
Abstract
Pt/C, Rh/C and PtRh/C electrocatalysts with different Pt:Rh atomic ratios supported on Vulcan XC 72 carbon were prepared using borohydride as reducing agent and tested for ethanol electro-oxidation in alkaline medium. X-ray diffraction patterns showed the formation of PtRh alloy. Transmission electron micrographs showed the nanoparticles with particle sizes between 3 and 6 nm for all materials. Electrochemical experiments showed PtRh/C (50:50) the most promising material for ethanol electro-oxidation; however, in situ ATR-FTIR experiments was observed that the ethanol oxidation is incomplete due to the formation of acetate and carbonate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The use of ethanol directly as a fuel for Proton Exchange Membrane Fuel Cells (PEMFC) has been studied extensively over the last years due to its large production from renewable sources, low toxicity and high-energy density of 8 kWh/kg; however, the breaking of the C–C bond of the ethanol molecule at low temperatures continues to be a challenge, and acetaldehyde and acetic acid are the principal products formed [1–7].
Recently, the development of anion exchange membranes for alkaline fuel cells [8–12] has attracted the interest in studying the ethanol electro-oxidation in alkaline medium [13–15]. Alkaline medium presents several advantages when compared to acidic medium, as an oxygen reduction reaction takes place with faster kinetic than in acid media, increasing the electric potential of the fuel cell [15] and many non-noble metals are stable for electrochemical applications, either as anode or a cathode [14]. Despite this, the break of C–C bond of ethanol molecule in alkaline medium is also a difficult task [16–18].
In acid medium, the addition of Rh to Pt catalysts increases the CO2 yield by facilitating C–C bond cleavage; however, the obtained currents do not increase [19–21]. Kowal et al. [4, 22] prepared PtRhSnO2/C electrocatalyst depositing Pt and Rh atoms on carbon-supported SnO2 nanoparticles and tested for ethanol electro-oxidation. They found that this electrocatalyst breaks the C–C bond in acid medium facilitating the oxidation of ethanol molecule to CO2. In a further work, Li et al. [4] studied Pt–Rh–SnO2 catalysts and concluded that the role of Rh is to break the C–C bond of ethanol molecule whereas the roles of Pt and SnO2 are to facilitate the oxidation of the reaction intermediates and avoid their bonding to Rh sites.
Comprehensive fundamental studies using in situ spectroscopic studies to provide molecular information about ethanol electro-oxidation on Pt-based catalysts are widely reported in the literature for the acid medium [3, 4, 23, 24], while only few studies were done involving the ethanol electro-oxidation in alkaline solutions [14, 15, 25]. On this basis, this work investigates the ethanol electro-oxidation in alkaline medium using PtRh/C electrocatalysts. Eletrochemical and in situ ATR-FTIR studies were performed to obtain information about the activity of the electrocatalysts and the intermediates and products formed.
Experimental
PtRh/C electrocatalysts (20 % of metal loading, with Pt:Rh ratio atomics of 100:0, 90:10, 70:30, 50:50, and 0:100) were prepared using H2PtCl6·6H2O (Chloroplatinic acid-Aldrich), and RhCl3·xH2O (rhodium chloride Aldrich) as metal sources, sodium borohydride (Aldrich) as reducing agent and Vulcan Carbon XC72 as support. In the borohydride reduction process, the metal sources were dissolved in a mixture of water/2-propanol (50/50, v/v) and the Vulcan XC72 support was dispersed in the solution. The resulting mixture was submitted to an ultrasonic bath for 10 min, where a solution of sodium borohydride was added under stirring in one portion at room temperature. After, the mixture was filtered and the solid was washed with water and dried at 70 °C for 2 h.
All electrocatalysts were characterized by transmission electron microscopy (TEM) analysis. These experiments were carried out using a JEOL JEM-2100 electron microscope operated at 200 kV, where it was determined the morphology, distribution, and size of the nanoparticles in the support. The mean particle sizes were determined by counting more than 200 particles from different regions of each sample. X-ray diffraction (XRD) analyses were performed on Rigaku diffractometer model Miniflex II using Cu Kα radiation source (λ = 0.15406 nm). The diffractograms were recorded from 2θ = 20° to 90° with a step size of 0.05° and a scan time of 2 s per step.
The cyclic voltammetry (CV) and chronoamperometry (CA) measurements were carried at room temperature using a Microquimica potenciostat, where the working electrodes (geometric area of 0.3 cm2 with a depth of 0.3 mm) were prepared using the thin porous coating technique [26], where an ink is prepared with 10 mg of catalyst and 100 μL of PTFE solution in 50 mL of water, then dispersed in ultrasonic bath for 5 min. After that, the ink is filtered and all the solids are deposited on the work electrode. The reference electrode was Ag/AgCl (3.0 mol L−1 KCl) and the counter electrode was a Pt plate. The electrochemical measurements were realized in the presence of 1.0 mol L−1 of ethanol + 1 mol L−1 KOH solutions saturated with N2.
Direct alkaline ethanol fuel cell test was realized using a single cell with 5 cm2 of area. The temperature was set to 60 °C for the fuel cell and 85 °C for the oxygen humidifier. All electrodes were constructed with 1 mg of metal per cm2 in the anode and the cathode. In all experiments, a commercial Pt/C (BASF) was used as cathode. All the electrocatalysts were painted over a carbon cloth in the form of a homogeneous dispersion prepared using Nafion solution (5 wt%, Aldrich). After the preparation, the electrodes were hot pressed on both sides of a Nafion 117 membrane at 125 °C for 3 min under a pressure of 247 kgf cm−2. Prior to use, the membranes were exposed to KOH 6 mol L−1 for 24 h as it has already been proposed by Hou et al. [27]. The fuel, ethanol 2.0 mol L−1 and 2.0 mol L−1 KOH were delivered at 1.0 mL min−1 and the oxygen flow was regulated at 150 mL min−1. Polarization curves were obtained using a potentiostat/galvanostat PGSTAT 302NAutolab.
The spectro-electrochemical ATR-FTIR in situ measurements were performed with a Nicolet 6700 FT-IR spectrometer equipped with an MCT detector cooled with liquid N2, ATR accessory (MIRacle with a Diamond/ZnSe Crystal Plate Pike®) and an electrochemical cell as explained better in the literature [23, 28, 29]. The working electrodes are the same that electrochemical experiments in presence of 1.0 mol L−1 ethanol + 1 mol L−1 KOH. The absorbance spectra were collected as the ratio R:R 0, where R represents a spectrum at a given potential and R 0 is the spectrum collected at −0.85 V. Positive and negative directional bands represent gain and loss of species at the sampling potential, respectively. The spectra were computed from 128 interferograms averaged from 3,000 to 850 cm−1 with the spectral resolution set to 8 cm−1. Initially, a reference spectrum (R 0) was measured at −0.85 V, and the sample spectra were collected after applying successive potential steps from 0.1 to −0.05 V.
Results and discussion
TEM micrographs and histograms of the particle size distribution for Pt/C, Rh/C and PtRh/C materials are shown in Fig. 1. All prepared materials showed the particles well dispersed on the support, even though some particle agglomerations can be observed. The nanoparticles’ mean diameter was determined by counting about 200 randomly chosen particles from the relevant TEM images [30, 31] and the particles are with an average diameter of 4 nm for all Rh content catalyst, and 6 nm for Pt/C. The particles sizes measured are in accordance with similar materials described in literature [20, 24, 32].


TEM micrographs and histograms of the particle size distribution for Pt/C, Rh/C and PtRh/C
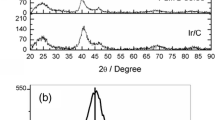
Figure 2a shows XRD patterns of Pt/C, Rh/C and PtRh/C electrocatalysts. In all diffractograms, it can be seen four peaks at 2θ = 40°, 47°, 67° and 82°, which are associated with the (111), (200), (220) and (311) planes, characteristic of face-centered cubic (fcc) structure of Pt and Pt alloys. The diffraction peaks of Pt(fcc) and Rh(fcc) phases are very close; however, it was observed that the diffraction peaks of the fcc phase of PtRh/C electrocatalyts were slightly shifted to higher 2θ values compared to Pt/C electrocatalyst and this shift increases with the increase of Rh content [32]. According to Gupta et al. [20], such evidence accounts for the presence of PtRh alloy formation. To obtain more information about the material structure, the diffractograms were refined using the Pawley method [33] carried out with Fityk 0.98 software, as reported by Wojdyr [34]. The peaks in this region are related to Pt (220) (JCPDF #04 802), Rh (220) (JCPDF # 88 2334) and PtRh (220) (JCPDF # 65 7938). In Fig. 2b, it is possible to observe the changes in the peak shape as a result of the contribution of Pt(fcc) (a = 0.392 nm), Rh(fcc) (a = 0.381 nm) and PtRhalloy(fcc) (a = 0.386 nm) phases. The presence of these three contributions was observed for all PtRh/C electrocatalysts and the peak related to PtRh alloy formation increases with the increase of Rh content.

a X-ray diffractograms of the Pt/C, Rh/C, and PtRh/C electrocatalysts. b Pawley refinement of peak (2 2 0) in all diffratograms with dot for experimental measure, line for modeled, and dash for residual line
Figure 3 shows the CV in alkaline media for Pt/C, Rh/C and PtRh/C electrocatalysts in 1 mol L−1 KOH. In the CVs, it was observed that with the increase of the Rh content the hydrogen adsorption–desorption region (−0.85 to −0.5 V) becomes less defined than the one observed for Pt/C electrocatalyst [35]. Also, the reverse scan of Pt/C electrocatalyst showed a peak around −0.3 V which was attributed to reduced PtOx species and with the increase in Rh content on PtRh/C materials, decrease the intensity of this peak. PtRh/C (50:50) and Rh/C electrocatalysts showed a redox pair in the current values of −0.3/−05 V and −0.3/−0.7 V, respectively, which may be attributed to the increase in the rhodium oxides species [35].

Cyclic voltammograms of Pt/C, Rh/C and PtRh/C electrocatalysts in 1 mol L−1 KOH solution with a scan rate of 10 mV s−1 at 25 °C
Figure 4 shows the liner sweep of Pt/C, Rh/C and PtRh/C electrocatalysts in the presence of 1.0 mol L−1 of ethanol and 1 mol L−1 of KOH. It was observed that the onset potential for ethanol electro-oxidation is very close (≈−0.65 V) for all eletrocatalysts as well as the current density values. It is interesting to note that Rh/C electrocatalyst showed a good activity for ethanol electro-oxidation in alkaline medium while in acid medium it was practically inactive [4, 22].

Linear sweep voltammograms of Pt/C, Rh/C and PtRh/C electrocatalysts in 1 mol L−1 ethanol solution in 1 mol L−1 KOH with a scan rate of 10 mV s−1 at 25 °C
Figure 5 shows the chronoamperometry curves for Pt/C, Rh/C and PtRh/C electrocatalysts in the potential of −0.3 V for 30 min. For all electrocatalysts, it was observed a faster decay of current values during the first 2 min followed by a slower decay. The final current values at −0.3 V were: PtRh/C (50:50) (7.9 A g−1) > Rh/C (6.2 A g−1) ≈ PtRh/C (90:10) (6.1 A g−1) > Pt/C (4.9 A g−1) > PtRh/C (70:30) (4.6 A g−1). It is worth noting that initially the Rh/C electrocatalyst presents higher current values than the other electrocatalysts; however, after 7 min its deactivation is faster than Pt/C and PtRh/C electrocatalysts,

Current–time curves at −0.3 V in 1 mol L−1 ethanol solution in 1 mol L−1 KOH for Pt/C, Rh/C and PtRh/C electrocatalysts at 25 °C
The decay for PtRh is slower than that for Rh/C indicating superior stability. As the consequence of the Rh incorporation, some modifications of structural, electronic and kinetic features on the PtRh/C catalysts were observed, such as changes in lattice parameter and charge transfers from Rh to Pt (Pt d-band vacancies electronically [32]). However, this effect does not occur in Pt or Rh alone.
Figure 6 shows the performances of single alkaline DEFC with Pt/C, Rh/C and PtRh/C as anode electrocatalysts and the following order of maximum power density was observed: PtRh/C (50:50) (5.9 mW cm−2) > Pt/C (5.5 mW cm−2) > PtRh/C 70:30 (4.9 mW cm−2) > PtRh (90:10) (4.7 mW cm−2) > Rh/C (4.4 mW cm−2).

I–V Curves and the power density at 60 °C of a 5 cm2 DAEFC using Pt/C, Rh/C and PtRh/C electrocatalysts anodes (1 mgmetal cm−2 catalyst loading) and Pt/C E-TEK electrocatalyst cathode (1 mgPt cm−2 catalyst loading, 20 wt% Pt loading on carbon), Nafion® 117 membrane KOH treated, ethanol (2.0 mol L−1) and oxygen pressure (2 bar)
A factor that can influence the activity of these materials may be caused by the manner in that ethanol adsorbs on the catalyst, as explained in the oxametallacyclic conformation model [36], which directly influences the kinetics of the reaction and the products formed. PtRh 50:50 alloy can be a conformation that favors the reactions kinetic [37], due to way of the ethanol adsorbs on catalyst surface, and influences too on OCV.
In situ ATR-FTIR experiments were performed to correlate the activity of ethanol electro-oxidation with the preferential mechanism (Fig. 7). The FTIR spectra in the region between 3,000 and 850 cm−1 were recorded during ethanol electro-oxidation for Pt/C, Rh/C and PtRh/C electrocatalysts as a function of the potential in ethanol 1 mol L−1 and KOH 1 mol L−1. Considering ATR-FTIR results, it was possible to observe the appearance of bands related to CO2 at 2,343 cm−1 formation [25].


In situ FTIR spectra taken at several potentials (indicated) in 1 mol L−1 KOH + 1.0 mol L−1 ethanol for Pt/C, Rh/C, and PtRh/C electrocatalysts. The backgrounds were collected at −0.85 V (Ag/AgCl scale)
A carbonate ion band at 1,370 cm−1 [14] was not clearly visible in the spectra, due to overlapping with the acetate band at 1,410 cm−1 [14], although its presence can be inferred by the deconvolution of the spectra, as reported by Neto et al. [38]. The acetate was displayed as two intense peaks at ≈1,553 and ≈1,410 cm−1, originating from asymmetric and symmetric C–O bond vibrations. The small peak at 926 cm−1 is associated with acetaldehyde C–C–O Stretching vibrations [15]. In the spectra, it is also possible to observe the presence of bands of ethanol that increase the intensity towards consumption of this species (1,080, 1,036, and 874 cm−1) [25].
The difference observed in the spectra can be seen more clearly after all the bands were deconvoluted to lorentzian line forms [38] of the CO2, acetate, carbonate ions, methyl groups, and an acetaldehyde as shown in Fig. 8.

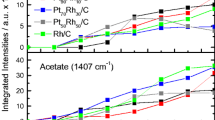
Integrated CO2, acetate, carbonate, methyl group, and acetaldehyde band intensity as a function of the electrode potential for Pt/C, Rh/C and PtRh/C electrocatalysts
As shown in Fig. 8, the PtRh/C (70:30) catalyst shows the most intense band of CO2 at all potentials, but it is important to note that PtRh/C 50:50 catalyst, the most active in EOR experiments, has an increase in signal intensity CO2 production from −0.35 V potential that coincides with the decrease in the signal of acetate and carbonate ions.
When we analyzed the production of acetate, it is possible to see that from −0.45 V the Rh/C catalyst presents higher intensity for this band than other materials; can indicate that in alkaline medium, this metal was not completely selective to C–C broken.
The production of carbonate ions, present in Fig. 8c, brings clues that may explain the loss of activity of the catalyst Rh/C, since this material continuously produces these ions even at high potentials as −0.05 V. Carbonate ions can create a physical barrier on the catalytic sites, thus reducing the ability to get more alcohol to oxidize. The production of acetaldehyde, it is passed over on all materials tested, except for PtRh/C 70:30 and is a another indication that the products formed depends on the structure of the catalyst and the way of the fuel is adsorbed.
In summary, Rh is an active metal for EOR, however, suffers very rapidly with the catalyst poisoning in alkaline medium, as seen in ATR-FTIR measurements, ethanol is oxidized preferentially over it by way of formation of carbonate ions, that may physically block the catalytic sites, other way is the acetate formation, indicating that reaction over Rh is not selective for C–C cleavage, as reported for acid medium [4, 21, 22]. Though when associated with Pt, 50:50, the resulting alloy has greater activity than other catalysts studied, which lower potentials oxidize the ethanol preferably by same way of Rh/C, but in potentials higher than −0.35 V decrease carbonate ions productions and increase the CO2 production, probably due to different types of ethanol adsorption on catalyst, this can be causes a increase in reaction kinetics as reported Shen et al. [37]. A factor that also can influence the activity of these materials may be caused by the manner that ethanol adsorbs on the active sites of the catalysts, as explained in the oxametallacyclic conformation model [36], which directly influences the kinetics of the reaction and the products formed. Therefore, PtRh/C 50:50 alloy could be a structure conformation that favors the kinetics reaction [37].
Conclusion
X-ray diffractograms of PtRh/C electrocatalysts showed the presence of Pt(fcc), Rh(fcc) and PtRh(fcc)alloy phases. The increase in Rh content on the PtRh/C electrocatalysts leads to an increase in PtRh(fcc)alloy phase. TEM micrographs showed a good distribution of the nanoparticles on the carbon support with average particle sizes in the range of 4–6 nm. Electrochemical and alkaline DEFC experiments showed that PtRh/C 50:50 was the best activity; however, Rh/C electrocalalyst showed a remarkable good activity in the first minutes but it deactivates more quickly than Pt/C and PtRh/C electrocatalysts. In situ ATR-FTIR experiments showed that ethanol oxidation reaction is incomplete; however, PtRu/C (50:50) is very promising due to the CO2 production (from −0.35 V potential) that coincides with the decrease in the signal of acetate and carbonate ions.
References
Zhou, Y., Xu, Q.J.: A review of Pt-based anode catalysts preparation for direct ethanol fuel cell. Adv. Mater. Res. 860–863, 797–800 (2014)
Beyhan, S., Léger, J.M., Kadirgan, F.: Pronounced synergetic effect of the nano-sized PtSnNi/C catalyst for ethanol oxidation in direct ethanol fuel cell. Appl. Catal. B 130–131, 305–313 (2013)
García-Rodríguez, S., Rojas, S., Peña, M.A., Fierro, J.L.G., Baranton, S., Léger, J.M.: An FTIR study of Rh–PtSn/C catalysts for ethanol electrooxidation: effect of surface composition. Appl. Catal. B 106(3–4), 520–528 (2011). doi:10.1016/j.apcatb.2011.06.011
Li, M., Zhou, W.P., Marinkovic, N.S., Sasaki, K., Adzic, R.R.: The role of rhodium and tin oxide in the platinum-based electrocatalysts for ethanol oxidation to CO2. Electrochim. Acta 104, 454–461 (2013). doi:10.1016/j.electacta.2012.10.046
Xu, C., Shen, P.K.: Electrochemical oxidation of ethanol on Pt–CeO2/C catalysts. J. Power Sources 142(1–2), 27–29 (2005)
Ribeiro, J., dos Anjos, D.M., Kokoh, K.B., Coutanceau, C., Léger, J.M., Olivi, P., de Andrade, A.R., Tremiliosi-Filho, G.: Carbon-supported ternary PtSnIr catalysts for direct ethanol fuel cell. Electrochim. Acta 52(24), 6997–7006 (2007)
Colmati, F., Antolini, E., Gonzalez, E.R.: Ethanol oxidation on a carbon-supported Pt75Sn25 electrocatalyst prepared by reduction with formic acid: effect of thermal treatment. Appl. Catal. B 73(1–2), 106–115 (2007)
Wang, G., Weng, Y., Chu, D., Xie, D., Chen, R.: Preparation of alkaline anion exchange membranes based on functional poly(ether-imide) polymers for potential fuel cell applications. J. Membr. Sci. 326(1), 4–8 (2009). doi:10.1016/j.memsci.2008.09.037
Wang, G., Weng, Y., Chu, D., Chen, R., Xie, D.: Developing a polysulfone-based alkaline anion exchange membrane for improved ionic conductivity. J. Membr. Sci. 332(1–2), 63–68 (2009). doi:10.1016/j.memsci.2009.01.038
Herman, H., Slade, R.C.T., Varcoe, J.R.: The radiation-grafting of vinylbenzyl chloride onto poly(hexafluoropropylene-co-tetrafluoroethylene) films with subsequent conversion to alkaline anion-exchange membranes: optimisation of the experimental conditions and characterisation. J. Membr. Sci. 218(1–2), 147–163 (2003). doi:10.1016/s0376-7388(03)00167-4
Danks, T.N., Slade, R.C.T., Varcoe, J.R.: Alkaline anion-exchange radiation-grafted membranes for possible electrochemical application in fuel cells. J. Mater. Chem. 13(4), 712–721 (2003). doi:10.1039/b212164f
Varcoe, J.R.: Investigations of the ex situ ionic conductivities at 30° C of metal-cation-free quaternary ammonium alkaline anion-exchange membranes in static atmospheres of different relative humidities. Phys. Chem. Chem. Phys. 9(12), 1479–1486 (2007). doi:10.1039/b615478f
Cai, J., Huang, Y., Guo, Y.: PdTex/C nanocatalysts with high catalytic activity for ethanol electro-oxidation in alkaline medium. Appl. Catal. B 150–151, 230–237 (2014)
Fang, X., Wang, L., Shen, P.K., Cui, G., Bianchini, C.: An in situ Fourier transform infrared spectroelectrochemical study on ethanol electrooxidation on Pd in alkaline solution. J. Power Sources 195(5), 1375–1378 (2010). doi:10.1016/j.jpowsour.2009.09.025
Geraldes, A.N., Da Silva, D.F., Pino, E.S., Da Silva, J.C.M., De Souza, R.F.B., Hammer, P., Spinacé, E.V., Neto, A.O., Linardi, M., Dos Santos, M.C.: Ethanol electro-oxidation in an alkaline medium using Pd/C, Au/C and PdAu/C electrocatalysts prepared by electron beam irradiation. Electrochim. Acta 111, 455–465 (2013)
Song, S.Q., Zhou, W.J., Zhou, Z.H., Jiang, L.H., Sun, G.Q., Xin, Q., Leontidis, V., Kontou, S., Tsiakaras, P.: Direct ethanol PEM fuel cells: the case of platinum based anodes. Int. J. Hydrogen Energy 30(9), 995–1001 (2005)
Coutanceau, C., Brimaud, S., Lamy, C., Léger, J.M., Dubau, L., Rousseau, S., Vigier, F.: Review of different methods for developing nanoelectrocatalysts for the oxidation of organic compounds. Electrochim. Acta 53(23), 6865–6880 (2008)
Neto, A.O., Farias, L.A., Dias, R.R., Brandalise, M., Linardi, M., Spinacé, E.V.: Enhanced electro-oxidation of ethanol using PtSn/CeO2-C electrocatalyst prepared by an alcohol-reduction process. Electrochem. Commun. 10(9), 1315–1317 (2008)
Lima, F.H.B., Gonzalez, E.R.: Ethanol electro-oxidation on carbon-supported Pt–Ru, Pt–Rh and Pt–Ru–Rh nanoparticles. Electrochim. Acta 53(6), 2963–2971 (2008). doi:10.1016/j.electacta.2007.11.007
Sen Gupta, S., Datta, J.: A comparative study on ethanol oxidation behavior at Pt and PtRh electrodeposits. J. Electroanal. Chem. 594(1), 65–72 (2006). doi:10.1016/j.jelechem.2006.05.022
de Souza, J.P.I., Queiroz, S.L., Bergamaski, K., Gonzalez, E.R., Nart, F.C.: Electro-oxidation of ethanol on Pt, Rh, and PtRh electrodes. A study using DEMS and in situ FTIR techniques. J. Phys. Chem. B 106(38), 9825–9830 (2002). doi:10.1021/jp014645c
Kowal, A., Li, M., Shao, M., Sasaki, K., Vukmirovic, M.B., Zhang, J., Marinkovic, N.S., Liu, P., Frenkel, A.I., Adzic, R.R.: Ternary Pt/Rh/SnO2 electrocatalysts for oxidizing ethanol to CO2. Nat. Mater. 8(4), 325–330 (2009). doi:10.1038/nmat2359
De Souza, R.F.B., Silva, J.C.M., Simões, F.C., Calegaro, M.L., Neto, A.O., Santos, M.C.: New approaches for the ethanol oxidation reaction of Pt/C on carbon cloth using ATR-FTIR. Int. J. Electrochem. Sci. 7(6), 5356–5366 (2012)
Silva-Junior, L.C., Maia, G., Passos, R.R., de Souza, E.A., Camara, G.A., Giz, M.J.: Analysis of the selectivity of PtRh/C and PtRhSn/C to the formation of CO2 during ethanol electrooxidation. Electrochim. Acta 112, 612–619 (2013). doi:10.1016/j.electacta.2013.09.030
Zhou, Z.-Y., Wang, Q., Lin, J.-L., Tian, N., Sun, S.-G.: In situ FTIR spectroscopic studies of electrooxidation of ethanol on Pd electrode in alkaline media. Electrochim. Acta 55(27), 7995–7999 (2010). doi:10.1016/j.electacta.2010.02.071
Neto, A.O., Brandalise, M., Dias, R.R., Ayoub, J.M.S., Silva, A.C., Penteado, J.C., Linardi, M., Spinacé, E.V.: The performance of Pt nanoparticles supported on Sb2O5·SnO2, on carbon and on physical mixtures of Sb2O5.SnO2 and carbon for ethanol electro-oxidation. Int. J. Hydrogen Energy 35(17), 9177–9181 (2010). doi:10.1016/j.ijhydene.2010.06.028
Hou, H., Wang, S., Jin, W., Jiang, Q., Sun, L., Jiang, L., Sun, G.: KOH modified Nafion112 membrane for high performance alkaline direct ethanol fuel cell. Int. J. Hydrogen Energy 36(8), 5104–5109 (2011). doi:10.1016/j.ijhydene.2010.12.093
Silva, J.C.M., Parreira, L.S., De Souza, R.F.B., Calegaro, M.L., Spinacé, E.V., Neto, A.O., Santos, M.C.: PtSn/C alloyed and non-alloyed materials: differences in the ethanol electro-oxidation reaction pathways. Appl. Catal. B 110, 141–147 (2011)
Henrique, R.S., De Souza, R.F.B., Silva, J.C.M., Ayoub, J.M.S., Piasentin, R.M., Linardi, M., Spinacé, E.V., Santos, M.C., Neto, A.O.: Preparation of Pt/C-In 2O3.SnO2 electrocatalysts by borohydride reduction process for ethanol electro-oxidation. Int. J. Electrochem. Sci. 7(3), 2036–2046 (2012)
Da Silva, S.G., Silva, J.C.M., Buzzo, G.S., De Souza, R.F.B., Spinacé, E.V., Neto, A.O., Assumpção, M.H.M.T.: Electrochemical and fuel cell evaluation of PtAu/C electrocatalysts for ethanol electro-oxidation in alkaline media. Int. J. Hydrogen Energy 39(19), 10121–10127 (2014)
Herranz, T., García, S., Martínez-Huerta, M.V., Peña, M.A., Fierro, J.L.G., Somodi, F., Borbáth, I., Majrik, K., Tompos, A., Rojas, S.: Electrooxidation of CO and methanol on well-characterized carbon supported PtxSn electrodes. Effect of crystal structure. Int. J. Hydrogen Energy 37(8), 7109–7118 (2012). doi:10.1016/j.ijhydene.2011.11.131
Kim, H.J., Choi, S.M., Nam, S.H., Seo, M.H., Kim, W.B.: Effect of Rh content on carbon-supported PtRh catalysts for dehydrogenative electrooxidation of cyclohexane to benzene over polymer electrolyte membrane fuel cell. Appl. Catal. A 352(1–2), 145–151 (2009). doi:10.1016/j.apcata.2008.10.014
Pawley, G.S.: Unit-cell refinement from powder diffraction scans. J. Appl. Crystallogr. 14(DEC), 357–361 (1981). doi:10.1107/s0021889881009618
Wojdyr, M.: Fityk: a general-purpose peak fitting program. J. Appl. Crystallogr. 43, 1126–1128 (2010). doi:10.1107/s0021889810030499
Zhang, Y., Gao, X.P., Weaver, M.J.: Nature of surface bonding on voltammetrically oxidized noble-metals in aqueous-media as probed by real-time surface-enhanced Raman-spectroscopy. J. Phys. Chem. 97(33), 8656–8663 (1993). doi:10.1021/j100135a020
Mavrikakis, M., Doren, D.J., Barteau, M.A.: Density functional theory calculations for simple oxametallacycles: trends across the periodic table. J. Phys. Chem. B 102(2), 394–399 (1998). doi:10.1021/jp971450p
Shen, S.Y., Zhao, T.S., Xu, J.B.: Carbon supported PtRh catalysts for ethanol oxidation in alkaline direct ethanol fuel cell. Int. J. Hydrogen Energy 35(23), 12911–12917 (2010). doi:10.1016/j.ijhydene.2010.08.107
Neto, A.O., Nandenha, J., Assumpção, M.H.M.T., Linardi, M., Spinacé, E.V., de Souza, R.F.B.: In situ spectroscopy studies of ethanol oxidation reaction using a single fuel cell/ATR-FTIR setup. Int. J. Hydrogen Energy 38(25), 10585–10591 (2013). doi:10.1016/j.ijhydene.2013.06.026
Acknowledgments
The authors thank the Laboratório de Microscopia do Centro de Ciências e Tecnologia de Materiais (CCTM) by TEM measurements, FAPESP (2012/03516-5, 2012/22731-4, 2013/01577-0 and 2011/18246-0) and CNPq (150639/2013-9) for the financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Fontes, E.H., Piasentin, R.M., Ayoub, J.M.S. et al. Electrochemical and in situ ATR-FTIR studies of ethanol electro-oxidation in alkaline medium using PtRh/C electrocatalysts. Mater Renew Sustain Energy 4, 3 (2015). https://doi.org/10.1007/s40243-015-0043-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-015-0043-z