
Abstract
Background
Persistent postoperative supraventricular tachyarrhythmias (SVTs) increase cardiac burden and aggravate cardiac hemodynamics. Therefore, for patients in unstable conditions after surgery, prompt and sustained control of heart rate is essential. The importance of β-adrenoceptor antagonists (β-blockers) in controlling such postoperative atrial fibrillation or atrial flutter has been established, and the usefulness of ultra-short-acting β1-blockers with high β1 selectivity has been suggested based on their safety and efficacy under such circumstances.
Objectives
Our objectives were to evaluate the effectiveness and safety of landiolol hydrochloride, an ultra-short-acting β1-selective blocker, in the treatment of postoperative SVT in patients with a high risk of myocardial ischemia, or in patients after highly invasive surgery, in a multicenter, randomized, double-blind, placebo-controlled, group-comparative study.
Methods
A total of 165 patients were randomly allocated to three groups and received LM or MH doses of landiolol hydrochloride or placebo. LM group: dose L (1-min loading dose at a rate of 0.03 mg/kg/min, followed by a 10-min infusion at 0.01 mg/kg/min) followed by dose M (1-min loading at a rate of 0.06 mg/kg/min, followed by a 10-min infusion at 0.02 mg/kg/min); MH group: dose M followed by dose H (1-min loading dose at a rate of 0.125 mg/kg/min, followed by a 10-min infusion at 0.04 mg/kg/min); placebo (PP) group: dose P (1-min loading dose at a rate of 0 mg/kg/min, followed by a 10-min infusion at 0 mg/kg/min) followed by another round of dose P. If the targeted heart-rate reduction was not obtained at the end of the first 10-min infusion, the higher dose was started. The primary endpoint was the percentage of patients who met the heart-rate reduction criteria (≥20 % reduction and <100 beats/min). The safety endpoint was the incidence of adverse events in each of the three groups.
Results
The percentages of patients who met the heart-rate reduction criteria (≥20 % reduction and <100 beats/min) were 0.0, 60.4, and 42.0 % in the PP, LM, and MH groups, respectively. There were significant differences in the LM and MH groups relative to the PP group, but there was no significant difference between the LM and MH groups. No significant difference was observed in the incidence of adverse events among the three groups: 29.6 % in the PP group, 45.5 % in the LM group, and 43.1 % in the MH group.
Conclusion
Landiolol hydrochloride is effective and safe for patients with postoperative SVT.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Persistent postoperative supraventricular tachyarrhythmias (SVTs) increase cardiac burden and aggravate cardiac hemodynamics. Hence, for patients in unstable conditions after surgery, prompt and sustained control of heart rate is essential. The importance of β-adrenoceptor antagonists (β-blockers) in controlling such postoperative atrial fibrillation or atrial flutter has been established [1–5], and the usefulness of ultra-short-acting β1-blockers with high β1 selectivity has been suggested based on their safety and efficacy under such circumstances. Esmolol hydrochloride, a prototype ultra-short-acting β1-blocker, has been shown to be effective for postoperative SVT [3] and atrial fibrillation or atrial flutter after open-heart surgery [4].
Landiolol hydrochloride, (–)-[(S)-2,2-dimethyl-1,3-dioxolan-4-yl]methyl 3-[4-[(S)-2-hydroxy-3-(2-morpholinocarbonylamino)ethylamino]propoxy]phenylpropionate monohydrochloride, is a newly developed ultra-short-acting selective β1-blocker with a short half-life (approximately 4 min) in healthy human subjects [6], and its efficacy for tachyarrhythmias during surgery has been confirmed [7].
In order to investigate the efficacy and safety of landiolol hydrochloride on postoperative SVT requiring emergency therapy, we conducted a multicenter, randomized, double-blind, placebo-controlled, three-group comparison study of landiolol hydrochloride in patients with complications of hypertension and ischemic cardiac disease and in patients who had undergone highly invasive sugary (cardiovascular surgery, resection of an esophageal cancer, thoracotomy, and upper abdominal surgery).
2 Methods
This study, conducted in Japan between January 2001 and December 2002, was designed as a central-registration, prospective, multicenter, double-blind, randomized, parallel-group study for the purpose of examining postoperative SVT. The study was conducted in accordance with the ethical principles of the Helsinki Declaration and Good Clinical Practice. Written informed consent was obtained, no later than the day before surgery, from all patients enrolled in the study, and the study protocol was approved by the Institutional Review Board of each study center.
2.1 Study Population
Patients aged 20 years or older who gave informed consent in writing before surgery were selected from 38 study centers in Japan. The selected subjects included those who developed postoperative tachycardia with an identifiable cause that could be relieved, such as hypovolemia, and who, even after treatment, still developed continuous SVT within 7 days postoperatively. Detailed selection criteria are shown in Fig. 1. Patients were excluded from the study if they met one of the following criteria: acute myocardial infarction (within 1 month after onset), preoperative severe heart failure (New York Heart Association [NYHA] functional class III or higher), atrioventricular block (grade II or higher), or sick sinus syndrome (including patients implanted with a pacemaker); treatment with tri- and tetracyclic psychotropic agents or oral β-blockers as concomitant postoperative drugs; a blood pressure decrease (<90/60 mmHg) during the run-in period; aspartate aminotransferase and alanine aminotransferase levels ≥2.5-fold higher than the normal values at the study center (or ≥100 U/L) and blood bilirubin ≥3.0 mg/dL within 2 weeks before surgery; ≥25 mg/dL blood urea nitrogen (BUN) and ≥2 mg/dL creatinine; the presence of drug hypersensitivity due to allergy, pregnancy, or lactation; and administration of any other investigational drug within 6 months prior to the start of the study. Patients judged ineligible by the investigator for any other reason were also excluded from the study.

Criteria for enrollment of patients in the study. *Patients for whom the therapeutic drug could be confirmed, or patients with SBP of 140 mmHg or higher and DBP of 90 mmHg or higher for ≥2 days, with reference to WHO hypertension criteria. †Patients for whom the therapeutic drug could be confirmed. ‡Patients with changes in ST segment of 0.1 mV or more (including a decrease in ST segment by 0.05 mV or more in a Master 2-step test), an abnormal Q wave, negative T wave, U wave, or other abnormal findings related to ischemic changes on the ECG in a resting state or after exercise; or patients with LVH, abnormal wall movement, or a right ventricular/right atrial load on echocardiography in a test performed within 1 month of surgery. §Patients with an abnormal change (≥0.1 mV) in ST segment from the value on ECG at rest and with an ST segment of ≥+0.1 mV or ≤−0.1 mV. ║Variation in HR immediately before administration is less than 10 % of that recorded 1 or 3 min earlier. DBP diastolic blood pressure, ECG electrocardiogram, HR heart rate, LVH left ventricular hypertrophy, SBP systolic blood pressure
2.2 Study Drugs and Study Design
Landiolol hydrochloride for injection (50 mg per vial) and a matching placebo were used in the study. Subjects were randomly allocated to the three groups: landiolol hydrochloride LM group, dose L (1-min loading dose at a rate of 0.03 mg/kg/min, followed by a 10-min infusion at 0.01 mg/kg/min) followed by dose M (1-min loading at a rate of 0.06 mg/kg/min, followed by a 10-min infusion at 0.02 mg/kg/min); MH group, dose M followed by dose H (1-min loading dose at a rate of 0.125 mg/kg/min, followed by a 10-min infusion at 0.04 mg/kg/min); and the placebo (PP) group. Because no ultra-short-acting drug indicated for postoperative SVT has been approved in Japan, placebo was selected as a comparator. After confirming that the selection criteria were met after surgery, four vials of the study drug were dissolved in physiological saline. After a 1-min intravenous infusion, the drug was continuously infused intravenously for 10 min using an infusion pump (the initial dose). If the targeted heart-rate reduction (≥20 % reduction from the baseline heart rate and a heart rate of <100 beats/min) was obtained after administration of the initial dose, the dose was not increased. If the targeted heart-rate reduction was not obtained, a second 1-min infusion and 10-min continuous intravenous infusions were given at an increased dose. If the targeted heart rate reduction was obtained during the period of increased-dose administration, administration was completed. If hypotension (a systolic blood pressure [SBP] decrease of ≥20 % from the baseline SBP and blood pressure ≤90/60 mmHg) or marked bradycardia (a heart rate of ≤60 beats/min) occurred, administration was discontinued.
2.3 Concomitant Drugs and Therapies
Concomitant treatment with β-blockers, calcium antagonists (diltiazem, verapamil, bepridil), other antiarrhythmic agents, adrenergic drugs, and other investigational drugs were prohibited throughout the run-in period and drug-administration period. For patients who had used a prohibited combination of these drugs, we provided a washout period of at least twice the half-life. No patients received intravenous administration of antiarrhythmic agents (propranolol, verapamil, diltiazem, procainamide, disopyramide, mexiletine, lidocaine, aprindine, cibenzoline, or pilsicainide) before or during the study for a period equal to two half-lives of the agent. Therapies that affect heart rate were also prohibited during the run-in and drug-administration periods. If dopamine/dobutamine had been administered by continuous drip (dose rate <10 μg/kg/min) before administration of the study drug, concomitant administration at the same dose was permitted. Digitalis medicines used before administration of the study drug were also permitted if the pharmacological effect was confirmed to have reached a steady state and the bradycardiac effect on the heart rate was constant.
2.4 Clinical Measurements
The following items were surveyed before initiation of the study: demographic characteristics, disease names, surgical techniques, date and time of surgery, type and onset time of SVT, preoperative findings (electrocardiogram [ECG], echocardiogram, other preoperative findings, NYHA classification of cardiac performance), complications of hypertension, previous and concurrent myocardial infarctions, previous and concurrent angina pectoris, other pertinent medical history, and other complications.
Heart rate, blood pressures (SBP/diastolic blood pressure [DBP]), rate-pressure product (RPP), and ECG were measured and calculated as efficacy parameters. ECG was recorded using 12 leads within 1 month before surgery. At least a II- or V5-lead ECG was recorded, and the RR interval, PQ interval, QRS duration, QT interval, corrected QT interval (QTc), and ST segment were measured. Laboratory tests were performed to assess safety, and adverse events were investigated for subjective symptoms and objective findings.
2.5 Efficacy Endpoints
When the heart rate reduction relative to the baseline heart rate was ≥20 % and the heart rate was <100 beats/min after completion or discontinuation of administration, the outcome was designated as ‘improved’. The primary endpoint, i.e., the improvement rate after the final dose, was calculated by the following equation:
In addition, the cumulative improvement rate, heart rate, blood pressure, RPP, and ECG parameters were evaluated as secondary endpoints. The cumulative improvement rate was calculated by the following equation:
2.6 Safety Variables
The incidence and nature of adverse events and adverse drug reactions, as well as abnormal changes in laboratory data, were investigated in comparison with the PP group.
2.7 Sample Size
According to the results of a late phase II study for postoperative SVT, improvement rates after the final dose were hypothesized to be 13.3, 51.1, and 73.3 % for the PP, LM, and MH groups, respectively. Chi-square tests were conducted for statistical analyses, aiming at a two-tailed significance level of 0.05 with a statistical power of 0.9. Multiplicity was adjusted by the Bonferroni correction, with a two-tailed significance level of 0.025 per test. To achieve this level of statistical precision, the necessary number of cases (taking dropouts and withdrawals into account) was determined to be 55 per group, leading to a total of 165 cases among the three groups.
2.8 Statistical Analysis
Statistical analyses were performed using SAS Version 6.12 or 8.2. For comparisons of demographic variables and patient characteristics, Fisher’s exact test or the Kruskal–Wallis test was used where appropriate. The significance level was set at 0.05 (two-tailed). The Chi-square test was used to evaluate differences in improvement rate after the final dose (the primary endpoint) between the PP and LM groups and between the PP and MH groups. The significance level was set at 0.025 (two-tailed), based on the Bonferroni inequality. Secondary endpoints were analyzed by Dunnett’s multiple comparison and the Chi-square test. Significance was defined as p ≤ 0.05 (two-tailed).
3 Results
3.1 Patient Characteristics
A total of 165 patients were enrolled in the study and randomly assigned to the PP group (54 patients), LM group (56 patients), and MH group (55 patients). Of these patients, five were completely excluded from any analyses, including safety evaluation, due to the absence of tachycardia prior to administration of the study drug. The remaining 160 patients are henceforth referred to as the ‘safety patients’ (PP group, n = 54; LM group, n = 55; MH group, n = 51). Protocol deviations and other factors led to exclusion of nine patients (PP group, n = 4; LM group, n = 5) from the full-analysis set (FAS), and 22 patients (PP group, n = 8; LM group, n = 9; MH group, n = 5) from the per-protocol set (PPS). Hence, 160 safety patients were used for safety analysis, and 151 ‘FAS patients’ (PP group, n = 50; LM group, n = 50; MH group, n = 51) and 138 ‘PPS patients’ (PP group, n = 46; LM group, n = 46; MH group, n = 46) were evaluated.
In the FAS patients, no significant differences in demographic characteristics were observed among the three groups (Table 1).
3.2 Primary Endpoint for Efficacy
The improvement rate after the final dose is shown for each group in Table 2. In the FAS patients—the primary population used in the efficacy analysis—the improvement rate was significantly higher in the LM group (60.4 %) and the MH group (42.0 %) than in the PP group (0.0 %) (Chi-square test with Bonferroni correction, p = 0.0001). No significant difference was observed between the LM and MH groups. Similar results were obtained in the PPS patients.
3.3 Secondary Endpoints for Efficacy
3.3.1 Cumulative Improvement Rate within Each Group
In the FAS patients, the cumulative improvement rates at the initial and increased doses were as follows: PP, 0.0 and 0.0 %; LM, 8.3 and 61.6 %; and MH, 22.0 and 43.1 %. Thus, the cumulative improvement rate increased with dose in both the LM and the MH groups. The results obtained from analysis of the cumulative improvement rates were consistent with the findings based on improvement rates after the final dose. Similar results were obtained in the PPS patients.
3.3.2 Tachycardia Improvement Rate after the Initial Dose
In the FAS patients, the tachycardia improvement rates after the initial dose were 0.0, 8.3, and 22.0 % in the PP group (initial dose: placebo [dose P]), LM (initial dose: dose L), and MH (initial dose: dose M) groups, respectively. The improvement rate was significantly higher with dose M than with dose P (Chi-square test, p = 0.0018), although no significant difference was observed between doses P and L or between doses L and M. Similar results were obtained in the PPS patients.
3.3.3 Heart Rate
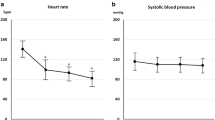
Heart rate time curves of the FAS patients are shown in Fig. 2. The percentage changes in heart rate from baseline (mean ± standard deviations) were −1.56 ± 6.80, −15.02 ± 7.41, and −13.68 ± 10.57 % in the PP, LM, and MH groups, respectively, following the initial dose. After completion of administration (or at discontinuation), the percentage changes (mean ± standard deviations) were −2.42 ± 7.89, −23.32 ± 7.96, and −18.93 ± 10.03 % in the PP, LM, and MH groups, respectively. Hence, after all dose intervals, a significant reduction in heart rate was observed in the LM and MH groups, compared with that in the PP group (Dunnett test, p = 0.0001). Regarding temporal changes in the heart rate, a significant reduction was observed within 6 min after initiation of administration in the LM and MH groups, compared with that in the PP group (Dunnett test: LM group, p = 0.0002; MH group, p = 0.0001), and the maximum bradycardiac effect was observed immediately after completion of administration (or at discontinuation) in the LM group, and 5 min after completion of administration (or at discontinuation) in the MH group. Heart rate increased with time thereafter, and no statistical differences among the three groups were observed 30 min after completion of administration.

Time course of percentage change in heart rate (mean ± standard deviation). Pre 1 measurement of heart rate 3 min (sinus tachycardia), 1 min (other than sinus tachycardia), or 1 min (tachycardia with abnormal ST segment) before initiation of infusion, Pre 2 measurement of heart rate conducted immediately before initiation of infusion, C sum of the result immediately after completion of infusion plus the results at discontinuation of infusion. Refer to Table 1 for treatment group definitions. *Dunnett test; significance was defined as p < 0.05 (two-tailed)
3.3.4 Blood Pressure
The changes in SBP from baseline were −4.2 ± 12.9, −11.2 ± 11.6, and −5.9 ± 14.1 mmHg in the PP, LM, and MH groups, respectively, after the initial dose. The changes in DBP were −1.4 ± 6.8, −4.2 ± 5.6, and −2.5 ± 6.0 mmHg in the PP, LM, and MH groups, respectively. After completion of administration (or at discontinuation), the changes in SBP from baseline were −6.2 ± 18.2, −15.7 ± 14.2, and −9.7 ± 14.7 mmHg in the PP, LM, and MH groups, respectively. Changes in DBP were −1.3 ± 9.3, −5.5 ± 6.4, and −3.6 ± 6.0 mmHg in the PP, LM, and MH groups, respectively.
The values of SBP and DBP were significantly decreased in the LM group at all dose intervals, compared with those in the PP group (Dunnett test: after the initial dose, p = 0.0147 for SBP and p = 0.0428 for DBP; after completion of administration (or at discontinuation), p = 0.0072 for SBP and p = 0.0112 for DBP), but no significant changes were observed in the MH group.
The changes in SBP and DBP in the FAS patients are shown in Fig. 3. Decreases in SBP and DBP were observed 6 min after initiation of administration in the LM and MH groups. After completion of administration, blood pressure increased with time, and the values were similar among the three groups 30 min after completion of administration.

Time course of changes in blood pressure (mean ± standard deviation). Pre 1 measurement of blood pressure 3 min (sinus tachycardia), 1 min (other than sinus tachycardia), or 1 min (tachycardia with abnormal ST segment) before initiation of infusion, Pre 2 measurement of blood pressure conducted immediately before initiation of infusion, C sum of the result immediately after completion of infusion plus the results at discontinuation of infusion, SBP systolic blood pressure, DBP diastolic blood pressure. Refer to Table 1 for treatment group. *Dunnett test; significance was defined as p < 0.05 (two-tailed)
3.3.5 Rate Pressure Product
In the FAS patients, baseline RPPs were 18,196 ± 4491, 16,784 ± 2570, and 16,690 ± 3,086 beats/min · mmHg in the PP, LM, and MH groups, respectively, i.e., the values were higher than 15,000 beats/min · mmHg in all groups. After completion of administration (or at discontinuation), significant decreases in RPP were observed in the LM group (11,255 ± 2,106 beats/min · mmHg) and the MH group (12,503 ± 3,206 beats/min · mmHg), compared with that in the PP group (16,994 ± 4,148 beats/min · mmHg) (Dunnett test, p = 0.0001).
In the RPP time-course, significant decreases were noted 6 min after initiation of administration in the LM and MH groups, compared with the PP group (Dunnett test: LM group, p = 0.0007; MH group, p = 0.0064), and the maximum change was observed immediately after completion of administration (or at discontinuation) in the LM group, and 5 min after completion in the MH group, as observed for the heart rate. RPP increased with time thereafter, and no differences relative to the PP group were detected 30 and 15 min after completion of administration in the LM and MH groups, respectively.
3.3.6 ECG Parameters
The ECG parameters are shown in Table 3. Compared with the PP group, the RR interval, PQ interval, QRS duration, and QT interval were significantly prolonged after completion of administration (or at discontinuation) in both the LM and the MH groups (Dunnett test: RR interval, p = 0.0001; PQ interval, p = 0.0160 and p = 0.0015, respectively; QRS duration, p = 0.0483; QT interval, p = 0.0001). No significant changes were observed in the ST segment or QTc.
3.3.7 Investigation of Subpopulations
The improvement rates after the final dose, stratified by background factors in the FAS population, are shown in Table 4. In the group stratified based on the baseline heart rate, the improvement rate decreased with an increase in the baseline heart rate. No meaningful bias was observed in the other stratifications.
3.4 Safety
3.4.1 Incidences of Adverse Events and Adverse Drug Reactions
Sixteen of 54 patients (29.6 %) in the PP group, 25 of 55 patients (45.5 %) in the LM group, and 22 of 51 patients (43.1 %) in the MH group experienced adverse events. Among these events, those in five patients (9.3 %) of the PP group, 13 patients (23.6 %) of the LM group, and 10 patients (19.6 %) of the MH group were judged by investigators to be related to administration of study drug, i.e., to be adverse drug reactions (ADRs). Although the incidences of adverse events and ADRs were higher in the LM and MH groups than in the PP group, no significant differences were observed among the three groups.
3.4.2 Subjective Symptoms and Objective Findings
There were 28 adverse events, based on subjective symptoms and objective findings, in 11 patients of the PP group, 21 events in 13 patients of the LM group, and 25 events in 12 patients of the MH group. Among these, two events in two patients of the PP group, six events in six patients of the LM group, and five events in four patients of the MH group were judged to be ADRs, as summarized in Table 5. All ADRs in all three groups were related to the cardiovascular system, with the most frequent ADR being hypotension (≤90/60 mmHg). One event of ventricular extrasystoles occurred in one patient of the PP group.
Of the six cases of hypotension in the LM group, the severity was mild in four patients, moderate in one patient, and severe in one patient. Moderate and severe cases of hypotension developed after initiation of the increased dose (dose M). Two cases of mild hypotension remitted without any action being taken, whereas the four other cases were resolved by discontinuation of study drug administration (three cases) and/or treatment with blood transfusion and plasma protein fraction (two cases). There were three mild and two moderate hypotension cases in the MH group. One moderate case developed after the initial dose (dose M), and another moderate case developed 7 min after initiation of the increased dose (dose H). All three cases of mild hypotension remitted without any action being taken, and the two cases of moderate hypotension were resolved by discontinuation of study drug administration (two cases) and/or treatment with procainamide hydrochloride (one case).
3.4.3 Laboratory Observations
Six events were judged to be ADRs based on abnormal changes in laboratory values in three patients of the PP group, 15 such events in seven patients of the LM group, and 14 such events in six patients of the LM group. The increased laboratory values were mainly related to hepatobiliary disorders. All abnormal changes in laboratory values were resolved or remitted without any action being taken.
4 Discussion
The mechanism underlying postoperative SVT is thought to be related to the action of catecholamines, resulting from increased adrenergic drive caused by various stresses during and after surgery [8–11]. In emergency treatment and acute management of postoperative SVT, the use of ultra-short-acting β1-blockers allows rapid onset of drug effect, as well as rapid dissipation of the effects when drug infusion is completed or when adverse events develop. Therefore, esmolol hydrochloride is very useful, and is effective for treatment of postoperative tachyarrhythmias [3] and atrial fibrillation and atrial flutter after open-heart surgery [4]. However, esmolol can be directly cardiodepressive and can cause hypotension and even congestive heart failure, limiting its usefulness [12–15]. Landiolol hydrochloride, a new ultra-short-acting β1-blocker, has a higher cardioselectivity (β1/β2 = 255) [16] and a shorter elimination half-life (4 min in healthy subjects) [6] than esmolol.
The results of this study demonstrate that landiolol hydrochloride is significantly more effective than placebo, based on a primary endpoint of the improvement rate after the final dose (the percentage of patients with ≥20 % reduction in the heart rate from the baseline heart rate and a heart rate of <100 beats/min after completion of administration). No significant difference was observed in the improvement rate after the final dose between the two landiolol hydrochloride-treated groups, although the rate was higher in the LM group than in the MH group. Based on investigations of subpopulations, only the baseline heart rate had a major influence on the improvement rate after the final dose. Therefore, the higher improvement rate after the final dose in the LM group cannot be clearly explained by patients’ background factors, and might be due to the influence of individual differences among the patients in the two groups. The response rate of esmolol in the postoperative SVT was different between the clinical study. We speculate that the absence of a significant difference in the dose response between the LM and MH groups is related to the fact that the postoperative systemic conditions, including hemodynamics, of the patients were highly unstable, and that this instability largely depended on individual patient characteristics; therefore, the response rate of the drug, i.e., the effective dose required to lower heart rate, may have differed markedly among individual patients. However, an increased dose was confirmed to be effective in both the LM and the MH groups. Consequently, we believe that it is appropriate to administer this drug in a regimen in which the dose is gradually increased from low dosage (dose L) to high dosage (dose H). Multiple factors may have contributed to this, including differences in patient demographics, diagnoses, clinical conditions, and, in particular, the types of arrhythmias [17]. Because the criterion “less than 100 beats/min” was used as one of the criteria for improvement, the improvement rate decreased with an increase in the baseline heart rate.
The RPP reflects myocardial oxygen consumption and is used as an index of cardiac load. In this study, the baseline RPP was ≥15,000 beats/min · mmHg in all three groups. There is a positive correlation between the incidence of ST-segment depression in an ECG, which reflects myocardial ischemia, and RPP in patients with RPP of ≥12,000 beats/min · mmHg during coronary-artery bypass graft surgery [18]; an RPP of 12,000 beats/min · mmHg is generally targeted for initiation of therapy for prevention of myocardial ischemia in the perioperative period. In both the LM and the MH groups, landiolol hydrochloride significantly decreased RPP to approximately 12,000 beats/min · mmHg, suggesting that the potency of the study drug was sufficient for avoidance of myocardial ischemia.
Regarding safety, hypotension (≤90/60 mmHg) was the most frequent ADR. However, because this hypotension was resolved or remitted rapidly (within 5 min to 1 h after onset), either without treatment, by discontinuation of study drug administration, or by treatment such as a blood transfusion, we concluded that hypotension could be controlled by appropriate adjustment of the dose or by other treatment. We observed no ADRs based on β2-blocking action, including asthma, peripheral vascular system disorders, or other reactions, reflecting the high β1-selectivity of landiolol hydrochloride.
Esmolol hydrochloride has been compared with placebo in a double-blind controlled study in SVT patients with heart rates of 120 beats/min or higher [19]. The improvement rate caused by esmolol hydrochloride was 66 %, based on a 20 % or greater heart rate reduction and a heart rate of less than 100 beats/min; the rate of recovery of sinus rhythm was 6 % [19]. In this study, the improvement rate in the landiolol hydrochloride-treated group was 62 %, a bradycardiac effect similar to that of esmolol hydrochloride. Regarding the relative safety of esmolol hydrochloride and landiolol hydrochloride, the incidence of hypotension (≤90/50 mmHg) at the effective dose of esmolol hydrochloride was reported by one study to be 52.4 % (13 of 24 patients) in patients with postoperative tachyarrhythmias [3]. By contrast, the incidence of hypotension with landiolol hydrochloride in this study was only 9.4 % (10 of 106 patients). Nonclinical comparison studies between esmolol hydrochloride and landiolol hydrochloride have been conducted in vitro and in vivo, and conflicting data regarding the half-lives and potencies of these drugs are available. Sasao et al. [20] reported that landiolol hydrochloride exhibited more potent negative chronotropic effects without a reduction in blood pressure relative to esmolol hydrochloride; furthermore, esmolol hydrochloride produced a dose-dependent decrease in mean arterial pressure in a rabbit model. Although there are currently no clinical data that directly compare esmolol hydrochloride and landiolol hydrochloride, the data obtained from this study appear to be similar to the results obtained in the rabbit model. This study had the following limitations: the study was conducted between January 2001 and December 2002 but these data could not be published earlier because of an internal company policy; although it would have been ideal to conduct a direct comparison with another short-acting beta-blocker such as esmolol, this could not be achieved as esmolol was not available in Japan at the time of this study; as landiolol is only available for clinical use in Japan, our results are of limited value to international readers.
In conclusion, landiolol hydrochloride can be used safely, and a sufficient therapeutic effect can be obtained by administration at an initial dose M, followed by an increase to dose H if the bradycardiac effect on postoperative SVT is insufficient, in patients with a high risk of myocardial ischemia and in patients who have undergone highly invasive surgery. Landiolol hydrochloride is superior in terms of controllability and safety, because of both the rapid attainment of a bradycardiac effect after initiation of administration and the rapid dissipation of this effect after administration is terminated. Thus, landiolol hydrochloride exhibits potency similar to that of esmolol hydrochloride, and has improved safety characteristics.
Abbreviations
- FAS:
-
Full-analysis set
- PPS:
-
Per-protocol set
- SBP:
-
Systolic blood pressure
- DBP:
-
Diastolic blood pressure
- RPP:
-
Rate pressure product
- ADR:
-
Adverse drug reaction
- NYHA:
-
New York Heart Association
- ECG:
-
Electrocardiogram
- SVT:
-
Supraventricular tachyarrhythmias
References
Matloff JM, Wolfson S, Gorlin R, Harken DE. Control of postcardiac surgical tachycardias with propranolol. Circulation. 1968;37(4 Suppl):II-133–8.
Balser JR, Martinez EA, Winters BD, Perdue PW, Clarke AW, Huang W, et al. β-Adrenergic blockade accelerates conversion of postoperative supraventricular tachyarrhythmias. Anesthesiology. 1998;89:1052–9.
Gray RJ, Bateman TM, Czer LSC, Conklin CM, Matloff JM. Esmolol: a new ultrashort-acting beta-adrenergic blocking agent for rapid control of heart rate in postoperative supraventricular tachyarrhythmias. J Am Coll Cardiol. 1985;5:1451–6.
Mooss AN, Wurdeman RL, Mohiuddin SM, Reyes AP, Sugimoto JT, Scott W, et al. Esmolol versus diltiazem in the treatment of postoperative atrial fibrillation/atrial flutter after open heart surgery. Am Heart J. 2000;140:176–80.
Daoud EG. Management of atrial fibrillation in the post-cardiac surgery setting. Cardiol Clin. 2004;22:159–66.
Nakashima M, Kanamaru M. Phase I study of ONO-1101, a new ultra short acting β1-blocking agent in healthy volunteers. Rinsho Iyaku. 2000;16(10):1531–56 (in Japanese).
Yoshiya I, Ogawa R, Okumura F, Shimada Y, Hanaoka K. Clinical evaluation of landiolol hydrochloride (ONO-1101) on perioperative supraventricular tachyarrhythmia—a phase III, double-blind study in comparison with placebo. Rinsho Iyaku. 1997;13(19):4949–78 (in Japanese).
Halter JB, Pflug AE, Porte D. Mechanism of plasma catecholamine increases during surgical stress in man. J Clin Endocrinol Metab. 1977;45:936–44.
Kalman JM, Munawar M, Howes LG, Louis WJ, Buxton BF, Gutteridge G, et al. Atrial fibrillation after coronary artery bypass grafting is associated with sympathetic activation. Ann Thorac Surg. 1995;60:1709–15.
Breslow MJ, Jordan DA, Christopherson R, Rosenfeld B, Miller CF, Hanley DF, et al. Epidural morphine decreases postoperative hypertension by attenuating sympathetic nervous system hyperactivity. JAMA. 1989;261:3577–81.
Reves JG, Karp RB, Buttner EE, Tosone S, Smith LR, Samuelson PN, et al. Neuronal and adrenomedullary catecholamine release in response to cardiopulmonary bypass in man. Circulation. 1982;66(1):49–55.
Kirshenbaum JM, Kloner RA, Antman EM, Braunwald E. Use of an ultra short-acting β-blocker in patients with acute myocardial ischemia. Circulation. 1985;72(4):873–80.
Jacobs JR, Maier GW, Rankin JS, Reves JG. Esmolol and left ventricular function in the awake dog. Anesthesiology. 1988;68:373–8.
Murthy VS, Hwang TF, Zagar ME, Vollmer RR, Schmidt DH. Cardiovascular pharmacology of ASL-8025, an ultra-short acting β blocker. Eur J Pharmacol. 1983;94:43–51.
Reilly CS, Wood M, Koshakji RP, Wood AJJ. Ultra-short-acting beta-blockade: a comparison with conventional beta-blockade. Clin Pharmacol Ther. 1985;38:579–85.
Iguchi S, Iwamura H, Nishizaki M, Hayashi A, Senokuchi K, Kobayashi K, et al. Development of a highly cardioselective ultra short-acting β-blocker, ONO-1101. Chem Pharm Bull (Tokyo). 1992;40:1462–9.
Schwartz M, Michelson EL, Sawin HS, Macvaugh H III. Esmolol: safety and efficacy in postoperative cardiothoracic patients with supraventricular tachyarrhythmias. Chest. 1988;93(4):705–11.
Kaplan JA, Dunbar RW, Jones EL. Nitroglycerin infusion during coronary artery surgery. Anesthesiology. 1976;45:14–21.
Anderson S, Blanski L, Byrd RC, Das G, Engler R, Laddu A, et al. Comparison of the efficacy and safety of esmolol, a short-acting beta blocker, with placebo in the treatment of supraventricular tachyarrhythmias. The Esmolol vs Placebo Multicenter Study Group. Am Heart J. 1986;111:42–8.
Sasao J, Tarver SD, Kindscher JD, Taneyama C, Benson KT, Goto H. In rabbits, landiolol, a new ultra-short-acting β-blocker, exerts a more potent negative chronotropic effect and less effect on blood pressure than esmolol. Can J Anesth. 2001;48:985–9.
Acknowledgments
The authors thank Dr. Hiroyuki Iinuma, Director of the Cardiovascular Institute, for ECG interpretation in his capacity as the central physician for the study center; and Chikuma Hamada, Associate Professor, Management Engineering, Department of Engineering, Tokyo University of Science, for statistical advice in his capacity as medical statistics advisor for the study. This study was supported by Ono Pharmaceutical Co., Ltd, Osaka, Japan.
Author information
Authors and Affiliations
Corresponding author
Appendix
Appendix
Trial members and Investigators and Study Sites are as follows.
1.1 Trial members
Jun Takezawa, Chikuma Hamada, Hiroyuki Iinuma, Nobuyuki Taenaka
1.2 Investigators and Study Sites
Doi Hirosato, Cardiovascular Center Hokkaido Ohno Hospital; Fumiyuki Okamoto, Teine Keijinkai Hospital; Hironori Ishihara, Hirosaki University School of Medicine & Hospital; Koukichi Andoh, Sendai City Hospital; Mitsukazu Gotoh, Fukushima Medical University Hospital; Kohjirou Urazumi, Ohta Atami Hospital; Hideaki Nakano, Tokyo Medical University Kasumigaura Hospital; Nobuhiro Saruki, Gunma Prefectural Cancer Center; Shunei Kyo, Saitama Medical University Hospital; Junzo Takeda, Keio University Hospital; Miyuki Yokota, Cancer Institute Hospital; Hideaki Miyamoto, Juntendo University Hospital; Takashi Hirotani, Tokyo Saiseikai Central Hospital; Yoshihiro Yagishita, International Medical Center of Japan; Kozo Hashimoto, Fujisawa City Hospital; Toshio Konishi, Yokohama Rosai Hospital; Hiroshi Kanazawa, Niigata City General Hospital; Shigetaka Kasuya, Tachikawa Medical Center; Tsutsumi Yasushi, Fukui Cardiovascular Center; Kousuke Baba, Hokushin General Hospital; Takahiro Takemura, National Nagano Hospital; Yoshito Shiraishi, Shizuoka General Hospital; Hiroshi Noguchi, Aichi Medical University Hospital; Tsutomu Ohi, Matsusaka Central General Hospital; Shinichi Nishi, Osaka City University Hospital; Hisao Kishida, Osaka Medical College Hospital; Masahiro Shinozaki, Wakayama Medical University Hospital; Hiroshi Katayama, Okayama University Hospital; Tatsuhiko Komiya, Kurashiki Central Hospital; Tsuyoshi Maekawa, Yamaguchi University Hospital; Yoshitoyo Miyauchi, Tokuyama Central Hospital; Yasutoshi Matayoshi, Yamaguchi Prefectural Central Hospital; Arifumi Kohyama, Tokushima Red Cross Hospital; Katsuhiro Seo, Kokura Memorial Hospital; Kazuhisa Matsuda, Saiseikai Fukuoka General Hospital; Koji Sumikawa, Nagasaki University Hospital; Shigenori Yoshitake, Oita Medical University Hospital; Yuichi Kanmura, Kagoshima University Medical and Dental Hospital.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Taenaka, N., Kikawa, S. The Effectiveness and Safety of Landiolol Hydrochloride, an Ultra-Short-Acting β1-Blocker, in Postoperative Patients with Supraventricular Tachyarrhythmias: A Multicenter, Randomized, Double-Blind, Placebo-Controlled Study. Am J Cardiovasc Drugs 13, 353–364 (2013). https://doi.org/10.1007/s40256-013-0035-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-013-0035-2