
Abstract
First principle calculations based on density functional theory have been performed on lithium containing transition metal sulfides Li2TiS3 and Li3NbS4 which are recently identified as novel positive electrode materials for rechargeable Li+ batteries. The calculations were performed to investigate the structural stability, electronic and transport properties of Li2TiS3 and Li3NbS4 along with their corresponding delithiated phases LiTiS3 and Li2NbS4. In this study it has been observed that these lithium containing sulfur materials maintain their face-centered cubic structure upon extraction of Li+. To calculate the structural stability and volume change due to lithium extraction, the total energies of Li2TiS3, Li3NbS4 and their corresponding delithiated phases LiTiS3 and Li2NbS4 have been computed by applying full potential linearized augmented plane wave (FP-LAPW) method implemented in WIEN2K. The equilibrium structural parameters for all the phases were determined by achieving total energy convergence. These electrode materials exhibit very small percentage of volume change with change in Li+ concentration which accounts for excellent structural stability. The computed band structure along high symmetry lines in the Brillouin zone, total and partial density of states clearly reveals that the extraction lithium from these electrode materials does not change their metallic nature. The electronic conductivities of both lithiated and delithiated phases have been calculated by employing BoltzTrap which can be interfaced with WIEN2K. The topological distributions of electron charge density at various critical points within the system were analyzed with the use of CRITIC code which is based on Bader’s theory of atoms in molecules (AIM). From the charge density calculations, it was observed that, there is strong ionic bond and weak covalent bond between atoms of the compounds Li2TiS3 and Li3NbS4. But the ionic bond nature was found to decrease in the delithiated phases LiTiS3 and Li2NbS4. The calculated values of electronic conductivities and discharge voltages for both electrodes are found to be in accordance with the recent experimental reports.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Li+ batteries are the most significant rechargeable power sources which are being successful in powering portable consumer electronic devices. The extensive research on Li+ batteries over the last few decades have tremendously increased the performance of Li+ battery, their safety, cost and environmental compatibility. In the Li+ battery technology, the cell voltage, Li+ transportation rate and specific capacities are mainly determined by the positive electrode materials [1].
The practical reversible capacities of conventional positive electrode materials with layered structure and olivine structure are usually limited by the intrinsic structural instability at low lithium concentration [2]. Even though the olivine LiMPO4 and spinel LiMn2O4 were proposed as the positive electrode materials with better cyclic performance and low cost, they have lower electronic conductivity [3–15]. Li2MnO3 [4], LixV2O5 (x = 1, 2, 3) [16], Li2FeS2 [17] and Li2FeSiO4 [18] are reported to have high specific capacity ranging from 300 to 400 mA h g−1 due to the transformation of more than one electron during charging and discharging. Also, Li2S, Li2Se and Li2O exhibit high capacities due to multi electron reaction [19–21]. Therefore, it is clear that electrodes involved in multi electron reactions exhibit high specific capacity.
The research on sulfur containing positive electrodes has been growing consistently in the last two decades. Among the high energy density storage systems, lithium sulfur batteries with energy density of 2600 W h kg−1 holds the potential to serve as next generation of high energy battery [23]. The remarkable advantages of lithium sulfur batteries are that the sulfur is abundant in nature, non-toxic, low cost and possess high theoretical capacity of 1673 mA h g−1. The limitations are that sulfur slowly dissolves in the electrolyte and slow down the ion movement result into poor cyclic performance and low electronic conductivity due to the insulator characteristic of elemental sulfur. To overcome these limitations, appropriate transition metals are added to prevent sulfur from dissolving in the electrolyte. Since Ti and Nb have very good mechanical strength, light in weight, ductile, corrosion resistive, abundant in nature, nontoxic and low cost, they received considerable attention to prepare the suitable electrode material. Lithium containing transition metal sulfides LiTiS2, Li2TiS3, Li3NbS4 have been reported experimentally and theoretically to have increased specific capacity and electronic conductivity [6, 22–28]. Among these lithium metal sulfides, Li2TiS3 and Li3NbS4 have been experimentally reported that they exhibit high specific capacity and very low volume expansion [25]. The unique advantage of these compounds is that the number of electrons in the valence state of transition metal will be increased due to multi electron reaction in charging and discharging which in turn increases the electronic conductivity of the positive electrode. Moreover, these compounds are found to crystallize in rock salt structure with face-centered cubic array which provides three dimensional interstitial spaces to accommodate large amount of lithium as well as it contributes to the excellent reversibility of the electrode compared to other structures [22, 28]. Hence, Li2TiS3 and Li3NbS4 have gained more attraction as potential materials for Li+ batteries with high energy and good electronic conductivity.
The computational methods by the application of first principle calculations based on density functional theory (DFT) with approximations such as local density approximation (LDA) and generalized gradient approximation (GGA) have been extensively applied in the field of Li+ batteries [26]. The aim of this work is to present a detailed theoretical description of electronic, structural and transport properties of positive electrode materials Li2TiS3, Li3NbS4 and their delithiated phases.
Method of calculation
In the present calculation, full potential linearized augmented plane wave (FP-LAPW) method within the generalized gradient approximation (GGA) is used as incorporated in WIEN2K code [29]. The principal motivation of this computational method is to obtain detailed information about the structural and electronic properties including discharge voltage and electronic conductivities of the positive electrodes Li2TiS3, Li3NbS4 and also for their corresponding delithiated phases LiTiS3 and Li2NbS4.To perform these calculations, the exchange–correlation potential according to the Perdew–Burke–Ernzerhof parameterization is used [30]. In this method, the space is divided into an interstitial region and non-overlapping muffin-tin (MT) spheres centered at the atomic sites. In the interstitial region, the basis set consists of plane waves. Inside the MT spheres, the basis set is described by radial solutions of the one particle Schrödinger equation (at fixed energy) and their energy derivatives multiplied by spherical harmonics. To determine the equilibrium volume (V 0) and the structural parameters for all the compounds the total energies are computed. For these calculations, self-consistency is obtained with the use of 5000 k points in the entire Brillouin zone [28]. The electronic properties are calculated within GGA approximation. The variations in electronic conductivity due to extraction of lithium are determined with the BoltzTrap code interfaced with WIEN2K [31].
Results and discussions
Structural properties
According to experimental investigations by Sakuda et al. on Li2TiS3 and Li3NbS4, the compounds crystallize in face-centered cubic structure belongs to the space group and they retain the same structure upon Li+ extraction process [22, 24]. To investigate the structural stability of these electrodes and to determine the equilibrium volume for the host compounds and their corresponding delithiated phases, a primitive unit cell is constructed. The self-consistent total energy calculations were performed by employing FP-LAPW method. The minimization of total energy of the system was achieved by computing the total energy for different unit cell volumes over a range ±15 % around experimental volume which is similar to our previous works [32–36]. The total energy calculations were converged within 10−5 Ry. The equilibrium volumes for all the investigated compounds were obtained by fitting the total energy as a function of volume to the Brich–Murnaghan’s equation of state [34]. The lattice parameters determined from the equilibrium volume are in good agreement with the experimental reports and listed in Table 1.
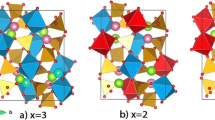
From the above calculations, a small percentage of volume change was observed due to Li+ extraction. It has been noted that the cubic array of the host material provides a three dimensional and isotropic interstitial space to accommodate Li+ ions without lattice strain to the host material. Hence, these compounds exhibit excellent structural stability which is a key factor in determining the volumetric and gravimetric capacity of the electrode material. The crystal structures of the compounds Li2TiS3, Li3NbS4 and their respective delithiated phases LiTiS3 and Li2NbS4 are illustrated in Figs. 1 and 2.

Crystal structures of a Li2TiS3 b LiTiS3

Crystal structures of a Li3NbS4 b Li2NbS4
Electronic properties
The preliminary idea to investigate the electronic properties of a potential electrode material is to determine its metallic, semiconducting or insulating character. Information about the energy gaps between valence and conduction bands can be deduced from the calculated density of states. To investigate the electronic properties of Li2TiS3, Li3NbS4 and their corresponding delithiated phases, the band structure calculations were performed within GGA along with high symmetry directions of Brillouin zone. The computed band structure of Li2TiS3, shown in Fig. 3a exhibits its metallic nature. The bands crossing Fermi level (E F) and the bands which are lying above E F are primarily originated from the valence electrons of Ti and S atoms. The band profile of delithiated LiTiS3 shown in Fig. 3b reveals that the extraction of lithium from Li2TiS3 pushed the conduction band to higher energy and valence band to lower energy with respect to E F. This is because of the removal of electrons from the host band structure due to the delithiation [37, 38]. The metallic nature of the host compound is retained in its delithiated phase which confirms that the bands crossing E F are from valence electrons of Ti. The metallic nature of Li3NbS4 is revealed from the calculated electronic band structure of the compound along the high symmetry directions of Brillouin zone which is presented in Fig. 3c. The valence electrons of transition metal atom Nb are the major contributors for the bands crossing E F and above Fermi level. However, from the sulfur atom, the p state electrons have coordinated with the electrons of Nb. The electronic band structure in Fig. 3d indicates the metallic character of Li2NbS4. The comparison of electronic band structures of the host compounds Li2TiS3 and Li3NbS4 with their respective delithiated phases LiTiS3 and Li2NbS4 concludes that extraction of Li+ does not imply any transition in the metallic nature of the host compounds.

Electronic band structures of a Li2TiS3 b LiTiS3 c Li3NbS4 and d Li2NbS4
To perform a widespread study on electronic properties, the partial and total DOS for all the investigated compounds were computed and are illustrated in Figs. 4,5,6 and 7. The partial and total DOS for Li2TiS3 presented in Fig. 4a indicated that the maximum number of electronic states obtained at E F was primarily originated from the d orbital electrons of transition metal atom. However, there is significant contribution from the p orbital electrons of chalcogenide atom. From the partial DOS of Li2TiS3 presented in Fig. 4a, it can be witnessed that the electronic states at −4 eV below E F are purely contributed by the p orbit electrons from the three sulfur atoms. All the electronic states lying above E F are absolutely raised from d state electrons of three S and Ti atoms. The contribution of electronic states of sulfur above this energy range is dominant. The contribution of electronic states of transition metal atom at this energy range is negligible. The DOS distribution for the delithiated phase LiTiS3 is presented in Fig. 4b. In the partial and total DOS, the electronic states at E F are completely due to the d state electrons of transition metal atom, and above E F the electronic states are mainly originated due to the chalcogenide atom. Thus, it is clearly evident that the distribution of electronic states at E F for the host compound is almost similar to its delithiated phase which implies that the transition metal atom is not affected by Li+ extraction. In the partial DOS of LiTiS3 given in Fig. 4b, the maximum number of electronic states at the energy −2.5 eV is increased compared to that of the host compound which might be due to the removal of electrons from host compound due to delithiation. From Fig. 5a, b it was observed that the electronic states at the energy 5 eV above E F for both compounds Li2TiS3 and LiTiS3 are attributed to the d state electrons of Ti and S atoms which is an evident for the covalent bonding nature of these atoms.

a Partial and total DOS of Li2TiS3 and b partial and total DOS of LiTiS3

a Partial DOS of sulfur in Li2TiS3 and b partial DOS of sulfur in LiTiS3

a Partial DOS of sulfur in Li3NbS4 and b partial DOS of sulfur in Li2NbS4

a Partial and total DOS of Li3NbS4 and b partial and total DOS of Li2NbS4
Figure 7a depicts the partial and total DOS for Li3NbS4. The electronic states at E F were raised from the d state electrons of transition metal atom and from p state electrons of chalcogenide atom. But the contribution of transition metal atom is much dominant at this level. Maximum contribution of chalcogenide atom is at the energy range −3 to −4 eV. This can be identified from the partial DOS of Li3NbS4 shown as Fig. 7a. The peaks obtained in the DOS at energies above E F were due to the d state electrons from the four sulfur atoms. Also, it has been observed that, valence electrons of niobium were not having significant importance at these energies. To identify the effect of Li+ extraction from Li3NbS4 in electronic structure, the partial and total DOS of Li2NbS4 was calculated and illustrated as Fig. 7b. The electronic states at E F for the delithiated phase Li2NbS4 in Fig. 7b was found to be identical with the states of Li3NbS4 at this level (Fig. 7a). This is quite normal since the role of transition metal atom was absolutely same in both phases. The partial DOS pattern in Fig. 6a, b clearly reveals that the electronic states below E F were purely originated from p state electrons of sulfur and above E F they were due to d state electrons of the four sulfur atoms. DOS observation infers that there is no significant difference in electronic states below E F, but it has been observed that there is substantial decrease in the electronics states at conduction band of delithiated phase which may have influence in electronic conductivity.
Bonding properties
To have a clear perception about the nature of chemical bonding between atoms, the valence charge density distribution for all the investigated compounds were calculated and plotted in two dimensional grid as shown in Figs. 8 and 9. The valence charge density contour plot for Li2TiS3 along (001) direction in Fig. 8a showing localized charge distribution which is a sign of ionic bonding between electropositive Li+, Ti4+ and electronegative S2−. It has also been identified that the isolines in charge density contour for the same compound Li2TiS3 along (112) direction in Fig. 8b reveal weak covalent bond between valence electrons of sulfur and titanium atoms. Therefore, the material is neither ionic nor covalent in nature as it exhibits the mixture of both. However, the charge density plots clearly reveal that the ionic bonding character dominates the covalent character in Li2TiS3. Since electrodes acquire complete ionic character at high concentration of lithium, may undergo a brittle fracture which may in turn decreases their life cycle[39]. This mixed bonding character of the electrode materials is an advantage over the electrodes with complete ionic nature at high lithium concentration. The valence charge density for the delithiated phase LiTiS3 along (001) direction is shown as Fig. 8c. At this direction, the isolines of charge density are not shared between Li, Ti and S atoms, which indicate ionic bond between these atoms. In Fig. 8d, the electronic charge density distribution of the same compound along (112) direction illustrates there is a strong covalent bonding between the valence electrons of Ti and S. Therefore, LiTiS3 show strong covalent nature and weak ionic nature due to extraction of Li+.

Valence charge density contour for a Li2TiS3 along (001) direction, b Li2TiS3 along (112) direction, c LiTiS3 along (001) direction and d LiTiS3 along (112) direction

Valence charge density contour for a Li3NbS4 along (100) direction, b Li3NbS4 along (112) direction, c Li2NbS4 along (100) direction and d Li2NbS4 along (112) direction
The valence charge distribution for Li3NbS4 was completely dominated by ionic bond nature between Li, Nb and S atoms (Fig. 9a) calculated along (100) direction. However, there is sharing of valence electrons between Nb and S atoms which has been identified through the charge density plot along (112) direction as in Fig. 9b. Hence, Li3NbS4 also exhibit mixed bonding character, whereas the ionic bonding nature weakens the covalent character. To investigate the impact of Li+ extraction in the nature of chemical bonding, the charge density contour plot for the delithiated phase Li2NbS4 was calculated. From Fig. 9c, it is observed that the isolines are not shared by Nb, S and Li atoms. Therefore, the bonding along (100) direction is completely ionic. The sharing of valence electrons between Nb and S atoms along (112) direction in Fig. 9d infer that the delithiated phase shows partially covalent and partially ionic bonding nature. From the electron charge density calculations, it is observed that the ionic bond character dominates the covalent nature in the host compounds Li2TiS3 and Li3NbS4, whereas covalent bonding nature dominates the ionic nature in their corresponding delithiated phases LiTiS3 and Li2NbS4. Hence, it is clearly evident that even though increase in Li+ concentration increases the ionic nature of the compound, there was not complete transfer of valence electrons between electropositive Li+, Ti4+ and electronegative S2−.
Electron charge distribution
Electron correlation effects are important for the electronic structure of transition metal compounds, thus we also tested two LDA + U corrections. The self-interaction corrected (SIC) LDA + U is a method going beyond the LDA by special treatment of a chosen set of states, which in our case are the 3 d states of Ti and 4 d states of Nb. To find the imbalanced redox potential in the delithiated phases, we have considered electron self-interaction (SIC) and employed LDA + U method with three different self-interaction corrections to obtain the ionic charge distribution inside the atomic basins for the electrodes Li2TiS3 and Li3NbS4 as well as for their delithiated phases. The topology of electron density (ρ) for all the compounds have been observed with the application of Bader’s theory based on atoms in molecules theory. For various self-interaction corrections, even though there is change in total energy of the compounds, there is a slight change in global minima of total energy curve which corresponds to equilibrium volume of the compounds. Table 2 illustrates the atomic charges within the atomic basins of Li, Ti, Nb and S obtained from GGA and LDA + U (U = 3, 5 and 7 eV). From the Table 2, it has been noted that removal of Li+ results in considerable change in the ionic charge distribution within the atomic basins of Ti, Nb and S, however, Li site has negligible change in charge distribution. This implies that extraction of Li has not much influence in the total charge distribution of the compound. The charge distribution in the delithiated phases are balanced between the transition metal atoms and sulfur atoms. In the delithiated phase LiTiS3, the ionic charge of Ti increases by +0.35 and the ionic charge of sulfur decreases by −0.45 In delithiated phase Li2NbS4, the ionic charge of Nb increases by +0.45 and the ionic charge of sulfur decreases by −0.35.
Moreover, to have a complete analysis of the chemical bonding of the compounds, topological analysis is applied with CRITIC program. The electron density is characterized by a well-defined set of critical points (CP). The existence of a (3, −1) CP indicates that electron density is accumulated between the nuclei that are linked by the associated atomic interaction line. Since the CP (3, −1) indicated the saddle point of ρ linked to nucleus and provides information about the bonding character of the atoms, it is known as bond CP. The calculated Hessian matrix of ρ is diagonalized to yield a set of eigenvalues. The eigenvalues correspond to the three principal curvatures of ρ. The CP (3, −1) has one positive and two negative eigenvalues. From these curvatures, the nature of chemical bonding can be known. Table 3 depicts the positions, curvatures, Laplacian of ρ and magnitude of ρ at the bond CP (3, −1). The ratio of principal curvatures h 1/h 2 together with the Laplacian provides information for a classification of chemical bonding. A small value h 1/h 2 ≪ 1 is typical for closed shell interactions. While for covalent bond this ratio increases with bond strength. The magnitude of ρ is positive and large for ionic bonding, and it is small or negative for covalent bonding [40–44]. From Table 3 it has been noted that there are two different topological distribution of ρ, which indicates the electrode materials exhibit two different bonding characters. Positions associated with Laplacian ≤0.3 Å−3 and other positions associated with Laplacian ≥1.5 Å−3 were attributed to strong and weak ionic bond, respectively. The analysis of distribution of ρ for the electrode materials and their corresponding delithiated structures indicates very little change in the electron charge density which is clearly evident for the structural stability of the compounds Li2TiS3 and Li3NbS4 during delithiation process.
The bonding CPs (3, 1) and (3, 3) are described in Table 4. The distribution of charge density obtained from the CPs (3, 1) and (3, 3) occurs as consequences of particular geometrical arrangement of bond paths, and they define the remaining elements of molecular structure rings and cages. (3, 1) CP is found at the interior of the ring. If the bond paths are arranged so as to enclose the interior of a molecule with the ring surfaces, then a (3, 3) cage CP is found. The distribution of ρ obtained for the ring and cage cps are identical for the electrodes Li2TiS3, Li3NbS4 with their corresponding delithiated phases. Therefore, the core electrons are not affected by the extraction of Li+ from the electrodes.
Transport properties
The calculations for the transport properties such as electronic conductivities are carried out using the BoltzTrap code which is interfaced with WIEN2K. BoltzTrap is a computational tool for evaluating the transport properties using the Boltzmann transport theory [45–49]. The band energies of the compounds Li2TiS3, Li3NbS4 and their respective delithiated phases LiTiS3 and Li2NbS4 are computed with self-consistent total energy calculation with the use of WIEN2K and given as input to BoltzTrap. The transport properties as a function of the carrier concentration are computed with the results of the electronic band structure calculated above. The Fourier expansion of band energies is carried out to determine the gradient along the energy bands to obtain the group velocities. The group velocities are calculated as derivatives of the energies. The transport property happens to be extremely dependent on the chemical potential. However, the chemical potential is related to the number of charge carriers.
Electronic conductivity
An accurate assessment of the electronic conductivity of electrodes is necessary for understanding and optimizing the battery performance. Electronic conductivity of a positive electrode material has significant effect on the performance of Li+ battery. The electronic conductivities for the electrode materials and their corresponding delithiated phases were calculated at room temperature and are listed in Table 5. The value of electronic conductivity found to have generous agreement with the recent experimental results available [22, 24].
Discharge voltage
The cell voltage is linearly related to the chemical potential of lithium within the positive electrode material. The average equilibrium voltage (V) is related to the difference in the Gibbs free energy (ΔG) between the delithiated phase at charged state and lithiated phase at discharged state. It has previously been reported that the average potential for Li+ extraction from a material is given by the following equations [7, 8]
where, G is the Gibbs free energy of the compound. F is the Faraday’s constant. The free energy change associated with the transfer of one mole of lithium between the two composition limits. The above Eq. (2) provides the average of the equilibrium potential between the lithium compositions x1 and x2. In DFT calculations, Free energies can be replaced by the ground state energies with very little error [50–54]. Therefore, the discharge voltage can be determined by computing the total energies of the compounds and their delithiated phases. For delithiation of Li2TiS3 and Li3NbS4, the voltage is calculated from the equations given below.
In both reactions (Eqs. 3 and 4), one electron transferred through outer circuit. Where, E total refers to the total energy per formula unit. The calculated voltage for the Li+ extraction process with Li2TiS3 is 2.35 V, and with Li3NbS4 it is 2.27 V. Both values are found to agree well with the experimental value of 2.2 V [22].
Volume change
The extraction of Li+ from the positive electrodes Li2TiS3 and Li3NbS4 during charging of battery was not accompanied by any structural change. But it has been observed that there was a small change in volume of the electrodes due to delithiation. First principle calculation can predict the volume change in the electrode material during Li+ insertion and extraction. The percentage of volume change is computed by comparing the computed equilibrium volumes of the two limiting structures of the electrode [10, 55]. The change in volume (∆v) for the positive electrodes Li2TiS3 and Li3NbS4 upon the extraction of Li+ have been calculated from Eq. (5), which is given by
The calculated volume change for Li2TiS3 with its delithiated phase LiTiS3 was 1.05 %, and for Li3NbS4 with Li2NbS4 was found to be 3.5 %. The low volume change is considered as an added advantage for the positive electrode materials to ensure very good structural stability.
Conclusions
In the present work, first principle calculations within GGA is carried out by employing WIEN2 K to investigate the structural parameters of the rock salt type ternary lithium metal sulfides Li2TiS3 and Li3NbS4 as positive electrodes for lithium battery. To acquire knowledge about the impact of lithium extraction in the electronic and bonding properties of the host compounds, the electronic band structure, density of electronic states and valence charge distribution are calculated. With a view to have a better insight into the transport properties due to the impact of Li+ extraction, the electronic conductivity has been calculated and found to agree well with the experimental reports. Discharge voltage obtained from the total energy difference for the investigated compounds were in significant agreement with the available experimental data. Li+ extraction from the electrode upon charging does not show any structural change. This structural similarity of the electrodes accounts for their high volumetric and gravimetric capacity. Moreover, the lithium extraction process with these electrodes was accompanied by relatively small changes in the unit cell parameters and volume. This scenario does not exist with metals because their dense structure does not provide favorable interstitial space for lithium diffusion. But the metal sulfides Li2TiS3 and Li3NbS4 with face-centered cubic array facilitate a three dimensional isotropic diffusion path for Li+ which does not strain the electrode during lithium extraction and insertion. It is very clear that the role of metals titanium and niobium is influenced in stabilizing the electrode structure. Finally, the most important property of Li2TiS3 and Li3NbS4 is their ability to change their lithium content within the same structure. These qualities identify these metal sulfides as novel positive electrodes for Li+ ion batteries.
References
Bo, Xu, Qian, Danna, Wang, Ziying: Ying Shirley Meng, Recent progress in cathode materials research for advanced lithium ion batteries. Mater. Sci. Eng. R73, 51–65 (2012)
Mizushima, K., Jones, P.C., Wiseman, P.J., Goodenough, J.B.: LixCoO2 (0 ≤ x ≥) A new cathode material for batteries of high energy density. Mater Res Bull. 15, 783–789 (1980)
Scrosati, B., Garche, J.: Lithium batteries status prospectus and future. J. Power Sources 195, 2419–2430 (2010)
Nitta, N., Wu, F., Lee, J.T., Yushin, G.: Li-ion battery materials: present and future. Mater. Today. 18, 252–264 (2015)
Padhi, A.K., Nanjundaswamy, K.S., Goodenough, J.B.: Phospho-olivines as positive-electrode materials for rechargeable lithium batteries. J. Electrochem. Soc. 144, 1188–1194 (1997)
Stanley, M.: Whittingham, lithium batteries and cathode materials. Chem. Rev. 104, 4271–4301 (2004)
Zhou, F., Cococcioni, M., Kang, K., Ceder, G.: The Li intercalation potential of LiMPO4 and LiMSiO4 Olivines with M = Fe, Mn Co, Ni. Electrochem. Commun 6, 1144–1148 (2004)
Saiful Islam, M., Craig, A.J.: Fisher, lithium and sodium battery cathode materials: computational insights into voltage, diffusion and nanostructural properties. Chem. Soc. Rev. 43, 185–204 (2014)
Lu, L.: Transition metal oxides, sulphite and sulphur composites of Lithium battery. PhD Thesis, University of Wollongong, December (2012)
Qi, Yue, Hector, Louis G., James, Christine, Kim, Kwang Jin: Lithium concentration dependent elastic properties of battery electrode materials from first principles calculations. J. Electrochem. Soc. 161, F3010–F3018 (2014)
Rastgoo-Deylami, M., Javanbakht, M., Ghaemi, M., Naji, L., Midvar, H., Ganjali, M.R.: Synthesis and electrochemical properties of rhombohedral LiFePO4/C microcrystals via a hydrothermal route for lithium ion batteries. Int. J. Electrochem. Sci. 9, 3199–3208 (2014)
Dathar, G.K.P., Sheppard, D., Stevenson, K.J., Henkelman, G.: Calculations of Li-ion diffusion in olivine phosphates. Chem. Mater. 23, 4032–4037 (2011)
Ojczyk, W., Marzec, J., Dygas, J., Krok, F., Liu, R.S., Molenda, J.: Structural and transport properties of LiFe0.45Mn0.55PO4 as a cathode material in Li-ion batteries. Mater. Sci. Pol 24, 161–167 (2006)
Goodenough, J.B., Thackeray, M.M., David, W.I.F., Bruce, P.G.: Lithium insertion into manganese spinels. Mater. Res. Bull. 18, 461–472 (1983)
Goodenough, J.B., Thackeray, M.M., David, W.I.F., Bruce, P.G.: Lithium insertion/extraction reactions with Manganese oxides. Revue de Chimie Minerale. 2, 435–455 (1984)
Li, Wang-Da, Cheng-Yan, Xu, Yue, Du, Fang, Hai-Tao, Feng, Yu-Jie: Liang Zhen Electrochemical lithium insertion behavior of β-Li x V2O5 (0 < x ≤ 3) as the cathode material for secondary lithium batteries. J. Electrochem. Soc. 161, A75–A83 (2014)
Kendrick, E., Barker, J., Bao, J., Swiatek, A.: The rate characteristics of lithium iron sulphide. J. Power Sources 196, 6929–6933 (2011)
Zhang, L.L., Sun, H.B., Yang, X.L., Wen, Y.W., Huang, Y.H., Li, M., Peng, G., Tao, H.C., Ni, S.B., Liang, G.: Study on electrochemical performance and mechanism of V-doped Li2FeSiO4. Electrochim. Acta 152, 496–504 (2015)
Poizot, P., Laruelle, S., Grugeon, S., Dupont, L., Tarascon, J.-M.: Nano-sized transition-metal oxides as negative-electrode materials for lithium-ion batteries. Nature 407, 496–499 (2000)
Zhang, K., Wang, L., Hu, Z., Cheng, F., Chen, J.: Ultrasmall Li2S nanoparticles anchored in graphene nanosheets for higher energy lithium batteries. Sci. Rep. 4, 6467 1–7 (2014)
Yi, Z., Yuan, L., Sun, D., Li, Z., Wu, C., Wen, Y.W., Shan, B.: High performance lithium-selenium batteries promoted by heteroatom doped microporous carbon. J. Mater. Chem. A 3, 3059–3065 (2015)
Sakuda, A., Takeuchi, T., Okamura, K., Kobayashi, H., Sakaebe, H., Tatsumi, K., Ogumi, Z.: Rock-salt-type lithium metal sulphides as novel positive-electrode materials. Sci. Rep 4, 4883 (2014)
Bing, X., Huang, J.Q., Zhang, Q., Peng, H.J., Zhao, M.Q., Wei, F.: Aligned carbon nanotube/sulphur composite cathodes with high sulphur content for lithium-sulfur batteries. Nano Energy 4, 65–72 (2014)
Sakuda, A., Takeuchi, T., Kobayashi, H., Sakaebe, H., Tatsumi, K., Ogumi, Z.: Preparation of novel electrode materials based on lithium niobium sulfides. Electrochemistry. 82, 880–883 (2014)
Sakuda, A., Taguchi, N., Takeuchi, T., Kobayashi, H., Sakaebe, H., Tatsumi, K., Ogumi, Z.: Amorphous niobium sulphides as novel positive-electrode. Mater. Electrochem. Lett. 3, A79–A81 (2014)
Nakhal, S., Lerch, M., Koopman, J., Islam, M., Bredow, T.: Crystal structure of 3R-LiTiS2 and its stability compared to other polymorphs. Z. Anorg. Allg. Chem. 639, 2822–2825 (2013)
Yersak, T.A., Yan, Y., Stoldt, C., Lee, S.H.: Ambient temperature and pressure mechanochemical preparation of nano-LiTiS2. ECS Electrochem. Lett 1, A21–A23 (2012)
Thackeray, M.M., Vaughey, J.T., Johnson, C.S., Kropf, A.J., Benedek, R., Fransson, L., Edstrom, K.: Structural considerations of intermetallic electrodes for lithium batteries. J. Power Sources 113, 124–130 (2003)
Blaha, P., et al.: WIEN2K an augmented plane wave + local orbital programme for calculating crystal properties. TU Wien, Austria (2001)
Perdew, J.P., Burke, K., Ernzerhof, M.: Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868 (1996)
Madsen, G.K.H., Singh, D.J.: BoltzTrap: a code for calculating band-structure dependent quantities. Comput. Phys. Commun 175, 67–71 (2006)
Christensen, NE., Svane A, Laskowski R, Palanivel B, Modak P, Chantis A.N, Van, Schilfgaarde and Kotari T, Electronic properties of 3R- CuAlO2 under pressure: three theoretical approaches, Phys Rev B 81, 045203-1-045203-9. (2010)
Jayalakshmi, V., Murugan, R., Palanivel, B.: Electronic and structural properties of CuMO2(M = Al, Ga, In). J. Alloys Compd. 388, 19–22 (2005)
Murnaghan, F.D.: The compressibility of media under extreme pressures. Proc. Natl. Acad. Sci. USA. 30, 244–246 (1994)
Reshak, A.H., Kamarudin, H.: Theoretical investigation for Li2CuSb as multifunctional materials: electrode for high capacity rechargeable batteries and novel materials for second harmonic generation. J. Alloys Compounds. 509, 78617869 (2011)
Gnanapoongothai, T., Murugan, R., Palanivel, B.: First principle study on lithium intercalated antimonides Ag3Sb and Mg3Sb2. Ionics (2014). doi:10.1007/s11581-014-1303-0
Menga, Y.S., Arroyo-de Dompablo, M.E.: First principles computational materials design for energy storage materials in lithium ion batteries. Energy Environ. Sci. 2, 589–609 (2009)
Holgate, S.A.: Understanding solid state physics. CRC Press (2009)
Khatun, F., Gafur, M.A., Ali, M.S., Islam, M.S., Sarker, M.A.R.: Impact of lithium composition on structural, electronic and optical properties of lithium cobaltite prepared by solid-state reaction. J. Sci. Res. 6, 217–231 (2014)
Bader, R.F.W.: Atoms in molecules—a quantum theory. Oxford University Press, Oxford (1990)
Jenkins, S., Ayers, P.W., Kirk, S.R., Mori-Sanchez, P., Martin, A.: Pendas bond metallicity of materials from real space charge density distributions. Chem. Phys. Lett. 471, 174–177 (2009)
Laskowski, R., Blaha, P., Schwarz, K.: Charge distribution and chemical bonding in Cu2O. Phy. Rev. B 67, 075102 (2003)
Kirfel, A., Krane, H.G., Blaha, P., Schwarz, K., Lippmann,T.: Electron density distribution in stihovite, SiO2:a new high-density synchrotron-radiation study. Acta Cryst. A57, 663–677 (2001)
Pendas, A.M., Costales, A., Luana, V.: Ions in crystals: the topology of the electron density in ionic materials. III. Geometry and ionic radii. J. Phys. Chem. B 102, 6937–6948 (1998)
Scheidemantel, T.J., Ambrosch-Drax, C., Thonhauser, T., Badding, J.V., Sofo, J.O.: Transport coefficients from first-principles calculations. Phy. Rev B. 68, 125210 (2003)
Sebastien L.: First-principles study of the electronic and thermoelectric properties of Ca3Co4O9. Ph.D Thesis University of Liege (2013)
Faghaninia, A., Cynthia, S.: Lo Electronic transport calculations for lightly-doped thermo electrics using densityfunctional theory: application to high-performing Cu-doped zinc antimonides. Condens. Matter Mater. Sci. arXiv:1401-2494v1 (2014)
Sharma, Yamini, Shukla, Seema, Dwivedi, Shalini: Ramesh Sharma Transport properties and electronic structure of intercalated compounds MTiS2 (M=Cr, Mn and Fe). Adv. Mater. Lett. 6, 294–300 (2015)
Aydinol, M.K., Kohan, A.F., Ceder, G.: Ab initio study of lithium intercalation in metal oxides and metal dichalcogenides. Phys. Rev. B 56, 1354–1364
Zhou, F., Cococcioni, M., Marianetti, A., Morgan, D., Ceder, G.: First principles prediction of redox potentials in transition metal compounds with LDA + U. Phys. Rev. B 70, 235121–235128 (2004)
Bruce, P.G.: Solid state chemistry of lithium power sources. Chem. Commun. 1817–1823 (1997)
Van der Ven, A., Marianetti, C., Morgan, D., Ceder, G.: Phase transformations and volume changes in spinel LixMn2O4. Solid State Ionics 135, 21–32 (2000)
Xiaohui, Z., Ning, C., Fang, L., Yaping, S., Yang, L.I.: First principle calculation of lithiation/delithiation voltage in Li ion battery materials. Chin. Sci. Bull. 56, 3229–3232 (2011)
Meng, Y.S., Wu, Y.W., Hwang, B., Li, Y., Ceder, G.: Combining Ab initio computation with experiments for designing new electrode materials for advanced lithium batteries. J. Electrochem. Soc. 151, A1134–A1140 (2004)
Benedek, R., Thackeray, M.M.: Lithium reactions with intermetallic compound electrodes. J. Power Sources 110, 406–411 (2004)
Acknowledgments
The authors are thankful to Professors Peter Blaha, Karlheinz Schwarz, Georg Madsen, Dieter K vasnicka and Joachim Luitz, Vienna University of Technology, Inst. of Physical and theoretical chemistry, Getreidemarkt 9/156, A-1060, Vienna/Austria.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Gnanapoongothai, T., Rameshe, B., Shanmugapriya, K. et al. First principle calculations on structural, electronic and transport properties of Li2TiS3 and Li3NbS4 positive electrode materials. Mater Renew Sustain Energy 5, 8 (2016). https://doi.org/10.1007/s40243-016-0072-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-016-0072-2