
Abstract
Hierarchical hollow urchin-like nickel cobaltite (NiCo2O4) was synthesized using a two-step hydrothermal method. The effects of metal composition and surfactant addition on the morphology, structure, and electrochemical performance toward oxygen evolution reaction (OER) were investigated. The addition of cetyltrimethylammonium bromide (CTAB) reduced particle aggregation, resulting in a higher electrochemical active surface area and electrical conductivity. Lowering the Ni content from 1.0 to 0.25 did not alter the morphology and structure of the product to any extent. However, the crystallite size slightly increased. Among the spinels with different Ni and Co compositions, NiCo2O4 exhibited a superior OER electrocatalytic activity, achieving a 380 mV overpotential at 10 mA/cm2 current density. It also delivered a good performance in an anion exchange membrane water electrolyzer (AEMWE) using 1 M NaOH at 60 °C, reaching a current density of about 420 mA/cm2 at a cell voltage of 1.95 V.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The rapid increase in the global population has led to a surge in energy demand and consumption. While fossil fuels remain the primary energy source, concerns over resource depletion and climate change have prompted many countries to incorporate renewable energy sources in their energy portfolios. One such promising energy carrier is hydrogen [1, 2]. Water can be electrochemically split to yield hydrogen and oxygen, theoretically requiring 1.23 V. However, additional voltage is necessary to overcome kinetic and resistance challenges within the electrolyte and electrodes [3]. In energy applications, the applied potential typically corresponds to the voltage needed to reach a specific current density, often set at 10 mA/cm2 for hydrogen fuel synthesis [4, 5]. A lower overpotential translates to better cell efficiency.
Over the years, various types of water electrolyzers have been developed. The alkaline water electrolyzer (AWE) represents the most mature technology, leveraging low-cost and abundant transition metals such as cobalt- and nickel-based oxides as cathode and anode materials, respectively [6, 7]. However, the major drawback of AWE lies in the sensitivity of liquid KOH electrolyte to atmospheric CO2, which can diminish cell performance by reducing hydroxyl ion availability for the anodic reaction [8]. On the other hand, proton exchange membrane water electrolyzers (PEMWE) offer an alternative technology that employs perfluorosulfonic acid (PFSA) polymer membranes like Nafion as solid electrolytes [9]. This design enhances safety by eliminating caustic electrolytes. However, the highly acidic nature of Nafion-based membranes limits the catalyst material options to noble and precious metals such as Pt, Ru, and Ir, making the technology more expensive.
In an effort to combine the advantages of both alkaline and PEM electrolysis while mitigating their disadvantages, the polymeric anion exchange membrane water electrolyzer (AEMWE) has been developed. AEMWE operates similarly to AWE but employs a polymeric anion exchange membrane (AEM), such as low-density polyethylene (LDPE), allowing for the use of a lower-concentration alkaline solution or even distilled water instead of concentrated KOH. This minimizes volumetric instability, leakage, and handling issues. Furthermore, AEMWE membranes are less costly than Nafion-based membranes used in PEMWE. The catalysts are based on inexpensive and abundant transition metals, including Li0.21Co2.79O4, Ni0.6Co0.2Fe0.2, NiFe2O4, and CuCoOx [10,11,12,13], rather than noble metals.
Among transition metal-based catalysts, nickel cobaltite (NiCo2O4) has demonstrated excellent electrochemical performance for OER [14,15,16]. Different preparation techniques, morphologies, and chemical compositions of NiCo2O4 yielded varying electrocatalytic activities. In our previous work, we demonstrated that changing the dopant metal (e.g., Ni, Cu, or Mn) in Co3O4 induces morphological and structural changes, leading to varying OER activities [17]. Ni-doped Co3O4 (NiCo2O4) exhibited superior performance due to its high specific surface area, large electrochemically active surface area (ECSA), and low charge-transfer resistance, facilitating electrochemical reactions. Additionally, the electronic, chemical, and structural properties that may influence OER activity can be tailored by changing metal dopant levels in NiCo2O4, affecting the activation energy required for electron transfer during OER [14].
The addition of surfactants during synthesis can also influence nucleation, growth processes [18, 19], particle dispersion, and charge carrier mobility. Using an ionic (e.g., cetyltrimethylammonium bromide) instead of an anion surfactant (e.g., polyvinylpyrrolidone and polyvinyl alcohol) resulted in better particle dispersion, smaller particles, and more porous structures, and, consequently, more efficient mass transport [20, 21]. However, these materials were primarily assessed for supercapacitor applications.
It is worth noting that most developed OER catalysts, including Ni-based ones, have only been tested in half-cell configurations. However, superior performance in a half-cell does not guarantee the same in a full cell, as complex mass and ion transport phenomena occur. Therefore, evaluating catalyst performance in full-cell configurations under various conditions is essential to assess their potential for industrial applications. In this study, NiCo2O4 was synthesized using a hydrothermal method followed by annealing. The effects of surfactants and metal composition on morphology, crystal structure, and OER activity were investigated. The performance of the best-performing catalyst in a half cell was further assessed in a single-cell AEMWE. The influence of varying electrolyte concentrations and operating temperatures on electrocatalytic activity was also explored.
Materials and methods
Synthesis of nickel cobaltite
A solution containing 20 mL distilled water and 10 mL absolute ethanol (CH3CH2OH, Univar) was mixed with 2.0 g urea (NH2CONH2, Techno Pharmchem). Urea serves as a hydrolyzing agent, releasing hydroxyl and carbonate ions which react with metal ions to initiate nucleation. For the surfactant investigation, this initial solution was mixed with 0.5 g cetyltrimethylammonium bromide (CTAB, Loba Chemie Pvt. Ltd.) or polyvinylpyrrolidone (10,000 MW PVP, Sigma-Aldrich). The resulting mixture was stirred for approximately 10 min until a clear solution was achieved. Cobalt(II) nitrate hexahydrate [Co(NO3)2·6H2O, Sigma-Aldrich] and nickel (II) nitrate hexahydrate [Ni(NO3)2·6H2O, Sigma-Aldrich] were then added, with the molar composition adjusted based on the stoichiometric formula NixCo3−xO4, where x = 0.25, 0.5, 0.75, 1. This mixture was stirred for a few minutes until a homogeneous solution formed. Subsequently, the solution was transferred into a 100-mL Teflon-lined stainless-steel autoclave reactor and placed in an oven at 100 °C for 10 h. The resulting precipitates were collected, washed multiple times with water and ethanol, and then dried at 60 °C for 6 h in air. The powder was then annealed at 400 °C for 6 h in air, with a heating rate of 1 °C/min.
Physicochemical characterization
The morphology was analyzed using field-emission scanning electron microscopy (FE-SEM, Hitachi SU-8230). The mean diameter of the structures was determined from the micrographs using Image Processing Analysis in Java software (ImageJ). Information regarding crystal structure, lattice constant, and crystallite size was obtained through X-ray diffraction (XRD, Shimadzu XRD-7000) analysis using Cu Kα (λ = 1.5406 Å). Elemental composition and specific surface area were determined using energy-dispersive X-ray spectroscopy (EDS, Phenom ProX) and Brunauer-Emmett-Teller (BET, Quantachrome Instruments), respectively. Organic impurities were detected using Attenuated Total Reflectance-Fourier-transform infrared spectroscopy (ATR-FTIR, Thermo Fisher Nicolet IS50).
Half-cell test
The electrochemical performance of the samples was evaluated using a conventional three-electrode system in a 1.0 M potassium hydroxide (KOH, Sigma-Aldrich, reagent grade, 90% purity) electrolyte. The counter, reference, and working electrodes were platinum wire, Ag/AgCl (Autolab 6.0726.107), and a catalyst on carbon paper, respectively. The working electrode was prepared by drop-casting the catalyst ink, containing catalyst powder, PVP binder, and a few drops of ethanol on C paper, followed by drying. The mass loading was approximately 0.25 mg/cm2.
Linear sweep voltammetry (LSV) was performed in the potential range of 0 to 0.8 V against Ag/AgCl at a scan rate of 1 mV/s, with potential values reported in the reversible hydrogen electrode (RHE) scale. Overpotential was calculated as the difference between the potential achieved at 10 mA/cm2 current density and theoretical thermodynamic potential for water oxidation. Tafel slope was derived from the polarization curve. Electrochemical impedance spectroscopy (EIS) was conducted in the frequency range of 10− 3 to 105 Hz to determine the charge transfer resistance (Rct) of the samples. The intrinsic activity of the catalysts was assessed using electrochemical active surface area (ECSA), estimated by performing cyclic voltammetry (CV) in the non-Faradaic region at an increasing scan rate from 10 to 50 mV/s.
Single-cell AEMWE test
Figure S1 shows the setup of the electrochemical performance test in the single-cell electrolyzer. The electrolyzer cell was made of titanium with gold-coated serpentine flow fields, having an active area of 1.0 cm2 and surrounded by an O-ring seal. An alkaline AEM based on LDPE-VBC-TMA membrane was used. The anode was prepared by spraying an ink containing nickel cobaltite catalyst, 12.5% wt (30% vol), ACTA I2 ionomer, and n-methyl-2pyrrolidone onto a 300 μm titanium fiber felt gas diffusion layer (Ti-felt GDL, Bekaert S.A. Fibre Technologies, Netherlands). The mass loading was approximately 2.0 mg/cm2. For the cathode, an ink composed of 20% Pt/C, 28% wt. ACTA I2 ionomer, and isopropanol were sprayed onto a non-wet-proof carbon GDL with a microporous layer (C GDL, Freudenberg Germany). The mass loading was about 0.4 mg/cm2.
The anode and cathode were sandwiched on either side of the membrane to assemble the cell, with the cathode side equipped with an ethylene tetrafluoroethylene (ETFE) gasket for proper sealing. The anode and cathode were separately supplied with 0.01 to 1.0 M NaOH (Sigma-Aldrich, reagent grade > 98% purity) electrolyte solution. Under thermostat control, cartridge heaters were inserted into the cell body to regulate the operating temperature from 20 to 60 °C. Electrochemical testing, including CV and EIS, was carried out using a potentiostat (Gamry IFC 5000E) at various intervals.
Results and discussion
Effects of surfactants on physicochemical properties
Surfactants were introduced during synthesis to manipulate particle size and oxide particle dispersion, thereby potentially impacting the electrochemical performance of the material. In this study, two surfactants, namely PVP (a non-ionic surfactant) and CTAB (an ionic surfactant), were employed to evaluate their influence on the morphology, structure, and electrochemical performance of nickel cobaltite (NCO). Figure 1 illustrates low- and high-magnification FESEM images of nickel cobaltite (NCO) prepared through different processes: without surfactant (NCO/BARE), with PVP (NCO/PVP), and with CTAB (NCO/CTAB).

Low- and high-magnification FESEM images of (a–c) NCO/BARE, (d–f) NCO/PVP, and (g–i) NCO/CTAB prepared by hydrothermal method at 180 °C for 10 h followed by calcination in air at 400 °C for 6 h
In the absence of surfactant, urchin-like NCO structures (Fig. 1a–c) were obtained, featuring microspheres and spikes with mean diameters of 3.38 μm and 15.4 nm, respectively. However, these microspheres exhibited aggregation despite a relatively high specific surface area of 59.34 m2/g. This aggregation reduces the exposed surface area as the pathway of ions, electrons, and evolved gases during electrochemical reactions is blocked. The addition of PVP resulted in similar microspheres but with reduced aggregation, having a mean diameter of about 3.33 μm and a slightly increased specific surface area of 63.31 m2/g. Notably, tiny particles adhered to the NCO/PVP surface (Fig. 1d–f).
Substituting PVP with CTAB further decreased microsphere aggregation (Fig. 1g–i). The microspheres appeared slightly smaller, with a mean diameter of about 2.70 μm, and the nanoparticles forming the nanorods measured approximately 16.5 nm. Interestingly, NCO/CTAB exhibited a specific surface area of 49.02 m2/g, slightly lower than NCO/BARE and NCO/PVP, which was attributed to the marginally larger particle size of the spikes of the urchin-like structure. Overall, the results indicate that adding surfactant reduces aggregation by lowering the surface energy of the particles [22]. CTAB proved more effective than PVP in minimizing aggregation, likely due to the formation of compact and electriferous films through electrostatic interactions [20]. The ionic nature of CTAB releases cetyltrimethylammonium ions (CTA+) that readily adhere to the metal oxide surface. It then easily attracts the oppositely charged groups, such as hydroxyl ions, to promote structure growth. A more detailed discussion of the formation mechanism can be found in our previous work [17]. PVP, a non-ionic surfactant, is adsorbed on the metal oxide surface and forms macromolecular protective films through hydrogen bonds [21], which might weaken or become unstable. This could potentially account for the presence of small particles alongside the urchin-like structures observed in the SEM images of NCO/PVP.
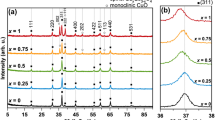
Shown in Fig. S2 are the XRD patterns of NCO/BARE, NCO/PVP, and NCO/CTAB. The peaks at 2θ = 31.00, 36.64, 38.36, 44.78, 55.35, 59.00, and 64.94º were indexed to the 220, 311, 222, 400, 422, 511, and 440 peaks of spinel NCO (JCPDS Card No. 002–8158). All the XRD patterns showed identical spinel NCO phase peaks, indicating that surfactant addition did not alter the phase composition. The absence of secondary phase peaks affirmed the high purity of the samples. Notably, NCO/CTAB exhibited slightly narrower peaks due to a larger crystallite size of about 12.39 ± 0.32 nm compared to NCO/BARE and NCO/PVP, which measured 11.85 ± 0.42 and 11.63 ± 0.34 nm, respectively.
Further analysis using FTIR spectra in Fig. 2 revealed distinct features. In the spectrum of NCO/PVP (Fig. 2a), peaks at 554 and 641 cm− 1 corresponded to Ni-O and Co-O stretching vibrations, confirming NiCo2O4 formation. Additional peaks at approximately 1092, 1592, and 2883 cm− 1 were attributed to the C-N, C = O, and C-H stretching vibrations within the PVP structure, which confirms the presence of residual PVP in NCO/PVP. Conversely, the spectrum of NCO/CTAB (Fig. 2b) displayed only peaks corresponding to Co-O and Ni-O stretching vibrations. This implies the complete removal of CTAB during calcination, resulting in pure nickel cobaltite.

[Color online] FTIR spectra of (a) NCO/PVP and (b) NCO/CTAB prepared by hydrothermal method at 180 °C for 10 h followed by calcination in air at 400 °C for 6 h
Effects of Ni concentration on physicochemical properties
Hydrothermal treatments of solutions containing CTAB and metal salt precursors, with different molar compositions adjusted according to the stoichiometric formula NixCo3-xO4 (x = 0.25, 0.5, 0.75, 1) were performed. The resulting products are depicted in Fig. S3, showing both low- and high-magnification FESEM images. When 0.417 mmol (x = 0.25) of Ni precursor was used, microspherical structures with a mean diameter of 6.46 μm were produced. Higher magnification images revealed similar hierarchical structures with nanorods having a mean diameter of about 29.0 nm. As shown in Fig. 3a, EDS analysis indicated that the precipitates contain 3.96 at% Ni, 39.25 at% Co, and 56.79 at% O. This composition is consistent with the most probable formula of Ni0.25Co2.75O4, suggesting the complete reaction of metal ions during the hydrothermal process. When 0.833 (x = 0.5), 1.250 (x = 0.75), and 1.667 (x = 1) mmol of Ni precursor were employed, similar structures were produced, corresponding to the chemical formulas Ni0.5Co2.5O4, Ni0.75Co2.25O4, and NiCo2O4, as confirmed by EDS analysis in Fig. 3b–d. This demonstrates that all the metal ions reacted, even with increased Ni concentration.

[Color online] EDX spectrum of (a) Ni0.25Co2.75O4, (b) Ni0.5Co2.5O4, (c) Ni0.75Co2.25O4, and (d) NiCo2O4 produced using hydrothermal synthesis at 180 °C for 10 h followed by calcination in air at 400 °C for 6 h
Figure S4 displays the XRD patterns of NixCo3-xO4 (x = 0.25, 0.50, 0.75, 1.0). All the patterns revealed peaks characteristic of spinel NCO (JCPDS Card No. 002–8158), suggesting spinel oxide formation irrespective of the metal composition. Notably, it appeared that the peaks slightly broadened as Ni content increased. This can be attributed to a slight reduction in crystallite size, from approximately 15.23 ± 0.53 to 12.88 ± 0.37 nm (See Table S1). This was likely influenced by introducing more dopant ions, increasing lattice disorder, and hindering crystallization [23]. This may also explain the observed reduction in the mean diameter of nanoparticles with higher Ni content. Furthermore, an increase in Ni content led to a leftward shift in the peaks, which can be associated with lattice expansion induced by the incorporation of more Ni ions into the cobaltosic structure (See Table S1), as Ni ions are slightly bigger than Co ions [24].
Half-cell electrochemical performance of nickel cobaltite
Table 1 summarizes key electrochemical data for NCO/BARE, NCO/PVP, and NCO/CTAB, and different NixCo3-xO4 (x = 0.25, 0.50, 0.75, 1.0), including overpotential at 10 mA/cm2, Tafel slope, charge-transfer resistance, and ECSA. These results provide insights into their OER efficiency.
Presented in Fig. 4a are the LSV polarization curves of NCO/BARE, NCO/PVP, and NCO/CTAB. NCO/BARE attained an overpotential of 399 mV at 10 mA/cm2. A slight decrease in overpotential was observed for NCO/PVP, with 387 mV. In contrast, NCO/CTAB achieved a slightly lower overpotential of 380 mV. Aside from overpotential, current density is also an essential parameter in OER. It indicates how effectively the catalyst can promote OER per unit electrode area. At 1.7 V, NCO/BARE, NCO/PVP, and NCO/CTAB obtained a current density of 27, 60, and 92 mA/cm2, respectively. This indicates a huge 61% increase in current density upon the addition of CTAB. This improvement in overpotential and current density can be attributed to the less aggregated and well-defined urchin-like structure of NiCo2O4, offering more active sites and allowing efficient ion and mass transport during electrochemical reactions. Additionally, residual PVP in NCO/PVP may have hindered performance by potentially blocking active sites. Figure 4b depicts the polarization curves of NixCo3−xO4 (x = 0.25, 0.50, 0.75, 1.0). Ni0.25Co2.75O4 recorded an overpotential of 399 mV. Increasing Ni content reduced overpotential to about 380 mV, achieved by NiCo2O4, indicating decreasing overpotential with higher Ni content. A substantial increase in current density from 21 to 92 mA/cm2 was also observed with increasing Ni content.

[Color online] Polarization curve of (a) NCO/BARE, NCO/PVP, and NCO/CTAB and (b) NixCo3-xO4 (x = 0.25, 0.50, 0.75, 1.0) in 1 M KOH at a scan rate of 1 mV/s, corresponding Tafel plots of (c) NCO/BARE, NCO/PVP, and NCO/CTAB and (d) NixCo3-xO4 (x = 0.25, 0.50, 0.75, 1.0), and Nyquist plots of (e) NCO/BARE, NCO/PVP, and NCO/CTAB and (f) NixCo3-xO4 (x = 0.25, 0.50, 0.75, 1.0)
In terms of the Tafel slope (Fig. 4c), NCO/CTAB achieved the lowest Tafel slope of 62 mV/dec, followed by NCO/PVP and NCO/BARE with 69 and 85 mV/dec, respectively. Among NixCo3-xO4 (x = 0.25, 0.50, 0.75, 1.0), NiCo2O4 had the lowest Tafel slope of about 62 mV/dec (Fig. 4d), suggesting faster kinetics. This aligns with our previous study [17] which showed a faster transition of Co3+ to Co4+, considered the rate-determining step in OER, with increasing Ni content.
Nyquist plots of electrocatalysts obtained through EIS and Randle equivalent circuit (inset) are shown in Fig. 4e–f. The equivalent circuit consists of solution resistance (Rs), internal resistance of the electrocatalyst (R1), constant-phase element for oxide electrocatalyst (CPE1), and charge-transfer resistance (Rct) and constant-phase element for electrocatalyst-electrolyte interface (CPE2). Two semicircles were observed in the Nyquist plots of the catalysts. The small semicircle in the high-frequency region corresponds to the internal resistance of the catalyst, while the large semicircle in the low-frequency region to the Rct for oxygen evolution at the electrocatalyst-electrolyte interface. The diameter of the semicircle corresponds to the resistance value. Hence, a smaller semicircle indicates lower charge-transfer resistance and faster charge transport at the electrocatalyst-electrolyte interface [25]. Based on the results, NCO/CTAB achieved the lowest Rct of about 7.2 Ω, followed by NCO/PVP and NCO/BARE with 8.5 and 27.8 Ω, respectively. On the other hand, Ni0.25Co2.75O4 had the highest Rct of about 15 Ω, followed by Ni0.5Co2.5O4, Ni0.75Co2.25O4, and NiCo2O4 with 14, 8.6, and 7.2 Ω, respectively. This suggests that the rate of kinetic reaction at the electrocatalyst-electrolyte interface affects OER activity.
In addition, electrochemically active surface area (ECSA) was estimated by dividing the double layer capacitance (Cdl) [Fig. S5] by the typical specific capacitance of catalysts in 1 M KOH (Cs = 40 µF/cm2). Unlike BET-specific surface area, ECSA could measure the extent of the catalyst surface that can participate in electrochemical reactions. NCO/CTAB attained the highest ECSA of 184 cm2, which is 10 and 55% higher than NCO/PVP (166 cm2) and NCO/BARE (82 cm2), respectively. This suggests that NCO/CTAB had the most exposed surface area, which could facilitate electrolyte penetration and oxygen gas desorption. The effects of factors, such as specific surface area, charge-transfer resistance, and ECSA, on the OER activity were elucidated in the following paragraph.
As our previous work [17] discussed, specific surface area, charge-transfer resistance, and ECSA significantly influence OER activity. A high specific surface area, low charge-transfer resistance at the electrocatalyst-electrolyte interface, and high ECSA can improve OER activity. However, as demonstrated in this study, NCO/CTAB achieved the lowest overpotential despite having the lowest specific surface area. It also had the lowest Rct and the highest ECSA. Therefore, this implies that a high specific surface area does not always equate to better OER performance [26]. The excellent performance of NCO/CTAB might result from the synergistic effect of ECSA and charge-transfer resistance. These findings further suggest that adding surfactant can effectively improve electrocatalytic performance towards OER. CTAB appeared more effective than PVP, possibly because it reduced particle aggregation more. A less-aggregated structure provides larger channels for efficient electrolyte penetration, facilitates electron transport and release of gas, and enhances active site utilization [27]. Furthermore, using CTAB during hydrothermal synthesis resulted in a pure NCO structure with a well-defined urchin-like morphology, less aggregation, and no impurities, which can expose more surface area, improve contact between particles, and prevent charge carrier scattering. These factors provide pathways for electrical conduction, facilitate the transport of carriers between neighboring particles, and enhance charge carrier mobility, ultimately leading to improved electrical conductivity [28].
It was also shown that the electrochemical performance improved with increasing amounts of Ni ions, which can be attributed to enhanced electrical conductivity. Incorporating more dopant ions into Co3O4 contributes to the creation of holes and increases the number and mobility of charge carriers (holes), thereby potentially improving electrical conductivity, as Co3O4 is a p-type semiconductor [29]. As indicated in the XRD analysis results, introducing more Ni ions led to lattice expansion, which could alter the electronic band structure and reduce charge carrier scattering. This, in turn, might enhance charge carrier mobility and, consequently, electrical conductivity.
In summary, this study demonstrated that an effective OER catalyst allows the rapid diffusion of charges, reactants, and products. This can be achieved by adding an optimal amount of dopant ions and designing structures with well-defined structures, minimal aggregation, and no impurities. Furthermore, the hydrothermal method used in this study can be employed to easily tailor the properties of nickel cobaltite for specific applications, such as water electrolyzers, which can potentially be extended to other energy storage devices, including batteries, fuel cells, and supercapacitors.
Single-cell AEMWE electrochemical performance of NiCo2O4/CTAB
Overall, NiCo2O4/CTAB achieved excellent half-cell performance towards OER. Interestingly, our material performed comparably or even better than other NiCo2O4 fabricated using different techniques (e.g., NiCo2O4 nanowires deposited on FTO by hydrothermal method) [16, 30,31,32,33,34,35]. This can be attributed to the differences in their morphologies. Compared with nanowire arrays, hierarchical structures offer a larger accessible surface area and more available sites for electrochemical reactions. This outstanding half-cell OER activity demonstrated by NiCo2O4/CTAB prompted an evaluation of its performance in a single-cell AEMWE under industrially relevant conditions.
Figure 5a presents the LSV polarization curves of NiCo2O4/CTAB in the AEMWE using 1.0 M NaOH electrolyte at operating temperatures ranging from 20 to 60 °C. NiCo2O4/CTAB exhibited an onset overpotential of about 326 mV at 20 °C, which slightly decreased to 306 mV at 40 °C and further reduced to 257 mV at 60 °C. A similar trend was observed for overpotential at 10 mA/cm2, with values decreasing from 379 to 336 mV with increasing temperature. Generally, higher temperatures typically result in improved electrical conductivity because of (1) increased rates of electrochemical reactions due to reduced energy barriers for OER, (2) improved mass transport due to lower electrolyte viscosity, and (3) favored forward reaction (i.e., oxygen gas formation) due to thermodynamic effects, as described in the Nernst equation [36]. Apart from overpotential, current density is a critical parameter in water electrolysis. At 1.95 V, NiCo2O4/CTAB achieved a current density of about 264 mA/cm2 at 20 °C, which increased to 318 and 420 mA/cm2 at 40 and 60 °C, respectively. This enhancement can be attributed to improved hydroxyl ion conductivity and material activity [37].

[Color online] (a) LSV polarization curve, (b) Tafel plot, (c) Nyquist plot at 1.7 V, and (d) Nyquist plot at 1.6–1.7 V of NiCo2O4 in AEMWE using 1 M NaOH at 20 to 60 °C
Figure 5b shows the Tafel slope derived from the polarization curves. The value decreased from 54 to 48 mV/dec as the operating temperature increased, indicating enhanced kinetics. The Nyquist plot in Fig. 5c also revealed a decrease in Rct from 1.92 to 1.04 Ω, with increasing temperature from 20 to 60 °C. This suggests improved charge-transfer kinetics at the electrolyte-electrode interface. In addition, the Nyquist plots generated at different applied potentials of 1.6 and 1.7 V were compared in Fig. 5d. At 20, 40, and 60 °C, the charge-transfer resistance decreased from 3.82 to 1.92, 2.64 to 1.44, and 2.00 to 1.04 Ω, respectively, when the applied potential was increased from 1.6 to 1.7 V. This can be attributed to the high redox reaction rate at higher potentials which decreases the charge-transfer resistance.
Figure 6 presents the LSV polarization curves of NiCo2O4/CTAB in an AEMWE operating at 60 °C, using NaOH electrolyte concentrations ranging from 0.01 to 1.0 M. At 0.01 M NaOH, an onset overpotential of 354 mV was recorded, which decreased to 299 and 257 mV at 0.1 and 1.0 M NaOH, respectively. The overpotential at 10 mA/cm2 decreased from 449 to 388 mV at 0.1 M and 336 mV at 1 M, respectively. Notably, at 1.95 V, an 85% rise in current density, from 63 to 420 mA/cm2, was observed with increasing electrolyte concentration. The improved electrochemical performance of NiCo2O4/CTAB can be attributed to the enhanced ionic conductivity at higher electrolyte concentrations [9]. Furthermore, as shown in Fig. 6b–c, the Tafel slope reduced from 104 to 48 mV/dec, and the charge-transfer resistance from 7.66 to 2.00 Ω with decreasing electrolyte concentration. These findings suggest slower kinetics at lower electrolyte concentrations because fewer ions are available to participate in the electrochemical reactions, resulting in slower ion transport and lower ionic conductivity [ref]. Moreover, lower concentrations of ions in the electrolyte can impede the adsorption of reactant molecules on the electrode surface, affecting the availability of active sites. Mass transport may also be hindered at lower electrolyte concentrations, leading to slower diffusion of reactants and products to and from the electrode surfaces [13].

[Color online] (a) LSV polarization curve, (b) Tafel plot, and (c) Nyquist plot of NiCo2O4 in AEMWE using 0.01 to 1 M NaOH at 60 °C
The electrochemical performance of NiCo2O4/CTAB at varying electrolyte concentrations and operating temperatures is summarized in Fig. S6, with relevant electrochemical data in Table 2. The results indicate that the electrochemical performance is most excellent at the highest electrolyte concentration and operating temperature and declines when these parameters are lowered. In 1 M NaOH at 60 °C, the catalyst achieved an overpotential at 10 mA/cm2 of 336 mV and a current density of approximately 420 mA/cm2 in 1 M NaOH. This performance aligns with the requirements of an AEMWE, which typically operates at 50 to 80 °C with a cell voltage and current density in the range of 1.8 to 2.2 V and 100 to 500 mA/cm2, respectively [9]. Therefore, this suggests its suitability for AEMWE applications.
Conclusion
Hierarchical nickel cobaltite structure was successfully prepared using a two-step hydrothermal method. The addition of surfactant resulted in less aggregated particles, with CTAB having a more substantial effect than PVP. Consequently, NCO/CTAB achieved a higher ECSA and electrical conductivity, hence the low overpotential reported. A decrease in Ni content from 1.0 to 0.25 did not change the morphology and structure of the material. However, the crystallite size slightly increased. Among the catalysts, NiCo2O4 exhibited the most excellent OER electrochemical performance. It also demonstrated a good performance in an AEMWE using 1 M NaOH at 60 °C, achieving a current density of about 420 mA/cm2 at a cell voltage of 1.95 V. Reducing the electrolyte concentration from 1 to 0.01 M and operating temperature from 60 to 20 °C led to a decline in electrocatalytic activity due to loss of ionic conductivity.
References
Khan, R., Arshad, J., Khan, S., Mansoor, M.A., Ali, S., Nisar, T., Wagner, V., Asghar, M.A., Haider, A.: Surfactant-assisted fabrication of prussian blue analogs as bifunctional electrocatalysts for water and hydrazine oxidation. Mol. Catal. (2023). https://doi.org/10.1016/j.mcat.2023.113415
Osman, A., Mehta, N., Elgarahy, A.M., Hefny, M., Al-Hinai, A., Al-Muhtaseb, A.H., Rooney, D.W.: Hydrogen production, storage, utilization, and environmental impacts: A review. Environ. Chem. Lett. (2022). https://doi.org/10.1007/s10311-021-01322-8
Suen, N.T., Hung, S.F., Quan, Q., Zhang, N., Xu, Y., Chen, H.M.: Electrocatalysis for the oxygen evolution reaction: Recent development and future perspectives. Chem. Soc. Rev. (2017). https://doi.org/10.1039/C6CS00328A
Karmakar, A., Srivastava, S.K.: Interconnected copper cobaltite nanochains as efficient electrocatalysts for water oxidation in alkaline medium. ACS Appl. Mater. Interfaces. (2017). https://doi.org/10.1021/acsami.7b03029
Ganley, J.: High temperature and pressure alkaline electrolysis. Int. J. Hydrog Energy. (2009). https://doi.org/10.1016/j.ijhydene.2009.02.083
Ursua, A., Gandia, L.M., Sanchis, P.: Hydrogen production from water electrolysis: Current status and future trends. Proc. IEEE. (2012). https://doi.org/10.1109/JPROC.2011.2156750
Wendt, H., Hofmann, H.: Ceramic diaphragms for advanced alkaline water electrolysis. J. Appl. Electrochem. 19, 605–610 (1989)
Gülzow, E.: Alkaline fuel cells: A critical view. J. Power Sources. (1996). https://doi.org/10.1016/S0378-7753(96)02344-0
Vincent, I., Bessarabov, D.: Low cost hydrogen production by anion exchange membrane electrolysis: A review. Renew. Sustain. Energy Rev. (2018). https://doi.org/10.1016/j.rser.2017.05.258
Wu, X., Scott, K.: CuxCo3–xO4 (0 ≤ x < 1) nanoparticles for oxygen evolution in high performance alkaline exchange membrane water electrolysers. J. Mater. Chem. (2011). https://doi.org/10.1039/C1JM11312G
Liu, S., Sajjad, S.D., Gao, Y., Kaczur, J., Masel, R.: An alkaline water electrolyzer with sustainion™ membranes: 1 a/cm² at 1.9v with base metal catalysts. ECS Trans. (2017). https://doi.org/10.1149/07709.0071ecst
Faid, A.Y.A.A., Barnett, A.O., Seland, F., Sunde, S.: Ternary NiCoFe nanosheets for oxygen evolution in anion exchange membrane water electrolysis. Int. J. Hydrog Energy. (2022). https://doi.org/10.1016/j.ijhydene.2022.05.143
Pavel, C.C., Cecconi, F., Emiliani, C., Santiccioli, S., Scaffidi, A., Catanorchi, S., Comotti, M.: Highly efficient platinum group metal free based membrane-electrode assembly for anion exchange membrane water electrolysis. Angew Chem. Int. Ed. (2014). https://doi.org/10.1002/anie.201308099
Osgood, H., Devaguptapu, S.V., Xu, H., Cho, J., Wu, G.: Transition metal (Fe, Co, Ni, and Mn) oxides for oxygen reduction and evolution bifunctional catalysts in alkaline media. Nano Today. (2016). https://doi.org/10.1016/j.nantod.2016.09.001
Béjar, J., Álvarez-Contreras, L., Ledesma-García, J., Arjona, N., Arriaga, L.G.: Electrocatalytic evaluation of Co3O4 and NiCo2O4 rosettes-like hierarchical spinel as bifunctional materials for oxygen evolution (OER) and reduction (ORR) reactions in alkaline media. J. Electroanal. Chem. (2019). https://doi.org/10.1016/j.jelechem.2019.113190
Wang, J., Qiu, T., Chen, X., Lu, Y., Yang, W.: Hierarchical hollow urchin-like NiCo2O4 nanomaterial as electrocatalyst for oxygen evolution reaction in alkaline medium. J. Power Sources. (2014). https://doi.org/10.1016/j.jpowsour.2014.06.034
Flores, C.L.I., Balela, M.D.L.: Electrocatalytic oxygen evolution reaction of hierarchical micro/nanostructured mixed transition cobalt oxide in alkaline medium. J. Solid State Electrochem. (2020). https://doi.org/10.1007/s10008-020-04530-4
Han, X., Wahl, S., Russo, P.A., Pinna, N.: Cobalt-assisted morphology and assembly control of co-doped ZnO nanoparticles. Nanomaterials. (2018). https://doi.org/10.3390/nano8040249
Caputo, G., Pinna, N.: Nanoparticle self-assembly using π-π interactions. J. Mater. Chem. A. (2013). https://doi.org/10.1039/C2TA00534D
Zhang, S.R., Hu, Z.B., Liu, K.Y., Liu, Y., Fang, H., Xie, Q.: Synthesis and characterization of porous cobalt oxide/copper oxide nanoplate as novel electrode material for supercapacitors. Trans. Nonferrous Met. Soc. China. (2015). https://doi.org/10.1016/S1003-6326(15)64055-9
Kong, L.B., Lu, C., Liu, M.C., Luo, Y., Kang, L.: Effect of surfactant on the morphology and capacitive performance of porous NiCo2O4. J. Solid State Electrochem. (2013). https://doi.org/10.1007/s10008-013-2016-4
Zhang, J., Lin, Z., Lan, Y., Ren, G., Chen, D., Huang, F., Hong, M.: A multistep oriented attachment kinetics: Coarsening of ZnS nanoparticle in concentrated NaOH. J. Am. Chem. Soc. (2006). https://doi.org/10.1021/ja062572a
Song, W., Ren, Z., Chen, S.Y., Meng, Y., Biswas, Y., Nandi, P., Elsen, H.A., Gao, P., Suib, S.L.: Ni- and Mn-promoted mesoporous Co3O4: A stable bifunctional catalyst with surface-structure-dependent activity for oxygen reduction reaction and oxygen evolution reaction. ACS Appl. Mater. Interfaces. (2016). https://doi.org/10.1021/acsami.6b06103
Abdelhak, L., Bedhiaf, B., Amar, B., Cherifa, D., Benhebal, H.: Tuning of the physical properties by various transition metal doping in Co3O4: TM (TM = ni, Mn, Cu) thin films: A comparative study. Chin. J. Phys. (2018). https://doi.org/10.1016/j.cjph.2018.08.012
Asghar, M.A., Ali, A., Haider, A., Zaheer, M., Nisar, T., Wagner, V., Akhter, Z.: Electrochemically deposited amorphous cobalt–nickel-doped copper oxide as an efficient Electrocatalyst toward Water Oxidation reaction. Int. J. Hydrogen Energy. (2021). https://doi.org/10.1021/acsomega.1c01251
Mamlouk, M.: 4.19 – Alkaline anion exchange membrane (AEM) water electrolysers—Current/future perspectives in electrolysers for hydrogen. Comprehensive Renewable Energy (2nd ed.) (2022). https://doi.org/10.1016/B978-0-12-819727-1.00103-5
Fu, H., Liu, Y., Chen, L., Shi, Y., Kong, W., Hou, J., Yu, F., Wei, T., Wang, H., Guo, X.: Designed formation of NiCo2O4 with different morphologies self-assembled from nanoparticles for asymmetric supercapacitors and electrocatalysts for oxygen evolution reaction. Electrochim. Acta. (2019). https://doi.org/10.1016/j.electacta.2018.11.103
Iskandar, F., Abdillah, O.B., Stavila, E., Aimon, A.H.: The influence of copper addition on electrical conductivity and charge transfer resistance of reduced graphene oxide (rGO). New. J. Chem. (2018). https://doi.org/10.1039/C8NJ03614D
Abdelhak, L., Bedhiaf, B., Amar, B., Benhebal, H., Ammari, A., Dalache, C.: Structural, optical, and electrical properties of Ni-doped Co3O4 prepared via sol-gel technique. Mat. Res. (2018). https://doi.org/10.1590/1980-5373-MR-2017-0545
Yu, X., Sun, Z., Yan, Z., Xiang, B., Liu, X., Du, P.: Direct growth of porous crystalline NiCo2O4 nanowire arrays on a conductive electrode for high-performance electrocatalytic water oxidation. J. Mater. Chem. A. (2014). https://doi.org/10.1039/C4TA05315J
Zheng, Z., Geng, W., Wang, Y., Huang, Y., Qi, T.: NiCo2O4 nanoflakes supported on titanium suboxide as a highly efficient electrocatalyst towards oxygen evolution reaction. Int. J. Hydrogen Energy. (2017). https://doi.org/10.1016/j.ijhydene.2016.11.187
He, J., Sun, Y., Wang, M., Geng, Z., Wu, X., Wang, L., Chen, H., Huang, K., Feng, S.: Direct growth of NiCo2O4 nanostructure on conductive substrate by electrospray technique for oxygen evolution reaction. J. Alloys Compd. (2018). https://doi.org/10.1016/j.jallcom.2018.04.132
Liu, X., Liu, J., Li, Y., Li, Y., Sun, X.: Au/NiCo2O4 arrays with high activity for water oxidation. ChemCatChem. (2014). https://doi.org/10.1002/cctc.201402217
Lu, B., Cao, D., Wang, P., Wang, G., Gao, Y.: Oxygen evolution reaction on Ni-substituted Co3O4 nanowire array electrodes. Int. J. Hydrogen Energy. (2011). https://doi.org/10.1016/j.ijhydene.2010.09.056
Umeshbabu, E., Ranga Rao, G.: NiCo2O4 hexagonal nanoplates anchored on reduced graphene oxide sheets with enhanced electrocatalytic activity and stability for methanol and water oxidation. Electrochim. Acta. (2016). https://doi.org/10.1016/j.electacta.2016.07.161
Sarkar, S., Peter, S.: An overview on Pd-based electrocatalysts for the hydrogen evolution reaction. Inorg. Chem. Front. (2018). https://doi.org/10.1039/C8QI00042E
Leng, Y., Chen, G., Mendoza, A.J., Tighe, T.B., Hickner, M.A., Wang, C.: Solid-state water electrolysis with an alkaline membrane. J. Am. Chem. Soc. (2012). https://doi.org/10.1021/ja302439z
Acknowledgements
This work was supported by the DOST-ERDT Program and CHED-Newton Agham Institutional Links under the project entitled “Affordable Electrolyzer Technology based on Transition Metal Catalysts for Energy Storage Applications.” Dr. Balela would like to acknowledge the Pablito R. Hao Chua Professorial Chair Award for Materials Engineering, College of Engineering, University of the Philippines-Diliman. The Engineering Research and Development for Technology (ERDT) of the Department of Science and Technology (DOST), Philippines, supported the publication of this article through the Faculty Research and Development Grant (FRDG).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
I declare that the work submitted for publication indicated above is original, has not been published before, and is not under consideration for publication anywhere. I declare that there is no conflict of interest concerning this publication. This statement and declaration have been approved by all co-authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Flores, C.L.I., Gupta, G., Mamlouk, M. et al. Enhancing the physicochemical properties of nickel cobaltite catalyst for oxygen evolution reaction in anion exchange membrane water electrolyzers. Mater Renew Sustain Energy (2024). https://doi.org/10.1007/s40243-024-00258-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-024-00258-7