
Abstract
This study has investigated the effect of the incorporation of graphene foam (GF) into the matrix of a ternary transition-metals hydroxide containing nickel, cobalt, and manganese for optimal electrochemical performances as electrodes for supercapacitors applications. An adopted simple, low-cost co-precipitation synthesis method involved the loading a mass of the ternary metal hydroxides (NiCoMn-TH) onto various GF mass loading so as to find ints effect on the electrochemical properties of the hydroxides. Microstructural and chemical composition of the various composite materials were investigated by employing scanning/transmission electron microscopy (SEM/TEM), x-ray diffraction (XRD), Raman spectroscopy, and N2 physisorption analysis among others. Electrochemical performances of the NiCoMn-TH/200 mg GF composite material evaluated in a three-electrode system using 1 M KOH solution revealed a maximum specific capacity around 178.6 mAh g−1 compared to 76.2 mAh g−1 recorded for the NiCoMn-TH pristine material at a specific current of 1 A g−1. The best mass loading of GF nanomaterial (200 mg GF), was then utilised as a positive electrode material for the design of a novel hybrid device. An assembled hybrid NiCoMn-TH/200 mg GF//CSDAC device utilizing the NiCoMn-TH/200 mg GF and activated carbon derived from the cocoa shell (CSDAC) as a positive and negative electrode, respectively, demonstrated a sustaining specific capacity of 23.4 mAh g−1 at a specific current of 0.5 A g−1. The device also yielded sustaining a specific energy and power of about 22.32 Wh kg−1 and 439.7 W kg−1, respectively. After a cycling test of over 15,000 cycles, the device could prove a coulombic efficiency of ~ 99.9% and a capacity retention of around 80% within a potential range of 0.0–1.6 V at a specific current of 3 A g−1. These results have demonstrated the prodigious electrochemical potentials of the as-synthesized material and its capability to be utilized as an electrode for supercapacitor applications.
Similar content being viewed by others


Introduction
The rapid increase of energy utilization worldwide has led to concerns such as energy shortages as well as severe environmental effects. These effects include the degradation of the ozone layer, climate change and global warming [1]. It is essential to explore efficient strategies to successfully diverge from fossil fuel to greener, cost-effective, and environmentally friendly energy sources and ensure adequate provision against the ever-increasing energy demand [2]. Renewable sources of energy (Solar, wind, etc.) represent true opportunities for the improvement of energy efficiency. These technologies are becoming very attractive for electricity generation. However, their durability does not seem to satisfy the demands of technical applications [3]. Nowadays, promising alternatives are being studied. These include chemical, electrochemical, mechanical, thermal, and physical energy generation and storage [3]. Supercapacitors and Batteries are energy storage devices standing at the core of these technologies. Batteries have high specific energy; this specification renders them very useful when requiring keeping devices charged throughout the day. When high specific power is needed, electrochemical capacitors are the best fit [4]. Both batteries and supercapacitors operate via electrochemical processes. However, these processes have different mechanisms that determine their relative specific power and energy [5].
Unlike batteries, supercapacitor devices are capable of giving off high specific power [4]. These devices have attracted attention as they could be used essentially in electric automobiles, backup memories, aircraft emergency doors, portable electronic devices, and micro-devices [3]. With their highly reversible energy storage paths, these devices also demonstrated excellent efficiency for long-standing applications [4]. One of the measures to reach advanced performances of the mentioned applications is to improve the supercapacitor's specific energy while keeping its high specific power. To do so, researchers strive to develop new materials while evolving the electrochemical knowledge of the interfaces at the nanoscale [3].
Interactions taking place within the supercapacitor’s active materials define their properties. The electrolyte, the positive, and the negative electrodes combination determine their functionalities, thermal as well as electrical features [6]. Metal oxides, metal chalcogenides, metal hydroxides, carbon-based materials, and polymers are commonly used materials for making electrodes for supercapacitors. These distinct materials display different electrochemical properties as they have inherent constituents’ properties [7]. For instance, transition metal hydroxides and oxides have the reputation of being battery-type (supercapacitors) materials because of their high specific capacity yield [3]. Their rich redox reactions result from a combination of various transitions metal ions [8,9,10]. However, most metal hydroxides are limited by their low conductivities and cyclic stabilities. Many scientists have investigated composite nanomaterials made from metal oxides, hydroxides, and carbon nanomaterials [11,12,13,14,15]. Having a high theoretical specific capacity and being highly conductive, cobalt oxide materials have attracted significant interest in electrode materials production. Nevertheless, their high cost and environmental pollution risk restrict their use for supercapacitor applications. To address this issue, scientists partly substitute the cobalt atoms with other elements while preserving their nanostructure. This technique appears to be very useful and promising [16]. Graphene foam sheets’ high conductivity trigger a fast transfer of electrons to and from the sites of active material intercalation as it allows the nanoparticles and sheets to be physically closely linked [17]. The graphene’s mechanical strength enables most of the expansion and contraction (associated with electrode mechanical loss and performance decrease) of the anchored nanoparticles to be absorbed during the material’s intercalation and deintercalation [18]. There have been many reports on the use of non-metallic, bimetallic oxides/hydroxide or even ternary oxides. To our awareness, there are very few publications on ternary metal hydroxides carbon composites. Merging ternary metal hydroxides and carbon nanoscale materials represent an opportunity to reach advanced supercapacitors’ designs [6].
This investigation's objective was to develop a composite material promoting synergy between the ternary hydroxide and the highly conductive graphene foam. Manganese, Nickel, and Cobalt elements were precipitated onto the graphene foam to obtain a Mn − Ni − Co ternary hydroxide/GF composite (NiCoMn-TH/GF). The optimisation consisted of varying the mass of GF in the composite. The NiCoMn-TH/GF composite electrode with the highest electrochemical performances was then assembled with an activated carbon derived from cocoa pod waste (CSDAC) in a two-electrode configuration. The assembled asymmetric NiCoMn-TH/200 mg GF//CSDAC hybrid device demonstrated a specific capacity of 23.4 mAh g−1 at a specific current of 0.5 A g−1 with sustaining specific power and energy of 439.7 W kg−1 and 22.3 Wh kg−1, respectively. The device revealed an excellent coulombic efficiency of 99.9% and a capacity retention of ~ 80% over a cycling test of over 15,000 cycles within at an operating potential of 1.6 V at a specific current of 3 A g−1 using 1 M KOH electrolyte. The various characterisation methods adopted in this investigation have demonstrated motivating and promising trends in implementing the NiCoMnTH/GF as suitable electrode materials for energy storage device applications.
Experiments details
Materials synthesis
Synthesis of NiCoMn-TH via co-precipitation method
The pristine nickel–cobalt-manganese ternary hydroxide (NiCoMn-TH) was synthesized via the co-precipitation of equimolar quantity (3 mmol) of nickel nitrate hexahydrate (Ni(NO3)2·6H2O; 548,109 mg), Cobalt nitrate hexahydrate (Co(NO3)2·6H2O; 548,829 mg) and manganese nitrate tetrahydrate (Mn(NO3)2·4H2O; 536,85 mg). Initially, the three chemical solutions were dripped into a beaker filled with 100 ml of deionized (DI) water and then stirred for about 30 min at 40 °C. Whilst stirring, 14 ml of 1 M NaOH was added dropwise to preserve a pH of 10 needed for polarization. Thereafter, the mixture was allowed to further stir for 2 h at the same temperature (40 °C). The resulting greyish precipitate was left to cool down naturally. The recovered sample was centrifuged, washed with DI water and ethanol until neutral. The final product was dried in an oven at 60 °C for 24 h.
Graphene foam (GF) synthesis
The GF was synthesised via a chemical vapour deposition technique (CVD). The procedure started by placing a polycrystalline Ni foam (3D scaffold template) template at the centre of the quartz tube. The Ni foam was then annealed for an hour at 1000 °C at a ramping rate of 2 °C min−1 with a combination of argon (Ar: 300 Sccm) and hydrogen (H2: 200 Sccm). Methane gas (CH4) was used as a source of carbon. The methane was flown through the tube reaction vessel during the last 10 min of the annealing process at the same 1000 °C at 10 sccm. This resulted in deposits of graphene on the nickel foam sheet, and the sample was quickly cooled by moving the tube at a cooler furnace section. To extract the GF, the sample was submerged in a 3 M HCl solution heated to 80 °C for 84 h to ensure a complete etching of the supporting nickel structure. Once the nickel was completely etched out, the recovered GF was cleaned multiple times with deionized water till the acid was drained and dried in an electric oven at 60 °C. Figure 1 shows a plot of Raman and XRD analysis, respectively, for the obtained GF. The Raman spectrum in Fig. 1a indicates the GF is formed by a few layers of graphene sheets, with two main peaks corresponding to G and 2D modes of typical graphene material. The observed symmetric shape/higher intensity of the 2D peak in relation to the G peak is evident of few layers and high crystalline graphene. Figure 1b shows the XRD pattern of the GF, indicating diffraction peaks at around 2θ = 31 and 64.5°, which correspond to the 002 and 004 reflections of hexagonal graphite, respectively. The XRD pattern displays no peaks that could be attributed to Ni, indicating the successful etching of Ni from the GF [26].

a Raman, and b XRD patterns of the as-synthesized GF sample, respectively
NiCoMn-TH/GF synthesis via a Co-precipitation-hydrothermal method
The NiCoMn-TH/GF was produced via a co-precipitation-hydrothermal method. A schematic description of the synthesis of NiCoMn-TH/GF is shown in Scheme 1.

Schematic of the synthesis route of NiCoMn-TH/GF composite
Initially, GF in a quantity of 100 mg was immersed in 100 ml of deionised water using an ultra-sonicator for 72 h at room temperature. Then 3 mmol of each cobalt nitrate hexahydrate (Co(NO3)2·6H2O; 548.829 mg), nickel nitrate hexahydrate (Ni(NO3)2∙6H2O; 548.109 mg), and manganese nitrate tetrahydrate (Mn(NO3)2·4H2O; 536.85 mg) were added to the GF dispersed in water and stirred at 200 rpm at a temperature of 40 °C. Droplets of 1 M NaOH (14 ml) were added to the mixture to preserve a pH of 10 required for polarisation after 30 min of magnetic stirring. The reagents were then kept at 40 °C for 2 h upon magnetic stirring. Thereafter, the product was moved into a stainless-steel Teflon-lined sealed autoclave and heated at 150 οC for 4 h. Lastly, the NiCoMn-TH/100 mg GF composite was exposed to air to cool to room temperature, rinsed with deionised water, and dried overnight at 60 °C. The synthesis of NiCoMn-TH/150 mg GF, NiCoMn-TH/200 mg GF, and NiCoMn-TH/250 mg GF composites were preceded in the same manner by varying the amount of graphene foam to 150 mg, 200 mg, and 250 mg, respectively.
Synthesis of cocoa pod waste derived activated carbon
The cocoa pod waste was obtained from a farmland disposal site. To produce the activated carbon, the pods were initially washed thoroughly using acetone and deionized water to remove the sand impurities and then dried for 24 h at a temperature of 60 οC. The washed and dried pods were cut into smaller pieces. Afterward, the raw material in a quantity of 10 g was soaked in a 100 ml 0.056 M sulphuric acid solution. The mixture was moved to a 100 ml sealed autoclave unit and heated up in an oven at a temperature of 160 °C for 12 h. The recovered product was allowed to cool down naturally to ambient temperature, washed, and dried for two days at 80 οC. The dried material was then mixed in a mass of ratio 1:1 with potassium hydroxide pellet in an agate mortar before being cautiously positioned inside a horizontal quartz tube furnace for further carbonization and activation. The process was carried out at 600 °C for a period of 1 h with a continuous flow of argon at 300 sccm using 5 °C min−1 as the ramping rate. The recovered substance was then treated with 3 M HCl and deionized water till reaching a neutral pH point. The finished product was rinsed with deionized water and dried in a furnace for 24 h at 60 °C.
Samples characterization
SEM images, as well as the EDX spectra of the samples in this study, were acquired by a Zeiss Ultra Plus 55 field emission scanning electron microscope (FE-SEM). The instrument was powered at 1.0 kV and fitted with an energy-dispersive X-ray spectrometer. TEM analysis was conducted on a JEOL JEM-2100F microscope with a field-emission gun at 200 kV. A theta/2theta-geometry XPERT PRO diffractometer (PANalytical BV, Netherlands), using a cobalt tube at 35 kV and 50 mA XRD was utilised to identify the synthesized materials phase and crystal structure. The samples Raman analysis was performed with a WITec confocal Raman microscope (WITec alpha 300 RAS + , Ulm, Germany) with a laser wavelength of 523 nm, laser power of 4 mW, and spectral acquisition time of 120 s. A NOVA-touch LX2 Quanta chrome was utilised to study the samples' surface area and pore size distribution (PSD). This analysis was performed after pre-degassing the samples under vacuum for 18 h at a temperature of 100 °C. The instrument was programmed with the BET method for the measurement of the specific surface area derived from the absorption/adsorption isotherms with the relative pressure (P/Po) ranging from 0.01 to 0.2. The pore size distribution was evaluated using the discrete Fourier transform (DFT) method.
Samples’ electrochemical analysis
To manufacture the electrodes, the active materials were mixed with polyvinylidene difluoride (PVDF) added as the binder and conductive carbon acetylene black (CAB) to compensate for the conductivity lost by adding the binder. The mixture was composed of 80 wt% of active material, 10 wt% of CAB, and 10 wt% of PVDF. N-methyl-2-pyrrolidone (NMP) solvent droplets were then added to the mixture, muddled together, and mixed evenly to make a paste. Afterwards, the mixture was pasted on a 1 × 1 cm2 cleaned Ni foam surface used as a current collector and then place to dry overnight in an electric furnace at 60 °C. The electrodes electrochemical performances were then analysed using the EC-Lab ® V11.33 software Bio-Logic VMP300 potentiostat (Knoxville TN 37.930, USA) in a three-electrode measurement setup. On the setup, the counter electrode used was glassy carbon, the reference electrode was Ag/AgCl, and the working electrode was the synthesised materials. The analysis was carried out with potassium hydroxide as the electrolyte having a concentration of 1 M at 25 °C. The active materials' masses pasted on the electrodes were in the range of 2.0–2.4 mg cm−2 for the pristine and all composite materials. The CV measurements of the samples were performed at various scan rates (mV s−1) in a working potential ranging from 0.0 to 0.5 V against the reference electrode. The GCD analysis was performed at specific currents varying from 1 to 10 A g−1 with operating potential ranging from 0.0 to 0.5 V. The samples EIS was controlled by the open-circuit potential at frequencies ranging from 10 MHz to 100 kHz. Using the GCD Curves, the specific capacity (Qs) and the Coulombic efficiency of the single electrode materials was determined using Eqs. 1 to 8 [18]:
\({Q}_{s}\) is the specific discharge capacity (mAh g−1); \(I\) is the applied current (A); \(t\) is the discharge time (s)); and \(m\) is the active material mass (g)).
\({\varepsilon }_{\mathrm{t}}\) is the columbic efficiency; \({t}_{\mathrm{D}}\) is the discharge time and \({t}_{\mathrm{C}}\) is the charge time.
The asymmetric device’s specific capacity (Qs) was also determined with Eq. 1 using the two-electrode discharge curves. Using the slope of the discharge pattern, the specific energy and power based on the specific current were calculated using the relations:
ED is the specific energy (Wh kg−1); \(V\) is the operating potential window (V); \(I\) is the discharge current (A); \(m\) is the total mass loading of both electrodes active materials in grams (g); \(t\) is the electrode discharge time (s)) and
\({P}_{D}\) is the specific power density (kW kg−1); \(\Delta t\) is the discharge time (s).
The fabricated hybrid device charges were balanced on the positive and negative electrodes using the equation [19]:
The positive electrode was made of faradic material, while the negative electrode was made of the carbon-based material. Therefore, the stored charges could be written as:
\(Q\) is the specific capacity of the positive electrode \((\mathrm{mAh g}-1)\);\({C}_{s}\) is the negative electrode specific capacitance (F g−1); \(m\) is the mass of active material (g) and \(\Delta V\) is the potential window (V).
The masses could then be determined, as displayed in Eqs. 7 and 8.
Results and discussion
Physical characterization of the materials
The SEM images of the as-prepared samples are shown in Fig. 2. Figure 2a displays an agglomerate flake-like surface representing the NiCoM-TH pristine. Figure 2b–e displays the NiCoM-TH/GF composites with mass loading ranging from 100 to 250 mg. All figures display an agglomeration of NiCoMn-TH particles on top of the graphene foam. However, Fig. 2d shows the NiCoMnTH/200 mg GF composite with particles appearing to garnish and evenly cover the carbon sheets reducing the self-aggregation of the NiCoMn-TH particles.

SEM images of a NiCoMn-TH (pristine), b NiCoMn-TH/100 mg GF, c NiCoMn-TH/150 mg GF, d NiCoMn-TH/200 mg GF, and e NiCoMn-TH/250 mg GF
Figure 3 illustrates the TEM results obtained for the different samples studied. Figure 3a displays the NiCoMn-TH pristine nanoparticles whereby single are particles stacked together. This phenomenon complements the argument on agglomerated flake-like structures observed with the SEM in Fig. 2a. Figure 3b displays the NiCoMn-TH/100 mg GF where the NiCoMn-TH particles and the GF could be distinguished from one another with the NiCoMn-TH particles agglomerating as noticed with the SEM. Figure 3c representing the NiCoMn-TH/150 mg GF composite illustrates more or less the same morphology as Fig. 3b with a poor distribution of the NiCoMn-TH nanoparticles on the graphene foam.

TEM micrographs of a NiCoMn-TH (pristine), b NiCoMn-TH/100 mg GF, c NiCoMn-TH/150 mg GF, d NiCoMn-TH/200 mg GF, and e NiCoMn-TH/250 mg GF
Additionally, the graphene foam seems to aggregate with the sheets restacking on themselves. Figure 3d displays the NiCoMn-TH/200 mg GF composite TEM image, where single NiCoMn-TH particles and a graphene layer could be seen to interact closely. This behaviour confirms the excellent distribution of the NiCoMn-TH on the graphene foam particles as noticed on the SEM in Fig. 2d. Figure 3e demonstrates the restacking of the NiCoMn-TH nanoparticles illustrated with the SEM analysis of the same material.
The sample EDX analysis results are illustrated in Fig. 4. Figure 4 proves the presence of Nickel, cobalt, and manganese components within all the various materials. These elements are depicted via their respective spectrums and weight percentage. The weight percentage of the material indicates the 1:1:1 ratio of the Nickel, cobalt, and manganese content within the material with the metal hydroxides having equal molarity as mentioned in the material synthesis. Figure 4 also displays the carbon spectrum in all the composites materials. The percentage of carbon is seen to increase as the graphene foam amount within the composite is increased from 100 to 250 mg with the highest carbon peak spectrum observed for the NiCoMn-TH/250 mg composite. Figure 5 is a display of EDX mapping for the materials. The figure shows the elemental distribution of the NiCoMn-TH (pristine), NiCoMn-TH/100 mg GF, NiCoMn-TH/150 mg GF, NiCoMn-TH/200 mg GF, and NiCoMn-TH/250 mg GF composites as observed in Fig. 5a–e, respectively. It is observed that the Ni, Co, and Mn elements are uniformly distributed throughout the interconnected porous carbon structure. This suggests that the agglomerated nanoparticles were composed of NiO, CoO and MnO embedded into the GF matrix.

EDX elemental composition of NiCoMn-TH (pristine), NiCoMn TH/100 mg GF, NiCoMn-TH/150 mg GF, NiCoMn-TH/200 mg GF, and NiCoMn-TH/250 mg

EDX mapping of a NiCoMn-TH (pristine), b NiCoMn TH/100 mg GF, c NiCoMn-TH/150 mg GF, d NiCoMn-TH/200 mg GF, and e NiCoMn-TH/250 mg, respectively
The Raman spectra for the pristine and all the composites are displayed in Fig. 6a. Figure 6a illustrates the modes characterising brucite-type hydroxides such as Ni(OH)2 and Co(OH)2 ranging from 310 to 530 cm−1 as previously reported for NiCoMnO composites [20]. The CoOH and Co(OH)2 Raman modes could also be identified for all the samples at 557 and 641 cm−1, respectively [20]. The vibration mode noticed at about 625 cm −1 for all the samples is in complete agreement with the previous peaks (500, 518, 590) displaying the significant vibrational features of MnO2 [21, 22]. The GF-based composites exhibit typical carbon materials bands, D, G, and 2D bands at about 1370 cm−1, 1640 cm−1, and 2790 cm−1, respectively [20]. It also reveals that the NiCoMn-TH/200 mg GF is formed with few-layer graphene sheets with more pronounced G and 2D peaks characteristic to a graphene material. Additionally, the NiCoMn-TH/200 mg GF displays a pronounced G' defect peak, which could be accredited to the strong interaction of the NiCoMn-TH nanoparticles with the GF material.

a Raman spectra, and b XRD patterns of as-synthesised samples, respectively
The powder X-ray spectroscopy data of the pristine as well as all the composite materials are displayed in Fig. 6b. The samples XRD spectra display characteristic diffraction patterns of the Ni(OH)2, Co(OH)2, and Mn(OH)2. These patterns are logged in the inorganic crystal structure database (ICSD) having ICSD card numbers of 28,101, 88,940, and JCPDS no. 73–1604 standards for Ni(OH)2, Co(OH)2, and Mn(OH)2 materials, respectively. The NiCoMn-TH key diffraction peaks at 2θ of 11.1° (003), 22.2° (006), 33.5° (101), 34.1° (012), 38.4° (015), 45.3° (018), 59.5° (110), and 60.8° (113) were also detected [23]. The GF presence in the composite materials could be depicted with the presence of the hexagonal graphite diffraction peak (002) at 2θ of approximately 27°. It can be noticed that this spectrum is highly pronounced for the composite NiCoMn-TH/200 mg GF. The narrow and strong diffraction peaks indicate a good crystallinity of the materials [24].
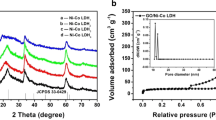
Figure 7a shows type IV isotherm having an H3 hysteresis loop for all the synthesised materials, and with a higher absorbed volume (cc g−1) for the NiCoMn-TH/200 mg GF suggesting a greater porosity of the material as compared to the pristine and other composites [25]. The BET specific surface areas (SSA) recorded for NiCoMn-TH, NiCoMn-TH/100 mg GF, NiCoMn-TH/150 mg GF, NiCoMn-TH/200 mg GF, and NiCoMn-TH/250 mg GF were 16.34 m2 g−1, 52.02 m2 g−1, 49.78 m2 g−1, 63.45 m2 g−1, and 32.67 m2 g−1, respectively. The higher pore volume recorded for the NiCoMn-TH/200 mg GF sample is supported by the larger specific surface area in comparison to the rest of the materials and its superior pore size distribution (PSD) displayed in Fig. 7b. The materials surface area features may also be related to the morphological traits (TEM and SEM) reported earlier whereby a flake-like, agglomerated morphology was shown with the NiCoMn-TH material opposite to the non-agglomerated layered sheet-like particle morphology of the NiCoMn-TH/200 mg GF. An average pore volume of ~ 0.05, 0.15, 0.10, 0.15, 0.08 cm3 g−1 was evaluated for the NiCoMn-TH, NiCoMn-TH/100 mg GF, NiCoMn-TH/150 mg GF, NiCoMn-TH/200 mg GF, and NiCoMn-TH/250 mg GF, respectively.

a N2 adsorption–desorption isotherms, and b pore size distribution of as-synthesised samples
Electrochemical analysis
Three-electrode measurements
Figure 8a illustrates the pristine and all composites’ CV patterns ran at a scan rate of 20 mV s−1. The pristine (NiCoMn-TH) appears to have a slightly different shape as compared to all the composites. The NiCoMn-TH/200 mg CV seemed to have a more pronounced shape with a higher current response indicating much better redox reactions as compared to the other samples. Fascinatingly, the surface of the composite sample can be expected to have a good electrical conductivity due to the excellent electrical conductivity of GF, compared to that of the pristine material [26]. It is suggested that the NiCoMn-TH/200 mg GF composite has an enhanced bounding of electrons between the redox sites. The graphene foam nanosheets, which improves the porosity of the material, contributes to a higher transport of charges [26], improves capacitance and enhances electrostatic charge storage of the electrode. Besides, such electrode utilizes the GF’s natural lightweight, elasticity alongside its mechanical strength [26]. The higher current response observed in Fig. 8a could also be related to the higher specific capacity for the NiCoMn-TH/200 mg as compared to the pristine and other composites. The oxidations and reductions peaks are principally caused by the electrode materials. The distinguished Faradaic reactions peaks illustrated on the CV curves in Fig. 8a could be associated with the anodic and cathodic peaks, respectively occurring during the charge–discharge [6]. These redox reactions are suggested as follow:

a CV patterns at 20 mV s−1, b GCD patterns at 1 A g−1, c EIS Nyquist plot, with the inset showing low-frequency region, d equivalent series resistance versus graphene foam mass loading, and e specific capacity versus specific current of different samples
Figure 8b illustrates the GCD patterns of the NiCoMn-TH, NiCoMn-TH/100 mg GF, NiCoMn-TH/150 mg GF, NiCoMn-TH/200 mg GF, NiCoMn-TH/250 mg GF materials at different specific currents delimited with a potential range varying from 0 to 0.5 V, respectively. The electrode materials Faradaic properties observed in the CV patterns are confirmed by the non-linear GCD pattern having noticeable plateaus. The patterns also display higher discharge times for the NiCoMn-TH/200 mg GF in comparison with the other samples. Figure 8c illustrates the Nyquist impedance plots for the as-prepared materials. This measurement was assessed at a potential of 0.0 V and frequencies varying from 10 MHz to 100 kHz. The low-frequency region displayed a semicircle for the pristine and all the composites. These semicircles occur as a result of high resistance in electron transfer from the electrode material to the electrolyte [6]. The NiCoMn-TH/200 mg GF semicircle curvature could be seen to be less pronounced as compared to the other electrodes indicating its lower electron transfer resistance.
From Fig. 8c, it could also be seen that the diffusion path length which provides information regarding the electrochemical conductivity of the material appears to be shorter and nearer to the ideal vertical line for the NiCoMn-TH/200 mg GF composite. The NiCoMn-TH/200 mg GF composite electrode as observed in the inset to Fig. 8c, showed the lowest values for equivalent series resistance, Rs as well as the charge transfer resistance, Rct. The composite electrode material, which combines the cutting-edge mechanical and electrical properties of conventional 2D graphene along with the properties of metallic foam, thus capable of providing high conductivity, excellent strength and flexibility. The GF’s unique structure in the material opens up the potential to store huge sums of energy, thus the enhanced electrochemical performances [26]. Figure 8d illustrates the plot of the electrical series resistance (ESR), which are the Nyquist plot’s x-intercept values. These values also provide information related to the electrolyte/solution resistance (Rs values). The low NiCoMn-TH/200 mg GF Rs value and its shorter diffusion path length demonstrate that is the most electrochemically conductive material as compared to the other electrode materials.
Figure 8e displays the evaluated specific capacities of the as-prepared nanomaterials as a function of their specific currents. Despite a low decrease of specific capacities as the current density is increased, all the materials retained most of their specific capacities (e.g. the specific capacity of the NiCoMn-TH/200 mg GF at 10 A g−1 has a value of 120.1 mAh g−1). The slight drop of the specific capacity is suggested to occur due to shorter interactions time between the electrolyte and the material when higher current densities are applied [24].
Driven by the exceptional electrochemical properties of the NiCoMn-TH/200 mg GF as compared to the pristine and the other composites displayed in Fig. 8, further electrochemical tests were done on the NiCoMn-TH/200 mg GF electrode material to have a better understanding of its behaviour. Figure 9a and b illustrate CV and GCD curves of the NiCoMn-TH/200 mg GF (composite with improved electrochemical performances) composite, respectively at different scan rates and specific currents. Figure 9 displays non-linear patterns for the CV and GCD as well as oxidation and reduction peaks at approximately 0.3 V and 0.45 V on the CV curves. As the scan rate values are increased, Fig. 9a illustrates that the oxidation–reduction peaks tend to be non-visible. These peaks at the anodes and cathodes are suggested to originate from the reversible redox Faradaic reaction of an assorted composition made of Ni3+, Ni4+, Co3+, Co4+, Mn3+, and Mn4+ emphasising the materials Faradaic behaviour [27, 28]. Figure 9b also displays prominent plateaus on the GCD complementing the Faradaic behaviour displayed with the peaks observed on the CV [28]. These oxidation and reduction peaks are noticed to be more pronounced at lower specific currents [6].

a CV curves at various scan rates and b GCD curves at distinct specific currents for NiCoMn-Th/200 mg
Two-electrode measurement
Encouraged by the improved electrochemical properties of the synthesised NiCoMn-TH/200 mg GF composite as illustrated earlier, an asymmetric hybrid device was assembled. Cocoa shell-derived activated carbon (CSDAC) which is being studied in detail for electrochemical properties in our research group was utilised as the negative electrode while NiCoMn-TH/200 mg GF was used as the positive electrode of the device. The NiCoMn-TH/200 mg GF//CSDAC device total mass balance ratio was determined with Eq. (8) to be 1–2.1. An effective mass loading of the electrodes was then determined to be approximately 2 mg cm−2 for the NiCoMn-TH/200 mg GF and 4.2 mg cm−2 for the CSDAC making an overall mass of the electrode materials equivalent to 6.2 mg per cm2. The device's electrodes were manufactured, as stated previously in the experimental procedure. The tests were completed in a Two-electrode setup made of a 1 M KOH electrolyte, a standard 2032 grade coin cell used as the casing, and a Watman Celgard paper used as the separator.
Figure 10a displays the individual CV patterns for the NiCoMn-TH/200 mg and the CSDAC electrodes measured in their respective steady potential ranges of − 0.9 to 0 V for the negative electrode and 0–0.5 V for the positive electrode both at a scan rate of 20 mV s−1. Figure 10b reveals the CV patterns of the full devices ran at several scan rates within a cell potential of 1.6 V. It appears that the constant shape of the device at several scan rates indicates no decay of capacity. This indicates a rapid mechanism of ion transport and excellent rate capability [29]. According to the individual potential windows, the device' maximum voltage should be approximated to 1.5 V. It was observed that the compatibility of the two electrodes enabled the device to operate at 1.6 V which is beyond its theoretical maximum potential window. Figure 10b also illustrates the CV patterns displaying the merging of both EDLC and Faradaic mechanisms of charge storage taking place in the assembled device with an extended voltage in aqueous electrolyte.

a CV curves at 20 mV s−1 of the positive and negative electrodes, b device’s CV curves at different scan rates, c device's GCD curves at different specific currents, d device's specific capacity versus specific current, e device's Ragone plot, f device's capacity retention and Coulombic efficiency, and g device's voltage holding
Figure 10c illustrates the GCD patterns of the assembled NiCoMn-TH/200 mg GF//CSDAC SC device at several specific currents in a Two-electrode setup. The distinct curves display a plateau (non-linear charge–discharge curves) which agrees with the Faradaic particularities detected in the CV curves of the device. Figure 10d illustrates the device-determined specific capacities at diverse specific currents. The highest specific capacity obtained with the device was approximately 23.4 mAh g−1 at 0.5 A g−1. The device sustaining specific power and energy values of 439.7 W kg−1 and 22.32 Wh kg−1 are also illustrated in Fig. 10e. These results could be well related to those previously reported by others working with similar materials. For example, Oyedotun et al. [6] reported a specific energy of 23.5 Wh kg−1 and power of 427 W kg−1 for a NiCoMn-TH/AEG//C-FPS device at 1 A g−1; Chen et al. [31] reported a Co2P nanostructure//graphene device with 8.8 Wh kg−1 at a power density of 6000 W kg−1 at 1 A g−1; Zhao et al. [32] assembled a Co(OH)2/GNS//AC/CFP device with an energy density of 19.3 Wh kg−1 at a power density of 187.5 Wkg−1, and a current density of 0.25 A g−1 and Singh et al. [33] assembled a NiCoMn-LDH (10%)//rGO device with an energy density of 57.4 Wh kg−1 at a power density of 749.9 W kg−1. The negative electrode material is an agricultural biomass waste representing a novel carbon source. This material makes part of the uniqueness of the study. The biomass waste was reused and transformed into a high-quality carbon material promoting the manufacture of a sustainable device having outstanding electrochemical properties.
A plot of the device Coulombic efficiency, together with capacity retention, could be seen in Fig. 10f. The figure illustrates the device's outstanding Coulombic efficiency of 99.9% and capacity retention of 80% at 3 A g−1 after 15,000 cycles. This exceptional performance could be assigned to the mechanical strength of the porous graphene foam nanosheet acting as a structural backbone for the NiCoMn-TH nanoparticles and enabling an exceptional cyclability [26]. Figure 10g illustrates the floating test (voltage holding test) performed by holding the cell at its peak potential of 1.6 V for a 10 h period. The device was then set to perform GCD cycles at 3 A g−1. This cycle was repeated for up to 9 series. The device capacity was then determined for each 10 h interval over the entire period. After a slight increase and minor reduction of the device’ specific capacity during the first 30 h of the test, the device constantly maintained a specific capacity of 9.74 mAh g−1 after 90 h as illustrated in Fig. 10g.
Figure 11a displays the device cyclic voltammetry curve before and after stability as well as voltage holding. The CV curve appears to maintain its shape after the stability cycles. It could also be observed that the CV became more pseudocapacitive after voltage holding. However, the current response is seen to reduce after both tests. The observation is similar to the CD patterns, as illustrated in Fig. 11b. The figure displays a reduced discharge time for both the post cycling stability and voltage holding tests with the post voltage holding test GCD being closer to an EDLC. Despite a lower specific capacity after the stability tests, the device displayed improvement on the pronounced redox peaks that were visible for the pristine sample on the three-electrode measurement. The enhanced linearity of the curves could be ascribed to a better polarity of the electrode within the cell. This is the result of greater accessibility of the electrolyte ions to pores that were unreachable before the floating test [30]

Electrochemical characterisation before and after stability and voltage holding: a CV curves, b CD curves, and c EIS Nyquist plot
Figure 11c illustrates the device EIS Nyquist plot before and after stability and voltage holding. The plot examined the diffusion kinetics of the electrode surface. The recorded Rs values before and after 15,000 cycling 0.9 Ω, 0.5 Ω, respectively. The Rs value after a 90-h floating test is 1.7 Ω. The device displayed no distinct semicircle in the low-frequency region for the curve after the 15,000 cycle’s stability test as well as the 90 h floating test. The semicircle is an indication of very a little charge transfer resistance and quick mass transport between its electrodes and the electrolyte after both tests [24].
Conclusion
The synthesis of a ternary metal hydroxide made from Nickel, cobalt, and manganese hydroxides was achieved via a co-precipitation method. Its composites made of graphene foam were then successfully synthesised via a cost-efficient co-precipitation-hydrothermal method resulting in a NiCoMn-TH/GF composite with improved electrochemical performances. Following a variation of GF mass, the optimal NiCoMnTH graphene foam composite was obtained with the NiCoMn-TH/200 mg GF. This composite displayed a specific capacity of 178.6 mAh g−1 in 1 M KOH when measured in a three-electrode configuration. This value was found to be higher than the pristine (NiCoMn-TH) having a specific capacity of 76.2 mAh g−1, the NiCoMn TH/100 mg GF: 38.1 mAh g−1, the NiCoMn TH/150 mg GF: 26.4 mAh g−1 and the NiCoMn TH/250 mg GF: 7.4 mAh g−1 when ran in the same configuration with the same parameters. An assembled asymmetric NiCoMn-TH/200mgGF//CSDAC device exhibited a sustaining specific capacity of 23.4 mAh g−1 at 0.5 A g−1. The device demonstrated a satisfying specific power and energy densities of values 22.3 Wh kg−1 and 439.7 W kg−1, respectively. It also displayed outstanding cycling stability with a Coulombic efficiency of 99.9% and capacity retention of 80% after a cycling test of up to 15,000 cycles within an operating potential of 1.6 V at a 3 A g−1.
References
Pérez-Lombard, L., Ortiz, J., Pout, C.: A review on buildings energy consumption information. Energy Build. 40, 394–398 (2008)
Zhao, C., Wang, Q., Zhang, H., Passerini, S., Qian, X.: Two-dimensional titanium carbide/RGO composite for high-performance supercapacitors. ACS Appl. Mater. Interfaces. 8, 15661–15667 (2016)
Simon, P., Gogotsi, Y.: Materials for electrochemical capacitors. Nat. Mater. 7, 845–854 (2008)
Anastas, P., Eghbali, N.: Green chemistry: principles and practice. R. Soc. Chem. 39, 301–312 (2010)
Yu, G., Xie, X., Pan, L., Bao, Z., Cui, Y.: Hybrid nanostructured materials for high-performance electrochemical capacitors. Nano Energy 2, 213–234 (2013)
Oyedotun, K.O., Masikhwa, T.M., Mirghni, A.A., Mutuma, B.K., Manyala, N.: Electrochemical properties of asymmetric supercapacitor based on optimized carbon-based nickel-cobalt-manganese ternary hydroxide and sulphur-doped carbonized iron-polyaniline electrodes. Electrochim. Acta 334, 135610 (2020)
Wang, G., Zhang, L., Zhang, J.: A review of electrode materials for electrochemical supercapacitors. Chem. Soc. Rev. 41, 797–828 (2012)
Wang, H., Gao, Q., Jiang, L.: Facile approach to prepare nickel cobaltite nanowire materials for supercapacitors. Small 7, 2454–2459 (2011)
Lu, X.F., Wu, D.J., Li, R.Z., Li, Q., Ye, S.H., Tong, Y.X., Li, G.R.: Hierarchical NiCo2O4 nanosheets@hollow microrod arrays for high-performance asymmetric supercapacitors. J. Mater. Chem. A. 2, 4706–4713 (2014)
Wu, N., Low, J., Liu, T., Yu, J., Cao, S.: Hierarchical hollow cages of Mn-Co layered double hydroxide as supercapacitor electrode materials. Appl. Surf. Sci. 413, 35–40 (2017)
Zhi, M., Xiang, C., Li, J., Li, M., Wu, N.: Nanostructured carbon-metal oxide composite electrodes for supercapacitors: a review. Nanoscale 5, 72–88 (2013)
Rantho, M.N., Madito, M.J., Ochai-Ejeh, F.O., Manyala, N.: Asymmetric supercapacitor based on vanadium disulfide nanosheets as a cathode and carbonized iron cations adsorbed onto polyaniline as an anode. Electrochim. Acta 260, 11–23 (2018)
Jiang, H., Ma, J., Li, C.: Mesoporous carbon incorporated metal oxide nanomaterials as supercapacitor electrodes. Adv. Mater. 24, 4197–4202 (2012)
Wang, X., Liu, W.S., Lu, X., Lee, P.S.: Dodecyl sulfate-induced fast faradic process in nickel cobalt oxide-reduced graphite oxide composite material and its application for asymmetric supercapacitor device. J. Mater. Chem. 22, 23114–23119 (2012)
Young, C., Salunkhe, R.R., Alshehri, S.M., Ahamad, T., Huang, Z., Henzie, J., Yamauchi, Y.: High energy density supercapacitors composed of nickel cobalt oxide nanosheets on nanoporous carbon nanoarchitectures. J. Mater. Chem. A. 5, 11834–11839 (2017)
Li, L., Zhang, Y., Shi, F., Zhang, Y., Zhang, J., Gu, C., Wang, X., Tu, J.: Spinel manganese-nickel-cobalt ternary oxide nanowire array for high-performance electrochemical capacitor applications. ACS Appl. Mater. Interfaces. 6, 18040–18047 (2014)
Zhu, N., Liu, W., Xue, M., Xie, Z., Zhao, D., Zhang, M., Chen, J., Cao, T.: Graphene as a conductive additive to enhance the high-rate capabilities of electrospun Li4Ti5O12 for lithium-ion batteries. Electrochim. Acta 55, 5813–5818 (2010)
Wang, G., Wang, B., Wang, X., Park, J., Dou, S., Ahn, H., Kim, K.: Sn/graphene nanocomposite with 3D architecture for enhanced reversible lithium storage in lithium ion batteries. J. Mater. Chem. 19, 8378–8384 (2009)
Tarimo, D.J., Oyedotun, K.O., Mirghni, A.A., Manyala, N.: Sulphur-reduced graphene oxide composite with improved electrochemical performance for supercapacitor applications. Int. J. Hydrogen Energy 45, 13189–13201 (2020)
Oyedotun, K.O., Madito, M.J., Momodu, D.Y., Mirghni, A.A., Masikhwa, T.M., Manyala, N.: Synthesis of ternary NiCo-MnO2 nanocomposite and its application as a novel high energy supercapattery device. Chem. Eng. J. 335, 416–433 (2018)
Ogata, A., Komaba, S., Baddour-Hadjean, R., Pereira-Ramos, J.P., Kumagai, N.: Doping effects on structure and electrode performance of K-birnessite-type manganese dioxides for rechargeable lithium battery. Electrochim. Acta 53, 3084–3093 (2008)
Xia, H., Wang, Y., Lin, J., Lu, L.: Hydrothermal synthesis of MnO2/CNT nanocomposite with a CNT core/porous MnO2 sheath hierarchy architecture for supercapacitors. Nanoscale Res. Lett. 7, 33 (2012)
Shang, Y., Ma, S., Wei, Y., Yang, H., Xu, Z.: Flower-like ternary metal of Ni-Co-Mn hydroxide combined with carbon nanotube for supercapacitor. Ionics 26, 3609–3619 (2020)
Mirghni, A.A., Madito, M.J., Masikhwa, T.M., Oyedotun, K.O., Bello, A., Manyala, N.: Hydrothermal synthesis of manganese phosphate/graphene foam composite for electrochemical supercapacitor applications. J. Colloid Interface Sci. 494, 325–337 (2017)
Ndiaye, N.M., Sylla, N.F., Ngom, B.D., Barzegar, F., Momodu, D., Manyala, N.: High-performance asymmetric supercapacitor based on vanadium dioxide/activated expanded graphite composite and carbon-vanadium oxynitride nanostructures. Electrochim. Acta 316, 19–32 (2019)
Oyedotun, K.O., Manyala, N.: Graphene foam–based electrochemical capacitors. Curr. Opin. Electrochem 21, 125–131 (2020)
Oyedotun, K.O., Momodu, D.Y., Naguib, M., Mirghni, A.A., Masikhwa, T.M., Khaleed, A.A., Kebede, M., Manyala, N.: Electrochemical performance of two-dimensional Ti3C2-Mn3O4 nanocomposites and carbonized iron cations for hybrid supercapacitor electrodes. Electrochim. Acta 301, 487–499 (2019)
Xiao, X., Han, B., Chen, G., Wang, L., Wang, Y.: Preparation and electrochemical performances of carbon sphere@ZnO core-shell nanocomposites for supercapacitor applications. Sci. Rep. 7, 1–13 (2017)
Barzegar, F., Bello, A., Momodu, D., Madito, M.J., Dangbegnon, J., Manyala, N.: Preparation and characterization of porous carbon from expanded graphite for high energy density supercapacitor in aqueous electrolyte. J. Power Sources 309, 245–253 (2016)
Fasakin, O., Dangbegnon, J.K., Momodu, D.Y., Madito, M.J., Oyedotun, K.O., Eleruja, M.A., Manyala, N.: Synthesis and characterization of porous carbon derived from activated banana peels with hierarchical porosity for improved electrochemical performance. Electrochim. Acta 262, 187–196 (2018)
Chen, X., Cheng, M., Chen, D., Wang, R.: Shape-controlled synthesis of Co2P nanostructures and their application in supercapacitors. ACS Appl. Mater. Interfaces. 8, 3892–3900 (2016)
Zhao, C., Ren, F., Xue, X., Zheng, W., Wang, X., Chang, L.: A high-performance asymmetric supercapacitor based on Co(OH)2/graphene and activated carbon electrodes. J. Electroanal. Chem. 782, 98–102 (2016)
Singh, S., Shinde, N.M., Xia, Q.X., Gopi, C.V.V.M., Yun, J.M., Mane, R.S., Kim, K.H.: Tailoring the morphology followed by the electrochemical performance of NiMn-LDH nanosheet arrays through controlled Co-doping for high-energy and power asymmetric supercapacitors. Dalton Trans. 46, 12876–12883 (2017)
Acknowledgements
The authors would like to acknowledge the South African Research Chairs Initiative (SARChI) of the Department of Science and Technology and the National Research Foundation (NRF) of South Africa (Grant No. 61056) for financial support towards this research. The opinions, observations, and conclusions set out herein are those of the author(s) and the NRF is not liable in this respect in any way. V. N. Kitenge also appreciates financial support from the University of Pretoria and the National Research Foundation through the SARChI chair in Carbon Technology and Materials.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kitenge, V.N., Oyedotun, K.O., Fasakin, O. et al. Enhancing the electrochemical properties of a nickel–cobalt-manganese ternary hydroxide electrode using graphene foam for supercapacitors applications. Mater Renew Sustain Energy 10, 7 (2021). https://doi.org/10.1007/s40243-021-00192-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-021-00192-y