19:1. https://doi.org/10.1186/s12944-019-1182-5.
Zhan S, Tang M, Liu F, et al. Ezetimibe for the prevention of cardiovascular disease and all-cause mortality events. Cochrane Database Syst Rev. 2018;11:CD012502. https://doi.org/10.1002/14651858.CD012502.pub2.
Ouchi Y, Sasaki J, Arai H, et al. Ezetimibe lipid-lowering trial on prevention of atherosclerotic cardiovascular disease in 75 or older (EWTOPIA 75). Circulation. 2019;140:992–1003. https://doi.org/10.1161/CIRCULATIONAHA.118.039415.
Abifadel M, Varret M, Rabès J-P, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6. https://doi.org/10.1038/ng1161.
Kathiresan S, Myocardial Infarction Genetics Consortium. A PCSK9 missense variant associated with a reduced risk of early-onset myocardial infarction. N Engl J Med. 2008;358:2299–300. https://doi.org/10.1056/NEJMc0707445.
Cohen JC, Boerwinkle E, Mosley TH, et al. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72. https://doi.org/10.1056/NEJMoa054013.
Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet Lond Engl. 2015;385:331–40. https://doi.org/10.1016/S0140-6736(14)61399-4.
Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–8. https://doi.org/10.1016/j.jacc.2014.03.019.
Koren MJ, Lundqvist P, Bolognese M, et al. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–40. https://doi.org/10.1016/j.jacc.2014.03.018.
Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–82. https://doi.org/10.1001/jama.2014.4030.
Blom DJ, Hala T, Bolognese M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19. https://doi.org/10.1056/NEJMoa1316222.
Schmidt AF, Carter J-PL, Pearce LS, et al. PCSK9 monoclonal antibodies for the primary and secondary prevention of cardiovascular disease. Cochrane Database Syst Rev. 2020;10:CD011748. https://doi.org/10.1002/14651858.CD011748.pub3.
Karatasakis A, Danek BA, Karacsonyi J, et al. Effect of PCSK9 inhibitors on clinical outcomes in patients with hypercholesterolemia: a meta-analysis of 35 randomized controlled trials. J Am Heart Assoc. 2017;6: e006910. https://doi.org/10.1161/JAHA.117.006910.
Sahebkar A, Di Giosia P, Stamerra CA, et al. Effect of monoclonal antibodies to PCSK9 on high-sensitivity C-reactive protein levels: a meta-analysis of 16 randomized controlled treatment arms. Br J Clin Pharmacol. 2016;81:1175–90. https://doi.org/10.1111/bcp.12905.
Monami M, Sesti G, Mannucci E. PCSK9 inhibitor therapy: a systematic review and meta-analysis of metabolic and cardiovascular outcomes in patients with diabetes. Diabetes Obes Metab. 2019;21:903–8. https://doi.org/10.1111/dom.13599.
Zhang X-L, Zhu Q-Q, Zhu L, et al. Safety and efficacy of anti-PCSK9 antibodies: a meta-analysis of 25 randomized, controlled trials. BMC Med. 2015;13:123. https://doi.org/10.1186/s12916-015-0358-8.
Talasaz AH, Ho A-CJ, Bhatty F, et al. Meta-analysis of clinical outcomes of PCSK9 modulators in patients with established ASCVD. Pharmacotherapy. 2021;41:1009–23. https://doi.org/10.1002/phar.2635.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J. 2015;169:906-915.e13. https://doi.org/10.1016/j.ahj.2015.03.004.
Nicholls SJ, Puri R, Anderson T, et al. Effect of evolocumab on progression of coronary disease in statin-treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373–84. https://doi.org/10.1001/jama.2016.16951.
Robinson JG, Farnier M, Krempf M, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99. https://doi.org/10.1056/NEJMoa1501031.
Ridker PM, Tardif J-C, Amarenco P, et al. Lipid-reduction variability and antidrug-antibody formation with bococizumab. N Engl J Med. 2017;376:1517–26. https://doi.org/10.1056/NEJMoa1614062.
Schmidt AF, Swerdlow DI, Holmes MV, et al. PCSK9 genetic variants and risk of type 2 diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2017;5:97–105. https://doi.org/10.1016/S2213-8587(16)30396-5.
Meade T, Zuhrie R, Cook C, et al. Bezafibrate in men with lower extremity arterial disease: randomised controlled trial. BMJ. 2002;325:1139. https://doi.org/10.1136/bmj.325.7373.1139.
Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet Lond Engl. 2005;366:1849–61. https://doi.org/10.1016/S0140-6736(05)67667-2.
Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention trial study group. N Engl J Med. 1999;341:410–8. https://doi.org/10.1056/NEJM199908053410604.
Ginsberg HN, Elam MB, ACCORD Study Group, et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med. 2010;362:1563–74. https://doi.org/10.1056/NEJMoa1001282.
Jun M, Foote C, Lv J, et al. Effects of fibrates on cardiovascular outcomes: a systematic review and meta-analysis. Lancet Lond Engl. 2010;375:1875–84. https://doi.org/10.1016/S0140-6736(10)60656-3.
Manson JE, Cook NR, Lee I-M, et al. Marine n-3 fatty acids and prevention of cardiovascular disease and cancer. N Engl J Med. 2019;380:23–32. https://doi.org/10.1056/NEJMoa1811403.
Nicholls SJ, Lincoff AM, Garcia M, et al. Effect of high-dose omega-3 fatty acids vs corn oil on major adverse cardiovascular events in patients at high cardiovascular risk: the STRENGTH randomized clinical trial. JAMA. 2020;324:2268–80. https://doi.org/10.1001/jama.2020.22258.
Zimmer R, Riemer T, Rauch B, et al. Effects of 1-year treatment with highly purified omega-3 fatty acids on depression after myocardial infarction: results from the OMEGA trial. J Clin Psychiatry. 2013;74:e1037-1045. https://doi.org/10.4088/JCP.13m08453.
Bosch J, Gerstein HC, ORIGIN Trial Investigators, et al. n-3 fatty acids and cardiovascular outcomes in patients with dysglycemia. N Engl J Med. 2012;367:309–18. https://doi.org/10.1056/NEJMoa1203859.
Bowman L, Mafham M, ASCEND Study Collaborative Group, et al. Effects of n-3 fatty acid supplements in diabetes mellitus. N Engl J Med. 2018;379:1540–50. https://doi.org/10.1056/NEJMoa1804989.
Kromhout D, Giltay EJ, Geleijnse JM, et al. n-3 fatty acids and cardiovascular events after myocardial infarction. N Engl J Med. 2010;363:2015–26. https://doi.org/10.1056/NEJMoa1003603.
Tavazzi L, Maggioni AP, Marchioli R, et al. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): a randomised, double-blind, placebo-controlled trial. Lancet Lond Engl. 2008;372:1223–30. https://doi.org/10.1016/S0140-6736(08)61239-8.
Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet Lond Engl 1999;354:447–55.
Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open-label, blinded endpoint analysis. Lancet. 2007;369:1090–8. https://doi.org/10.1016/S0140-6736(07)60527-3.
Bhatt DL, Steg PG, Miller M, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. 2019;380:11–22. https://doi.org/10.1056/NEJMoa1812792.
Khan SU, Lone AN, Khan MS, et al. Effect of omega-3 fatty acids on cardiovascular outcomes: a systematic review and meta-analysis. EClinicalMedicine. 2021;38:100997. https://doi.org/10.1016/j.eclinm.2021.100997.
Rizos EC, Ntzani EE, Bika E, et al. Association between omega-3 fatty acid supplementation and risk of major cardiovascular disease events: a systematic review and meta-analysis. JAMA. 2012;308:1024–33. https://doi.org/10.1001/2012.jama.11374.
Mason RP, Libby P, Bhatt DL. Emerging mechanisms of cardiovascular protection for the omega-3 fatty acid eicosapentaenoic acid. Arterioscler Thromb Vasc Biol. 2020;40:1135–47. https://doi.org/10.1161/ATVBAHA.119.313286.
Zhang X, Ritonja JA, Zhou N, et al. Omega-3 polyunsaturated fatty acids intake and blood pressure: a dose-response meta-analysis of randomized controlled trials. J Am Heart Assoc. 2022;11: e025071. https://doi.org/10.1161/JAHA.121.025071.
Sarajlic P, Artiach G, Larsson SC, et al. Dose-dependent risk reduction for myocardial infarction with eicosapentaenoic acid: a meta-analysis and meta-regression including the STRENGTH trial. Cardiovasc Drugs Ther. 2021;35:1079–81. https://doi.org/10.1007/s10557-021-07212-z.
Bernasconi AA, Wiest MM, Lavie CJ, et al. Effect of omega-3 dosage on cardiovascular outcomes: an updated meta-analysis and meta-regression of interventional trials. Mayo Clin Proc. 2021;96:304–13. https://doi.org/10.1016/j.mayocp.2020.08.034.
Kastelein JJP, Stroes ESG. FISHing for the miracle of eicosapentaenoic acid. N Engl J Med. 2019;380:89–90. https://doi.org/10.1056/NEJMe1814004.
Tokgözoğlu L, Libby P. The dawn of a new era of targeted lipid-lowering therapies. Eur Heart J. 2022. https://doi.org/10.1093/eurheartj/ehab841.
European Medicines Agency (EMA). Assessment report: Vazkepa. 2021.https://www.ema.europa.eu/en/documents/assessment-report/vazkepa-epar-public-assessment-report_en.pdf. Accessed 24 Sep 2022.
Gaba P, Bhatt DL, Steg PG, et al. Prevention of cardiovascular events and mortality with icosapent ethyl in patients with prior myocardial infarction. J Am Coll Cardiol. 2022;79:1660–71. https://doi.org/10.1016/j.jacc.2022.02.035.
Ademi Z, Ofori-Asenso R, Zomer E, et al. The cost-effectiveness of icosapent ethyl in combination with statin therapy compared with statin alone for cardiovascular risk reduction. Eur J Prev Cardiol. 2020. https://doi.org/10.1177/2047487319896648.
Lachaine J, Charron J-N, Gregoire JC, et al. Cost-Effectiveness of icosapent ethyl (IPE) for the reduction of the risk of ischemic cardiovascular events in Canada. Clin Outcomes Res CEOR. 2023;15:295–308. https://doi.org/10.2147/CEOR.S377935.
Michaeli DT, Michaeli JC, Boch T, et al. Cost-effectiveness of icosapent ethyl for primary and secondary cardiovascular prevention in the UK. Eur Heart J. 2022;43:ehac544.2845. https://doi.org/10.1093/eurheartj/ehac544.2845.
Weintraub WS, Bhatt ZZ, et al. Cost-effectiveness of icosapent ethyl in us reduce-it patients. J Am Coll Cardiol. 2020;75:1914–1914. https://doi.org/10.1016/S0735-1097(20)32541-9.
Ollendorf DA, McQueen RB, Campbell JD, et al. Additive therapies for cardiovascular disease: effectiveness and value. 2019.http://icer-review.org/material/cvd-final-evidence-report/. Accessed 22 Mar 2021.
Kodera S, Morita H, Kiyosue A, et al. Cost-effectiveness of statin plus eicosapentaenoic acid combination therapy for cardiovascular disease prevention in Japanese patients with hypercholesterolemia—an analysis based on the Japan Eicosapentaenoic acid Lipid Intervention Study (JELIS). Circ J Off J Jpn Circ Soc. 2018;82:1076–82. https://doi.org/10.1253/circj.CJ-17-0995.
Ross S, D’Mello M, Anand SS, et al. Effect of bile acid sequestrants on the risk of cardiovascular events: a mendelian randomization analysis. Circ Cardiovasc Genet. 2015;8:618–27. https://doi.org/10.1161/CIRCGENETICS.114.000952.
Boden WE, Probstfield JL, HIGH Investigators, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67. https://doi.org/10.1056/NEJMoa1107579.
Landray MJ, Haynes R, HPS2-THRIVE Collaborative Group, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12. https://doi.org/10.1056/NEJMoa1300955.
Merćep I, Strikić D, Slišković AM, et al. New therapeutic approaches in treatment of dyslipidaemia—a narrative review. Pharm Basel Switz. 2022;15:839. https://doi.org/10.3390/ph15070839.
Bilen O, Ballantyne CM. Bempedoic acid (ETC-1002): an investigational inhibitor of ATP citrate lyase. Curr Atheroscler Rep. 2016;18:61. https://doi.org/10.1007/s11883-016-0611-4.
Laufs U, Banach M, Mancini GBJ, et al. Efficacy and safety of bempedoic acid in patients with hypercholesterolemia and statin intolerance. J Am Heart Assoc. 2019;8: e011662. https://doi.org/10.1161/JAHA.118.011662.
Bays HE, Banach M, Catapano AL, et al. Bempedoic acid safety analysis: pooled data from four phase 3 clinical trials. J Clin Lipidol. 2020;14:649-659.e6. https://doi.org/10.1016/j.jacl.2020.08.009.
Goldberg AC, Leiter LA, Stroes ESG, et al. Effect of bempedoic acid vs placebo added to maximally tolerated statins on low-density lipoprotein cholesterol in patients at high risk for cardiovascular disease: the CLEAR Wisdom randomized clinical trial. JAMA. 2019;322:1780–8. https://doi.org/10.1001/jama.2019.16585.
Ray KK, Bays HE, Catapano AL, et al. Safety and efficacy of bempedoic acid to reduce LDL cholesterol. N Engl J Med. 2019;380:1022–32. https://doi.org/10.1056/NEJMoa1803917.
Nicholls S, Lincoff AM, Bays HE, et al. Rationale and design of the CLEAR-outcomes trial: evaluating the effect of bempedoic acid on cardiovascular events in patients with statin intolerance. Am Heart J. 2021;235:104–12. https://doi.org/10.1016/j.ahj.2020.10.060.
Ballantyne CM, Banach M, Mancini GBJ, et al. Efficacy and safety of bempedoic acid added to ezetimibe in statin-intolerant patients with hypercholesterolemia: a randomized, placebo-controlled study. Atherosclerosis. 2018;277:195–203. https://doi.org/10.1016/j.atherosclerosis.2018.06.002.
Nissen SE, Lincoff AM, Brennan D, et al. Bempedoic acid and cardiovascular outcomes in statin-intolerant patients. N Engl J Med. 2023;388:1353–64. https://doi.org/10.1056/NEJMoa2215024.
Banach M, Penson PE, Farnier M, et al. Bempedoic acid in the management of lipid disorders and cardiovascular risk. 2023 position paper of the International Lipid Expert Panel (ILEP). Prog Cardiovasc Dis. 2023. https://doi.org/10.1016/j.pcad.2023.03.001.
Novartis Pharmaceuticals. A two-part (double-blind placebo controlled/open-label) multicenter study to evaluate safety, tolerability, and efficacy of inclisiran in subjects with Homozygous Familial Hypercholesterolemia (HoFH) (ORION-5). clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT03851705. Accessed 22 Aug 2022.
University of Oxford. HPS-4/TIMI 65/ORION-4: a double-blind randomized placebo-controlled trial assessing the effects of inclisiran on clinical outcomes among people with atherosclerotic cardiovascular disease. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT03705234. Accessed 22 Aug 2022.
Ray KK, Wright RS, Kallend D, et al. Two phase 3 trials of inclisiran in patients with elevated LDL cholesterol. N Engl J Med. 2020;382:1507–19. https://doi.org/10.1056/NEJMoa1912387.
Raal FJ, Kallend D, Ray KK, et al. Inclisiran for the treatment of heterozygous familial hypercholesterolemia. N Engl J Med. 2020;382:1520–30. https://doi.org/10.1056/NEJMoa1913805.
Hovingh GK, Lepor NE, Kallend D, et al. Inclisiran durably lowers low-density lipoprotein cholesterol and proprotein convertase subtilisin/kexin type 9 expression in homozygous familial hypercholesterolemia: the ORION-2 pilot study. Circulation. 2020;141:1829–31. https://doi.org/10.1161/CIRCULATIONAHA.119.044431.
Wright RS, Ray KK, Raal FJ, et al. Pooled patient-level analysis of inclisiran trials in patients with familial hypercholesterolemia or atherosclerosis. J Am Coll Cardiol. 2021;77:1182–93. https://doi.org/10.1016/j.jacc.2020.12.058.
Khan SA, Naz A, Qamar Masood M, et al. Meta-analysis of inclisiran for the treatment of hypercholesterolemia. Am J Cardiol. 2020;134:69–73. https://doi.org/10.1016/j.amjcard.2020.08.018.
Landmesser U, Haghikia A, Leiter LA, et al. Effect of inclisiran, the small-interfering RNA against proprotein convertase subtilisin/kexin type 9, on platelets, immune cells, and immunological biomarkers: a pre-specified analysis from ORION-1. Cardiovasc Res. 2021;117:284–91. https://doi.org/10.1093/cvr/cvaa077.
Köster A, Chao YB, Mosior M, et al. Transgenic angiopoietin-like (angptl)4 overexpression and targeted disruption of angptl4 and angptl3: regulation of triglyceride metabolism. Endocrinology. 2005;146:4943–50. https://doi.org/10.1210/en.2005-0476.
Fujimoto K, Koishi R, Shimizugawa T, et al. Angptl3-null mice show low plasma lipid concentrations by enhanced lipoprotein lipase activity. Exp Anim. 2006;55:27–34. https://doi.org/10.1538/expanim.55.27.
Shimamura M, Matsuda M, Yasumo H, et al. Angiopoietin-like protein3 regulates plasma HDL cholesterol through suppression of endothelial lipase. Arterioscler Thromb Vasc Biol. 2007;27:366–72. https://doi.org/10.1161/01.ATV.0000252827.51626.89.
Dewey FE, Gusarova V, Dunbar RL, et al. Genetic and pharmacologic inactivation of ANGPTL3 and cardiovascular disease. N Engl J Med. 2017;377:211–21. https://doi.org/10.1056/NEJMoa1612790.
Athyros VG, Katsiki N, Dimakopoulou A, et al. Drugs that mimic the effect of gene mutations for the prevention or the treatment of atherosclerotic disease: from PCSK9 inhibition to ANGPTL3 inactivation. Curr Pharm Des. 2018;24:3638–46. https://doi.org/10.2174/1381612824666181009100517.
Banerjee P, Chan K-C, Tarabocchia M, et al. Functional analysis of LDLR (low-density lipoprotein receptor) variants in patient lymphocytes to assess the effect of evinacumab in homozygous familial hypercholesterolemia patients with a spectrum of LDLR activity. Arterioscler Thromb Vasc Biol. 2019;39:2248–60. https://doi.org/10.1161/ATVBAHA.119.313051.
Gaudet D, Gipe DA, Pordy R, et al. ANGPTL3 Inhibition in homozygous familial hypercholesterolemia. N Engl J Med. 2017;377:296–7. https://doi.org/10.1056/NEJMc1705994.
Raal FJ, Rosenson RS, Reeskamp LF, et al. Evinacumab for homozygous familial hypercholesterolemia. N Engl J Med. 2020;383:711–20. https://doi.org/10.1056/NEJMoa2004215.
Rosenson RS, Burgess LJ, Ebenbichler CF, et al. Evinacumab in patients with refractory hypercholesterolemia. N Engl J Med. 2020;383:2307–19. https://doi.org/10.1056/NEJMoa2031049.
Regeneron Pharmaceuticals. A three-part, single-arm, open-label study to evaluate the efficacy, safety, and pharmacokinetics of evinacumab in pediatric patients with homozygous familial hypercholesterolemia. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT04233918. Accessed 22 Aug 2022.
Regeneron Pharmaceuticals. A randomized, double-blind, placebo-controlled study to evaluate the efficacy and safety of evinacumab in patients with severe hypertriglyceridemia for the prevention of recurrent acute pancreatitis. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT04863014. Accessed 22 Aug 2022.
Regeneron Pharmaceuticals. An open-label study to evaluate the long-term safety and efficacy of evinacumab in patients with homozygous familial hypercholesterolemia. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT03409744. Accessed 22 Aug 2022.
Alonso R, Cuevas A, Mata P. Lomitapide: a review of its clinical use, efficacy, and tolerability. Core Evid. 2019;14:19–30. https://doi.org/10.2147/CE.S174169.
Goulooze SC, Cohen AF, Rissmann R. Lomitapide. Br J Clin Pharmacol. 2015;80:179–81. https://doi.org/10.1111/bcp.12612.
Cuchel M, Bloedon LT, Szapary PO, et al. Inhibition of microsomal triglyceride transfer protein in familial hypercholesterolemia. N Engl J Med. 2007;356:148–56. https://doi.org/10.1056/NEJMoa061189.
Cuchel M, Meagher EA, du Toit TH, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet Lond Engl. 2013;381:40–6. https://doi.org/10.1016/S0140-6736(12)61731-0.
Amryt Pharma. Phase III, single arm, open label, international, multi centre study to evaluate the efficacy and safety of lomitapide in paediatric patients with Homozygous Familial Hypercholesterolaemia (HoFH) on stable lipid lowering therapy. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT04681170. Accessed 30 Aug 2022.
Amryt Pharma. Global lomitapide pregnancy exposure registry. clinicaltrials.gov 2020. https://clinicaltrials.gov/ct2/show/NCT02399839. Accessed 30 Aug 2022.
Amryt Pharma. LOWER: Lomitapide observational worldwide evaluation registry. clinicaltrials.gov 2020. https://clinicaltrials.gov/ct2/show/NCT02135705. Accessed 30 Aug 2022.
Parham JS, Goldberg AC. Mipomersen and its use in familial hypercholesterolemia. Expert Opin Pharmacother. 2019;20:127–31. https://doi.org/10.1080/14656566.2018.1550071.
Fogacci F, Ferri N, Toth PP, et al. Efficacy and safety of mipomersen: a systematic review and meta-analysis of randomized clinical trials. Drugs. 2019;79:751–66. https://doi.org/10.1007/s40265-019-01114-z.
Chan DC, Chen MM, Ooi EMM, et al. An ABC of apolipoprotein C-III: a clinically useful new cardiovascular risk factor? Int J Clin Pract. 2008;62:799–809. https://doi.org/10.1111/j.1742-1241.2007.01678.x.
Sacks FM. The crucial roles of apolipoproteins E and C-III in apoB lipoprotein metabolism in normolipidemia and hypertriglyceridemia. Curr Opin Lipidol. 2015;26:56–63. https://doi.org/10.1097/MOL.0000000000000146.
Varbo A, Benn M, Nordestgaard BG. Remnant cholesterol as a cause of ischemic heart disease: evidence, definition, measurement, atherogenicity, high risk patients, and present and future treatment. Pharmacol Ther. 2014;141:358–67. https://doi.org/10.1016/j.pharmthera.2013.11.008.
Ito Y, Azrolan N, O’Connell A, et al. Hypertriglyceridemia as a result of human apo CIII gene expression in transgenic mice. Science. 1990;249:790–3. https://doi.org/10.1126/science.2167514.
Ooi EMM, Barrett PHR, Chan DC, et al. Apolipoprotein C-III: understanding an emerging cardiovascular risk factor. Clin Sci Lond Engl. 1979;2008(114):611–24. https://doi.org/10.1042/CS20070308.
Nordestgaard BG, Varbo A. Triglycerides and cardiovascular disease. Lancet Lond Engl. 2014;384:626–35. https://doi.org/10.1016/S0140-6736(14)61177-6.
Pollin TI, Damcott CM, Shen H, et al. A null mutation in human APOC3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–5. https://doi.org/10.1126/science.1161524.
Jørgensen AB, Frikke-Schmidt R, Nordestgaard BG, et al. Loss-of-function mutations in APOC3 and risk of ischemic vascular disease. N Engl J Med. 2014;371:32–41. https://doi.org/10.1056/NEJMoa1308027.
Crosby J, Peloso GM, TG and HDL Working Group of the Exome Sequencing Project, National Heart, Lung, and Blood Institute, et al. Loss-of-function mutations in APOC3, triglycerides, and coronary disease. N Engl J Med. 2014;371:22–31. https://doi.org/10.1056/NEJMoa1307095.
Graham MJ, Lee RG, Bell TA, et al. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ Res. 2013;112:1479–90. https://doi.org/10.1161/CIRCRESAHA.111.300367.
Gaudet D, Brisson D, Tremblay K, et al. Targeting APOC3 in the familial chylomicronemia syndrome. N Engl J Med. 2014;371:2200–6. https://doi.org/10.1056/NEJMoa1400284.
Gaudet D, Alexander VJ, Baker BF, et al. Antisense inhibition of apolipoprotein c-iii in patients with hypertriglyceridemia. N Engl J Med. 2015;373:438–47. https://doi.org/10.1056/NEJMoa1400283.
Tardif J-C, Karwatowska-Prokopczuk E, Amour ES, et al. Apolipoprotein C-III reduction in subjects with moderate hypertriglyceridaemia and at high cardiovascular risk. Eur Heart J. 2022;43:1401–12. https://doi.org/10.1093/eurheartj/ehab820.
Gouni-Berthold I, Alexander VJ, Yang Q, et al. Efficacy and safety of volanesorsen in patients with multifactorial chylomicronaemia (COMPASS): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet Diabetes Endocrinol. 2021;9:264–75. https://doi.org/10.1016/S2213-8587(21)00046-2.
Fruchart J-C, Santos RD, Aguilar-Salinas C, et al. The selective peroxisome proliferator-activated receptor alpha modulator (SPPARMα) paradigm: conceptual framework and therapeutic potential: a consensus statement from the International Atherosclerosis Society (IAS) and the Residual Risk Reduction Initiative (R3i) Foundation. Cardiovasc Diabetol. 2019;18:71. https://doi.org/10.1186/s12933-019-0864-7.
Pradhan AD, Paynter NP, Everett BM, et al. Rationale and design of the pemafibrate to reduce cardiovascular outcomes by reducing triglycerides in patients with diabetes (PROMINENT) study. Am Heart J. 2018;206:80–93. https://doi.org/10.1016/j.ahj.2018.09.011.
Das Pradhan A, Glynn RJ, Fruchart J-C, et al. Triglyceride lowering with pemafibrate to reduce cardiovascular risk. N Engl J Med. 2022;387:1923–34. https://doi.org/10.1056/NEJMoa2210645.
Karwatowska-Prokopczuk E, Clouet-Foraison N, Xia S, et al. Prevalence and influence of LPA gene variants and isoform size on the Lp(a)-lowering effect of pelacarsen. Atherosclerosis. 2021;324:102–8. https://doi.org/10.1016/j.atherosclerosis.2021.03.036.
Tsimikas S, Karwatowska-Prokopczuk E, Gouni-Berthold I, et al. Lipoprotein(a) reduction in persons with cardiovascular disease. N Engl J Med. 2020;382:244–55. https://doi.org/10.1056/NEJMoa1905239.
Reyes-Soffer G, Ginsberg HN, Berglund L, et al. Lipoprotein(a): a genetically determined, causal, and prevalent risk factor for atherosclerotic cardiovascular disease: a scientific statement from the American Heart Association. Arterioscler Thromb Vasc Biol. 2022;42:e48-60. https://doi.org/10.1161/ATV.0000000000000147.
Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, et al. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9. https://doi.org/10.1001/jama.2009.801.
Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–28. https://doi.org/10.1056/NEJMoa0902604.
Burgess S, Ference BA, Staley JR, et al. Association of LPA variants with risk of coronary disease and the implications for lipoprotein(a)-lowering therapies: a mendelian randomization analysis. JAMA Cardiol. 2018;3:619–27. https://doi.org/10.1001/jamacardio.2018.1470.
Langsted A, Nordestgaard BG, Kamstrup PR. Low lipoprotein(a) levels and risk of disease in a large, contemporary, general population study. Eur Heart J. 2021;42:1147–56. https://doi.org/10.1093/eurheartj/ehaa1085.
Satterfield BA, Dikilitas O, Safarova MS, et al. Associations of genetically predicted Lp(a) (Lipoprotein [a]) levels with cardiovascular traits in individuals of european and african ancestry. Circ Genomic Precis Med. 2021;14: e003354. https://doi.org/10.1161/CIRCGEN.120.003354.
Patel AP, Wang(汪敏先) M, Pirruccello JP, et al. Lp(a) (lipoprotein[a]) concentrations and incident atherosclerotic cardiovascular disease: new insights from a large national biobank. Arterioscler Thromb Vasc Biol. 2021;41:465–74. https://doi.org/10.1161/ATVBAHA.120.315291.
Emdin CA, Khera AV, Natarajan P, et al. Phenotypic characterization of genetically lowered human lipoprotein(a) levels. J Am Coll Cardiol. 2016;68:2761–72. https://doi.org/10.1016/j.jacc.2016.10.033.
Erqou S, Kaptoge S, Emerging Risk Factors Collaboration, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–23. https://doi.org/10.1001/jama.2009.1063.
Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–53. https://doi.org/10.1093/eurheartj/ehq386.
O’Donoghue ML, Fazio S, Giugliano RP, et al. Lipoprotein(a), PCSK9 inhibition, and cardiovascular risk. Circulation. 2019;139:1483–92. https://doi.org/10.1161/CIRCULATIONAHA.118.037184.
Schettler VJJ, Neumann CL, Peter C, et al. Lipoprotein apheresis is an optimal therapeutic option to reduce increased Lp(a) levels. Clin Res Cardiol Suppl. 2019;14:33–8. https://doi.org/10.1007/s11789-019-00094-4.
Roeseler E, Julius U, Heigl F, et al. Lipoprotein apheresis for lipoprotein(a)-associated cardiovascular disease: prospective 5 years of follow-up and apolipoprotein(a) characterization. Arterioscler Thromb Vasc Biol. 2016;36:2019–27. https://doi.org/10.1161/ATVBAHA.116.307983.
Parish S, Hopewell JC, Hill MR, et al. Impact of apolipoprotein(a) isoform size on lipoprotein(a) lowering in the HPS2-THRIVE study. Circ Genomic Precis Med. 2018;11: e001696. https://doi.org/10.1161/CIRCGEN.117.001696.
Merki E, Graham MJ, Mullick AE, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–53. https://doi.org/10.1161/CIRCULATIONAHA.108.786822.
Merki E, Graham M, Taleb A, et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J Am Coll Cardiol. 2011;57:1611–21. https://doi.org/10.1016/j.jacc.2010.10.052.
Viney NJ, van Capelleveen JC, Geary RS, et al. Antisense oligonucleotides targeting apolipoprotein(a) in people with raised lipoprotein(a): two randomised, double-blind, placebo-controlled, dose-ranging trials. Lancet Lond Engl. 2016;388:2239–53. https://doi.org/10.1016/S0140-6736(16)31009-1.
Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet Lond Engl. 2015;386:1472–83. https://doi.org/10.1016/S0140-6736(15)61252-1.
Novartis Pharmaceuticals. A randomized double-blind, placebo-controlled, multicenter trial assessing the impact of lipoprotein (a) lowering with pelacarsen (TQJ230) on major cardiovascular events in patients with established cardiovascular disease. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT04023552. Accessed 18 Sep 2022.
Maki K, Keane W, Bouhajib M, et al. Pharmacokinetics of MAT9001, an omega-3 fatty acid medication, compared with eicosapentaenoic acid ethyl esters in hypertriglyceridemic subjects. FASEB J. 2016;30(suppl):1198.7. https://doi.org/10.1096/fasebj.30.1_supplement.1198.7.
Dai Perrard X-Y, Lian Z, Bobotas G, et al. Effects of n-3 fatty acid treatment on monocyte phenotypes in humans with hypertriglyceridemia. J Clin Lipidol. 2017;11:1361–71. https://doi.org/10.1016/j.jacl.2017.08.011.
Maki KC, Bobotas G, Dicklin MR, et al. Effects of MAT9001 containing eicosapentaenoic acid and docosapentaenoic acid, compared to eicosapentaenoic acid ethyl esters, on triglycerides, lipoprotein cholesterol, and related variables. J Clin Lipidol. 2017;11:102–9. https://doi.org/10.1016/j.jacl.2016.10.010.
Maki KC, Bays HE, Ballantyne CM, et al. A head-to-head comparison of a free fatty acid formulation of omega-3 pentaenoic acids versus icosapent ethyl in adults with hypertriglyceridemia: the ENHANCE-IT study. J Am Heart Assoc. 2022;11: e024176. https://doi.org/10.1161/JAHA.121.024176.
Alexander VJ, Xia S, Hurh E, et al. N-acetyl galactosamine-conjugated antisense drug to APOC3 mRNA, triglycerides and atherogenic lipoprotein levels. Eur Heart J. 2019;40:2785–96. https://doi.org/10.1093/eurheartj/ehz209.
Ionis Pharmaceuticals, Inc. A randomized, double-blind, placebo-controlled, phase 3 study of ISIS 678354 administered subcutaneously to patients with severe hypertriglyceridemia. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT05079919. Accessed 18 Sep 2022.
Ionis Pharmaceuticals, Inc. A randomized, double-blind, placebo-controlled, phase 2b study of ISIS 678354 in patients with hypertriglyceridemia and atherosclerotic cardiovascular disease (established or at increased risk for), and/or with severe hypertriglyceridemia. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT05355402. Accessed 18 Sep 2022.
Ionis Pharmaceuticals, Inc. A randomized, double-blind, placebo-controlled, phase 3 study of AKCEA-APOCIII-LRx administered subcutaneously to patients with familial chylomicronemia syndrome (FCS). clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT04568434. Accessed 18 Sep 2022.
Ionis Pharmaceuticals, Inc. An open-label safety study of AKCEA-APOCIII-LRX administered subcutaneously to patients with familial chylomicronemia syndrome (FCS) previously treated with volanesorsen (ISIS 304801). clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT05185843. Accessed 18 Sep 2022.
Ionis Pharmaceuticals, Inc. An Open-Label Extension Study of AKCEA-APOCIII-LRx Administered Subcutaneously to Patients With Familial Chylomicronemia Syndrome (FCS). clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT05130450. Accessed 18 Sep 2022.
Novo Nordisk A/S. Dose response and safety of an oral PCSK9i, NNC0385-0434, in patients with established atherosclerotic cardiovascular disease (ASCVD) or ASCVD risk on maximally tolerated statin dose and other lipid-lowering therapy requiring further LDL-C reduction. clinicaltrials.gov 2022. https://clinicaltrials.gov/ct2/show/NCT04992065. Accessed 18 Sep 2022.
Koren MJ, Moriarty PM, Baum SJ, et al. Preclinical development and phase 1 trial of a novel siRNA targeting lipoprotein(a). Nat Med. 2022;28:96–103. https://doi.org/10.1038/s41591-021-01634-w.
O’Donoghue ML, López GJA, Knusel B, et al. Study design and rationale for the Olpasiran trials of cardiovascular events and lipoproteiN(a) reduction-DOSE finding study (OCEAN(a)-DOSE). Am Heart J. 2022;251:61–9. https://doi.org/10.1016/j.ahj.2022.05.004.
Amgen. Amgen announces positive topline phase 2 data for investigational olpasiran in adults with elevated lipoprotein(a). 2022. https://www.amgen.com/newsroom/press-releases/2022/05/amgen-announces-positive-topline-phase-2-data-for-investigational-olpasiran-in-adults-with-elevated-lipoproteina. Accessed 21 Sep 2022.
Bellosta S, Rossi C, Alieva AS, et al. Cholesterol lowering biotechnological strategies: from monoclonal antibodies to antisense therapies. A pre-clinical perspective review. Cardiovasc Drugs Ther. 2022. https://doi.org/10.1007/s10557-021-07293-w.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
Open Access funding enabled and organized by Projekt DEAL. No external funding was used for the preparation of this manuscript.
Conflict of interest
Daniel Tobias Michaeli, Julia Caroline Michaeli, Sebastian Albers, Tobias Boch and Thomas Michaeli declare that they have no potential conflicts of interest that might be relevant to the contents of this manuscript.
Ethics Approval
None needed.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
Code Availability
Not applicable.
Author Contributions
Daniel Tobias Michaeli was responsible for conceptualization, data curation, analysis, investigation, methodology, project administration, resources, supervision, validation, visualization, writing, reviewing and editing; Julia Caroline Michaeli for supervision, reviewing and editing; Sebastian Albers for reviewing and editing; Tobias Boch for supervision, reviewing and editing; and Thomas Michaeli for supervision, reviewing and editing.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Michaeli, D.T., Michaeli, J.C., Albers, S. et al. Established and Emerging Lipid-Lowering Drugs for Primary and Secondary Cardiovascular Prevention. Am J Cardiovasc Drugs 23, 477–495 (2023). https://doi.org/10.1007/s40256-023-00594-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-023-00594-5