
Abstract
Lithium manganese spinels tend to aggregate upon annealing and do not allow for attaining high discharge rates when used as cathodes in lithium-ion batteries. To obtain spinel samples of lower aggregation and better high-rate properties, precursors synthesized by means of a citric acid-aided route are suggested to be pyrolyzed in an inert atmosphere, instead of pyrolysis in air. The synthesis of nanosized Li[Li0.033Mn1.967]O4 is described, and its characteristics including X-ray diffraction, scanning electron microscopy, and porosity, as well as electrochemical test results are presented. The particle size of the materials obtained is smaller, the degree of aggregation is lower, and high-rate properties are better than for analogues pyrolyzed in air. In particular, sample Li||Li[Li0.033Mn1.967]O4 cells deliver ~60 mAh g−1 at the current loads of 4,000 mA g−1 (30 C). After a sequence of 62 charge/discharge cycles with the currents growing from 0.1 to 100 C the specific capacity retains its initial value.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lithium manganese spinel LiMn2O4 as a cathode material for lithium-ion batteries has been suggested in 1983 [1] and since that time attracts much attention of experimentalist [2]. Compared to currently employed LiCoO2 it is cheap, non-toxic, has a high potential against lithium electrode (3.0–4.5 V) and the theoretical capacity of 148 mAh g−1 [1, 3, 4]. Properties of this and related spinel compounds and advances in their synthesis are reviewed in a recent paper [5].
LiMn2O4 belongs to the cubic \(Fd\bar{3}m\) space group, Z = 8 [4, 6]. In this cubic structure, oxygen ions occupy the tetrahedral positions (32e) and form the cubic dense packing. The octahedral positions (16d) are inhabited by manganese ions (Mn3+/Mn4+). Lithium ions hold the tetrahedral (8a) positions. Each of them is separated from four neighboring ions by voids (16c). This means that three-dimensional channels (8a–16c–8a–16c) exist for possible insertion and transport of lithium ions.
Electrochemical intercalation/deintercalation of Li+ into/from this material occurring in the potential range of 3.0–4.5 V can be expressed as
In the composition range of 0 ≤ x ≤ 0.5 and 0.5 ≤ x ≤ 1.0, reaction (1) occurs in two stages easily discernible in both galvanostatic and potentiodynamic regime, however, the spinel structure of LiMn2O4 remains cubic keeping the lattice parameter a close to ca. 8.248 Å.
Much work has been done so as to understand the nature of the Li0.5Mn2O4 phase and positioning of the lithium ions in it. Careful electrochemical, X-ray and neutron diffraction studies have shown that the charge/discharge process occurs in three steps, hence up to three phases coexist in the 0 ≤ x ≤ 1 range [7–9]. The structure of Li0.5Mn2O4 has been determined as belonging to the modulated \(R\bar{3}m\) space group, Z = 6, with the O2− ions in the 6c and 18h positions, the Mn3+ ions in the 3b positions, the Mn4+ ions in the 9e positions, and the Li+ ions partially filling 6c sites. In a cubic setting, this cell has a = 8.142 Å. Its remarkable feature is an opportunity to derive superlattices corresponding to the two “missing” phases having the predicted Li0.25Mn2O4 and Li2/3Mn2O4 compositions [9].
If x exceeds 1.0, i.e. at potentials of ~3.0 V against Li/Li+, a tetragonal LiMnO2 phase with a slightly increased unit volume (a = 8.007 Å, c = 9.274 Å) appears,
Furthermore, tetragonal distortions in lithium–manganese spinels are favored by the Jahn–Teller effect which facilitates lowering the symmetry of the structure if more than 50 % of Mn3+ is accumulated in the product of reaction (2) [10].
An effect of solid state synthesis conditions and annealing of LiMn2O4 on its stoichiometry and electrochemical properties is studied in detail [11, 12]. It is found that annealing at temperatures not exceeding 850 °C and slow cooling ensure the minimal loss of oxygen and the maximal specific capacity. However, major drawbacks of the material are inability to gain the theoretical capacity values and a significant capacity fading on cycling (up to 10 % after 50 cycles).
There are at least two possible directions in improving the electrochemical properties of the lithium manganese spinel electrode materials. One of them is based on the replacement of a part of the manganese ions in the 16d positions by excess lithium ions and obtaining substituted spinels of a general formula Li[Li q Mn2−q]O4 [13, 14]. It should be mentioned that annealing in air often leads to an oxygen loss and to a non-stoichiometricity of the spinels whose formula can be written as Li[Li q Mn2−q]O4-δ. A detailed investigation by Paulsen and Dahn [15] reveals that between 400 and 880 °C only the mentioned spinel is stable having a negligible oxygen nonstoichiometric range, δ < 0.02. The lithium stoichiometry can vary from 0.05 < q < 0.13 and increases with decreasing temperature and increasing oxygen partial pressure. Upon the substitution, the fraction of Mn3+ decreases and the specific capacity diminishes (e.g., to 117 mAh/g for Li1.063Mn1.932O4 [13] and to 98 mAh/g for Li1.12Mn1.88O4 [14]). However, in this case, the mean degree of oxidation of manganese exceeds 3.5 and thus prevents of arising the Jahn–Teller effect and capacity-fading phenomena.
Another way for improving the electrochemical properties of electrode materials is obtaining them in the form of nanosized crystals. Decreasing the size of particles leads to their better cycling ability and makes it possible to discharge lithium ion batteries with higher currents, i.e. increases their rate [16]. To do so, significant efforts are undertaking in the field of sol–gel synthesis of electrode materials with improved electrochemical parameters [17–26], in particular, using oxyacid metal salts (citrates on the first place) as precursors [21–27]. High homogeneity of final products and lower thermal treatment temperatures decrease the probability of particle growth and are considered to be advantages of such approaches.
In a previous paper [28], we have described the high-rate properties of the lithium substituted spinel of the Li[Li0.033Mn1.967]O4 composition with the lithium to manganese ratio equal to 1.05:2 and the theoretical capacity of 133 mAh g−1. It has been obtained according to the aforementioned directions by means of an optimized citric acid-aided route. In terms of Paulsen and Dahn [15], this composition is structurally stable and does not tend to lose oxygen upon heating. As follows from Ref. [28], sample cells with cathodes made of the material in question evidence the absence of discharge capacity fading within 100 cycles and the capability to be discharged with great currents. In particular, at the current density of 1,480 mA g−1 (~11 C) the discharge capacity equals to 58 mAh g−1 or almost the half of the theoretical capacity of the material, and at the highest current load of 2,220 mA g−1 (~16.5 C) applied, the capacity of 33 mAh g−1 has been retained.
On the other hand, the data presented in Ref. [15] and our practice have shown that even being synthesized at mild conditions, these and other lithium manganese spinel samples demonstrate a clear trend to the fast aggregation of particles upon the thermal treatment of a pyrolyzed material. This trend levels the advantages of the citric acid-aided route where pyrolysis gives nanosized powders with crystallites of ~15 nm size.
To avoid this obstacle and to obtain spinel samples of the lower degree of aggregation and better high-rate properties the following idea is suggested in this paper. Instead of the pyrolysis in air (a ‘standard’ scheme), citrate precursors are pyrolyzed in an inert atmosphere (a proposed scheme). In this case, two circumstances are considered to favor the smaller degree of aggregation of the target product. First of all, no chemical reactions between lithium carbonate and manganese-II oxide obtained in the course of pyrolysis shall occur. Second, a great amount of carbon shall be formed covering the products of pyrolysis. This means that upon further annealing, carbon-coated Li2CO3 and MnO will react, and the coating, even being burnt, will prevent the rapprochement of the spinel particles. In what follows, we describe the physicochemical and electrochemical properties of the lithium-substituted spinel of the Li[Li0.033Mn1.967]O4 composition supporting this idea. Its particle size is smaller, the degree of aggregation is lower, and high-rate properties are better that for its analogue pyrolyzed in air. In particular, model Li||Li[Li0.033Mn1.967]O4 cells deliver ~60 mAh g−1 at the current loads of 4,000 mA g−1 (30 C); at the highest current load of 13,300 mA g−1 (100 C), the capacity 11 mAh g−1 is retained; after a sequence of 62 charge/discharge cycles with the currents varied from 0.1 to 100 C the specific capacity returns at its initial value.
Experimental
Citrate precursors have been obtained in the following way [27–31]. Solutions of lithium nitrate, manganese-II nitrate, and citric acid (all of analytical grade) of approx. 1 M concentration have been analyzed with the four significant digits accuracy, mixed using calibrated pipettes in the 1.05:2:6 ratio, and evaporated up to a resin state (80 °C). The resins have been dried at 120 °C giving the precursors for all further operations.
According to the ‘standard’ scheme, the precursors have been pyrolyzed and annealed in air. By the proposed scheme, pyrolysis has been performed in the argon atmosphere and after that, pyrolyzed samples have been annealed in air. The temperature of pyrolysis in both cases has been equal to 400 °C; the duration of pyrolysis has been taken equal to 0.5 h. Subsequent operations with the samples pretreated in air have been made according to Ref. [28] and consisted of 24 h annealing in air at 700 °C with heating and cooling rates taken equal to 5 and 2 °C·min−1, respectively. The samples pretreated in argon have been annealed in air at two different regimes: for 24 h at 700 °C and for 10 h at 800 °C; the heating and cooling rates have been kept as the same as in the case of the samples pretreated in air.
Since the content of lithium and manganese is crucial for the correct comparison of the properties of the samples pretreated at different conditions, it has been controlled by means of atomic absorption spectrophotometry (AA-8500, Nippon Jarrell–Ash, Japan). The mean Li:2Mn ratio of 1.0503 has been determined for thermally treated samples with standard deviation of 0.00082 and standard error of 0.00033 thus signifying that no lithium loss occurs. This Li:2Mn ratio perfectly matches the title composition, Li[Li0.033Mn1.967]O4. To check the thermal decomposition process, the differential thermal analysis has been employed (Q-1500 D, MOM, Hungary). Porosity studies have been utilized on an ASAP 2000 device (Micromeritics, USA). The phase composition of the samples, their morphology and the particle size have been studied by means of X-ray diffraction (DRON-4-07, LOMO, Russia, CoКα and CuКα radiation) and scanning electron microscopy (JSM 6700F, JEOL, Japan). To qualitatively determine the phase composition of samples JCPDS-ICDD database has been used.
Electrochemical tests have been performed in sample Li||Li[Li0.033Mn1.967]O4 cells on a home-made versatile workstation. Working electrodes have consisted of 82 wt. % of the studied material, 10 wt. % of a conductive additive (soot) and 8 wt. % of a binder (poly (vinyliden) difluoride). A 20 µm layer of the slurry has been put on an aluminum foil with a doctor blade and then dried at 120 °C for 18-21 h under an IR radiator. The quantity of Li[Li0.033Mn1.967]O4 in a dried remainder was of 3–8 mg cm−2. The electrodes were rolled so as to decrease the thickness of the layer from 70–80 to 25–35 μm. Sample coin-type cells have been assembled in 2016 cases with Celgard 2500 separators and an electrolyte containing the 1 M solution of LiPF6 in a mixture of ethylene carbonate and dimethyl carbonate taken in the 1:1 mass ratio. The reference and counter electrode has been made of lithium metal. Potentiodynamic tests have been performed within the voltage range of 3.4–4.5 V at the scanning speed of 0.1 mV s−1. High rate tests have been carried out in a galvanostatic mode within the same voltage range using a CCCV regime with the charge current of 1C and a trickle charge at 0.1 C; discharge currents varied from 0.1C to 100C. As noticed above, the theoretical specific capacity of the compound studied, Li[Li0.033Mn1.967]O4 is 133 mAh g−1, therefore, 1C = 133 mA g−1.
Results and Discussion
As follows from Refs. [29–31], citrate precursors after drying at 120 °C (dried resins) consist of stoichiometric hydrous citrates and upon pyrolysis, decompose giving the target products. In the case of lithium manganese spinels, the precursors contain the mixture of dihydrates of lithium citrate, manganese-II citrate, and two excess moles of citric acid, and their decomposition in air is described by the following reaction [28],
Unlike pyrolysis in air, the decomposition of the spinel precursor in argon atmosphere should proceed according the following scheme,
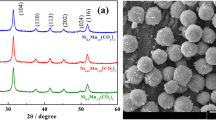
In this case, the reaction products are lithium carbonate and manganese-II oxide. On further annealing in air, the excess carbon should serve as a coating preventing the aggregation of the spinel particles. The calculated mass loss in reaction (4) is 60.9 %. In accord with this estimate, pyrolysis at 400 °C for 0.5 h leads to the experimental mass loss of 62.0 %. X-ray diffraction data evidence the formation of MnO. A diffraction pattern obtained (Fig. 1a) well agrees with existing data (JCPDS 07-0230). The lithium salt formed is not recognizable due to low scattering abilities of lithium, carbon and oxygen.

X-ray diffraction pattern of the decomposition products of the precursor treated in argon atmosphere (a) and the spinel obtained upon further thermal treatment (b)
Upon heating the mixture of Li2CO3, MnO and carbon in air, the following reaction accompanied by the mass loss of 51.0 % should occur,
Respective thermal analysis data are given in Fig. 2. The mass loss of 47.0 % registered is in a good agreement with the calculations by reaction (5). On the other hand, the burning of carbon and accompanying exothermic effect makes any attempts to interpret the DTA data difficult. An X-ray analysis proves that the product obtained is the lithium manganese spinel (Fig. 2b).

Thermal analysis data for the mixture of Li2CO3, MnO and carbon obtained in the course of its heating in air
To determine the crystallite size d of spinels the Scherrer formula has been employed [32],
where λ is the wavelength of X-ray radiation, B is the line width at the half maximum of the diffraction peak, and θ is the angle of diffraction. The lattice parameters a (Å) for the spinel samples have been calculated by the Holland-Redfern method using a UnitCell 1997 software [33]. The values of d and a are summarized in Table 1. As follows from these data, the samples consist of nanosized crystallites of comparable dimensions.
Scanning electron microscopy supports a conclusion regarding the nanosized nature of the spinels under investigation (Fig. 3). Comparing the micrographs one easily reassures that the particle size dpart. in the sample pyrolyzed in argon is at least twice smaller than in the sample pyrolyzed in air (50–70 nm against 70–100 nm, respectively). Moreover, the former sample demonstrates a less significant tendency to aggregation. Combining these data with the crystallite size d obtained by means of X-ray diffraction, Table 1, one can speculate that the particles in the argon-pyrolyzed samples are single crystals, whereas the particles in the air-pyrolyzed samples are aggregates of single crystals.

Scanning electron micrographs (×100,000) of Li[Li0.033Mn1.967]O4 treated in air (left) and in argon (right)
Unlike lithium manganese spinels obtained by means of nanochemistry routes [14, 34] and showing high surface areas and clear hysteresis loops on nitrogen adsorption/desorption curves, porosity data for our samples are much simpler (Fig. 4). They reveal low surface areas (~11 m2/g) and do not enable one to perform the calculation of pore size distributions. This means that the materials obtained have no micro- and mesopores, and their porosity, as follows from SEM micrographs, is determined by voids between particles.

Nitrogen adsorption/desorption isotherm for Li[Li0.033Mn1.967]O4 treated in argon
After physico-chemical characterization the samples have been subjected to electrochemical tests. In Fig. 5, stationary voltammetric curves corresponding to the 3rd cycle for sample Li||Li[Li0.033Mn1.967]O4 cells are presented. The term ‘stationary’ means the complete reproducibility of voltammetric data in preceding and following measurements. Peaks in the cathodic and anodic regions reflect processes occurring upon insertion/deinsertion of lithium ions into/from the lithium manganese spinel in the composition range of 0 ≤ x ≤ 0.5 and 0.5 ≤ x ≤ 1.0. It should be mentioned that the low-voltage insertion and deinsertion peaks are clearly asymmetric possibly signifying the presence of a third phase, as described in Introduction. These features, as well as an analysis of diffusion coefficients in Li[LiqMn2-q]O4 spinels (0 ≤ q ≤ 0.065) determined from voltammetric and impedance measurements will be described elsewhere.

Stationary voltammetric characteristics of Li[Li0.033Mn1.967]O4–based electrodes. Scanning speed 0.1 mV/s. aThin black curve Li[Li0.033Mn1.967]O4 treated in air. bThick red curve Li[Li0.033Mn1.967]O4 treated in argon
Discharge curves for sample Li||Li[Li0.033Mn1.967]O4 cells at various discharge current densities are given in Fig. 6, and dependences of specific capacities on current densities are shown in Fig. 7. At minimal current loads, the specific capacities of materials under investigation are lower than the theoretical values. For the samples fired in air discharge capacities \(Q_{\text{d}}^{\hbox{max} .}\) at minimal discharge currents Imin. = 0.1C are equal to 103 mAh g−1; for those fired in argon \(Q_{\text{d}}^{\hbox{max} .}\) = 113 mAh g−1. As follows from Figs. 6 and 7, the cells with the cathode materials fired in argon retain their working ability at the currents growing from 14.8 to 14,800 mA g−1 (from 0.1 to 100 C). After heavy-duty tests during 62 charge/discharge cycles their specific capacity returns to its initial value. The materials treated in air remain stable at much lower current loads and do not retain their initial capacity after loading.

Discharge curves for sample Li||Li[Li0.033Mn1.967]O4 cells at various discharge current densities: a Li[Li0.033Mn1.967]O4 treated in air; b Li[Li0.033Mn1.967]O4 treated in argon

Dependences of discharge capacity on cycle number for Li||Li[Li0.033Mn1.967]O4 cells. Left Li[Li0.033Mn1.967]O4 treated in air. Right Li[Li0.033Mn1.967]O4 treated in argon. Numeric values mean discharge current densities in C units
It is tempting to compare these results with those existing in the literature. Since the rate capability of Li-substituted spinels has not been widely studied we present Table 2 where selected latest data on LiMn2O4 are summarized. This seems appropriate because the lithium excess in the sample is very small. More detailed structural and electrochemical information about these and other LiMn2O4 samples demonstrating good high-rate properties can be found in Ref. [5]. For the sake of comparison, the high-rate parameters are characterized by the minimal capacity losses per the unit current rate, \(dQ_{\text{d}} /dI = (Q_{\text{d}}^{\hbox{max} .} - Q_{\text{d}}^{\hbox{min} .} )/(I_{\hbox{max} .} - I_{\hbox{min} .} )\), where \(Q_{\text{d}}^{\hbox{max} .}\) and \(Q_{\text{d}}^{\hbox{min} .}\) are the discharge capacities at the minimal and maximal discharge currents applied, Imin. and Imax.. For our samples these values have been determined from Fig. 8.

Capacities retained by Li||Li[Li0.033Mn1.967]O4 cells upon various current loads. Squares, black Li[Li0.033Mn1.967]O4 treated in air. Circles, red Li[Li0.033Mn1.967]O4 treated in argon
The inspection of Table 2 reveals that electrode materials demonstrating good high-rate properties have low initial capacities, except the sample obtained by means of a resorcinol-formaldehyde route and exhibiting exceptional results. It should be mentioned parenthetically that, to our surprise, numerous materials with high initial capacities (130–133 mAh g−1 [19, 41], 135 mAh g−1 [42–45], 145 mAh g−1 [46]) have never been treated at high current loads. On the other hand, it is well-seen that pyrolyzing the citrate precursor in argon one can attain better high-rate capabilities and significantly improve the initial capacity of the spinel material.
In our opinion, this promising result is bound not only to a decrease in the size and degree of aggregation of the spinel particles, but to the nature of the crystallites in the argon-treated sample, each of which, according to XRD and SEM data, can be considered as a separate single crystal. As follows from Ref. [5], just the perfectness of the particles, together with their size and degree of aggregation, is crucial for the accessibility of theoretical capacities and high speed of discharge of lithium batteries.
Conclusion
In this paper, we have synthesized two samples of the nanosized lithium-doped lithium manganese spinel, Li[Li0.033Mn1.967]O4 using a citric acid-aided route, performed their characterization by means of X-ray diffraction, scanning electron microscopy, and porosity studies, and carried out their electrochemical tests in Li||Li[Li0.033Mn1.967]O4 cells. The difference between these samples lies in the pyrolysis regime, viz., one has been pyrolyzed in air and another in argon. The particle size in the sample pyrolyzed in argon (50–70 nm) is at least twice smaller than in the sample treated in air (70–100 nm), and the former demonstrates a smaller tendency to aggregation. In high-rate tests, the argon-fired samples overwhelm the air-fired ones retaining the working ability at the currents growing from 14.8 to 14,800 mAh g−1 (from 0.1 to 100C), and after 63 charge/discharge cycles their specific capacity returns to its initial value with no capacity fading. This result may by prospective for lithium-ion batteries for heavy-duty applications.
References
Thackeray, M.M., David, W.I.F., Bruce, P.G., Goodenough, J.B.: Lithium insertion into manganese spinels. Mater. Res. Bull. 18, 461–472 (1983)
Whittingham, M.S.: Lithium batteries and cathode materials. Chem. Rev. 104, 4271–4302 (2004)
Thackeray, M.M., Johnson, P.J., de Piciotto, L.A., Bruce, P.G., Goodenough, J.B.: Electrochemical extraction of lithium from LiMn2O4. Mater. Res. Bull. 19, 179–187 (1984)
Rossouw, M.H., de Kock, A., de Piciotto, L.A., Thakeray, M.M., David, W.I.F., Ibberson, R.M.: Structural aspects of lithium-manganese-oxide electrodes for rechargeable lithium batteries. Mater. Res. Bull. 25, 173–182 (1990)
Potapenko, A.V., Kirillov, S.A.: Lithium manganese spinel materials for high-rate electrochemical application. J. Energy Chem. 23, 543–588 (2014)
Ohzuku, T., Kitagawa, M., Hirai, T.: Electrochemistry of manganese dioxide in lithium nonaqueous cell III. X-Ray diffractional study on the reduction of spinel-related manganese dioxide. J. Electrochem. Soc. 137, 769–775 (1990)
Liu, W., Kowal, K., Farrington, G.C.: Mechanism of the electrochemical insertion of lithium into LiMn2O4 spinels. J. Electrochem. Soc. 145, 459–465 (1998)
Lee, Y.J., Wang, F., Mukerjee, S., McBreen, J., Grey, C.P.: 6Li and 7Li magic-angle spinning nuclear magnetic resonance and in situ x-ray diffraction studies of the charging and discharging of LixMn2O4 at 4 V. J. Electrochem. Soc. 147, 803–812 (2000)
Björk, H., Gustafsson, T., Thomas, J.O., Lidin, S., Petříĉek, V.: Long-range ordering during delithiation of LiMn2O4 cathode material. J. Mater. Chem. 13, 585–589 (2003)
Yamada, A.: Lattice instability in Li(LixMn2−x)O4. J. Solid State Chem. 122, 160–165 (1996)
Tarascon, J.M., McKinnon, W.R., Coowar, F., Bowmer, T.N., Amatucci, G., Guyomard, D.: Synthesis conditions and oxygen stoichiometry effects on Li insertion into the spinel LiMn2O4. J. Electrochem. Soc. 141, 1421–1431 (1994)
Tarascon, J.M., Wang, E., Shokoohi, F.K., McKinnon, W.R., Colson, S.: The spinel phase of LiMn2O4 as a cathode in secondary lithium cells. J. Electrochem. Soc. 138, 2859–2864 (1991)
Xia, Y., Yoshio, M.: An investigation of lithium ion insertion into spinel structure Li-Mn-O compounds. J. Electrochem. Soc. 143, 825–833 (1996)
Jiao, F., Bao, J., Hill, A.H., Bruce, P.G.: Synthesis of ordered mesoporous Li–Mn–O spinel as a positive electrode for rechargeable lithium batteries. Angew. Chem. 47, 9711–9716 (2008)
Paulsen, J.M., Dahn, J.R.: Phase diagram of Li-Mn-O spinel in air. Chem. Mater. 11, 3065–3079 (1999)
Aricò, A.S., Bruce, P., Scrosati, B., Tarascon, J.M., Van Schalkwijk, W.: Nanostructured materials for advanced energy conversion and storage devices. Nat. Mater. 4, 366–377 (2005)
Tang, W., Wang, X.J., Hou, Y.Y., Li, L.L., Sun, H., Zhu, Y.S., Bai, Y., Wu, Y.P., Zhu, K., Van Ree, T.: Nano LiMn2O4 as cathode material of high rate capability for lithium ion batteries. J. Power Sources 198, 308–311 (2012)
Mao, J., Dai, K.H., Zhai, Y.C.: High rate capability and cycling stability of Li1.07Mn1.93O4 nanoflakes synthesized via gel-combustion method. Acta Phys. Chim. Sin. 28, 349–354 (2012)
Feng, C., Li, H., Zhang, C., Guo, Z., Wu, H., Tang, J.: Synthesis and electrochemical properties of non-stoichiometric Li–Mn-spinel (Li1.02MxMn1.95O4 − yF y ) for lithium ion battery application. Electrochim. Acta 61, 87–93 (2012)
Chen, Y., Xie, K., Pan, Y., Zheng, C.: Effect of calcination temperature on the electrochemical performance of nanocrystalline LiMn2O4 prepared by a modified resorcinol–formaldehyde route. Solid State Ionics 181, 1445–1450 (2010)
Liu, W., Farrington, C.C., Chaput, F.: Synthesis and electrochemical studies of spinel phase LiMn2O4 cathode materials prepared by the Pechini process. J. Electrochem. Soc. 143, 879–884 (1996)
Hwang, B.J., Santhanam, R., Liu, D.G.: Characterization of nanoparticles of LiMn2O4 synthesized by citric acid sol–gel method. J. Power Sources 97–98, 443–446 (2001)
Hwang, B.J., Santhanam, R., Liu, D.G.: Effect of various synthetic parameters on purity of LiMn2O4 spinel synthesized by a sol–gel method at low temperature. J. Power Sources 101, 86–89 (2001)
Hwang, B.J., Santhanam, R., Hu, S.G.: Synthesis and characterization of multidoped lithium manganese oxide spinel, Li1.02Co0.1Ni0.1Mn1.8O4, for rechargeable lithium batteries. J. Power Sources 108, 250–255 (2002)
Wang, X., Chen, X., Gao, L., Zheng, H., Ji, M., Shen, T., Zhang, Z.J.: Citric acid-assisted sol–gel synthesis of nanocrystalline LiMn2O4spinel as cathode material. Cryst. Growth. 256, 123–127 (2003)
Yi, T., Dai, C., Gao, K., Hu, X.: Effects of synthetic parameters on structure and electrochemical performance of spinel lithium manganese oxide by citric acid-assisted sol–gel method. J. Alloys Compd. 425, 343–347 (2006)
Potapenko, A.V., Chernukhin, S.I., Romanova, I.V., Rabadanov, KSh, Gafurov, M.M., Kirillov, S.A.: Citric acid aided synthesis, characterization, and high-rate electrochemical performance of LiNi0.5Mn1.5O4. Electrochim. Acta 134, 442–449 (2014)
Potapenko, A.V., Chernukhin, S.I., Romanova, I.V., Kirillov, S.A.: Physico-Chemical and Electrochemical Properties of Nanosized Li[Li0.033Mn1.967]O4. Chem. Phys. Tech. Surf. (In Russian) 2, 175–181 (2011)
Farbun, I.A., Romanova, I.V., Terikovskaya, T.E., Dzanashvili, D.I., Kirillov, S.A.: Complex formation in the course of synthesis of zinc oxide from citrate solutions. Russ. J. Appl. Chem. 80, 1798–1803 (2007)
Romanova, I.V., Farbun, I.A., Khainakov, S.A., Kirillov, S.A., Zazhigalov, V.A.: Investigaon fcatlyic proetis ofmaterils baed ontrasion metals oxides and Ce oxide. Rep. Nat. Acad. Sci. Ukraine. (In Russian) 10, 154–159 (2008)
Farbun, I.A., Romanova, I.V., Kirillov, S.A.: Optimal design of powdered nanosized oxides of high surface area and porosity using a citric acid aided route, with special reference to ZnO. J. Sol Gel Sci. Technol. 68, 411–422 (2013)
Patterson, A.: The diffraction of X-rays by small crystalline particles. Phys. Rev. 56, 978 (1939)
Holland, T.J.B., Redfern, S.A.T.: Unit cell refinement from powder diffraction data: the use of regression diagnostics. Mineral. Mag. 61, 65–77 (1997)
Wang, Y., Shao, X., Xu, H., Xie, M., Deng, S., Wang, H., Liu, J., Yan, H.: Facile synthesis of porous LiMn2O4 spheres as cathode materials for high-power lithium ion batteries. J. Power Sources 226, 140–148 (2013)
Huang, S., Wen, Z., Yang, X., Zhu, X., Lin, B.: Synthesis and the improved high-rate performance of LiMn2O4/Ag composite cathode for lithium-ion batteries. Electrochem. Solid State Lett. 9, A443–A447 (2006)
Lanz, M., Kormann, C., Steininger, H., Heil, G., Haas, O., Novák, P.: Large-agglomerate-size lithium manganese oxide spinel with high rate capability for lithium-ion batteries. J. Electrochem. Soc. 147, 3997–4000 (2000)
Tang, W., Liu, L.L., Tian, S., Li, L., Li, L.L., Yue, Y.B., Bai, Y., Wu, Y.P., Zhu, K., Holze, R.: LiMn2O4 nanorods as a super-fast cathode material for aqueous rechargeable lithium batteries. Electrochem. Commun. 13, 1159–1162 (2011)
Kim, J.S., Kim, K.S., Cho, W., Shin, W.H., Kanno, R., Choi, J.W.: A truncated manganese spinel cathode for excellent power and lifetime in lithium-ion batteries. Nano Lett. 12, 6358–6365 (2012)
Hosono, E., Kudo, T., Honma, I., Matsuda, H., Zhou, H.: Synthesis of single crystalline spinel LiMn2O4 nanowires for a lithium ion battery with high power density. Nano Lett. 9, 1045–1051 (2009)
Chen, Y.C., Xie, K., Pan, Y., Zheng, C.M., Wang, H.L.: High power nano-LiMn2O4 cathode materials with high-rate pulse discharge capability for lithium-ion batteries. Chin. Phys. B 20, 028201–028206 (2011)
Sun, Y.K., Kim, D.W., Jin, S.H., Hyung, Y.E., Moon, S.I., Park, D.K.: Synthesis and cycling behavior of LiMn2O4cathode materials prepared by glycine-assisted sol-gel method for lithium secondary batteries. Korean J. Chem. Eng. 15, 64–70 (1998)
Suryakala, K., Marikkannu, K.R., Paruthimal Kalaignan, G., Vasudevan, T.: Synthesis and rlectrochemical characterization of LiMn2O4 and LiNd0.3Mn1.7O4 as cathode for lithium ion batteryIntern. J. Electrochem. Sci. 3, 136–144 (2008)
Thirunakaran, R., Sivashanmugam, A., Gopukumar, S., Dunnill, C.W., Gregory, D.H.: Studies on chromium/aluminium-doped manganese spinel as cathode materials for lithium-ion batteries-A novel chelated sol-gel synthesis. J. Mater. Proc. Tech. 208, 520–531 (2008)
Thirunakaran, R., Sivashanmugam, A., Gopukumar, S., Dunnill, C.W., Gregory, D.H.: Phthalic acid assisted nano-sized spinel LiMn2O4 and LiCr x Mn2 − xO4 (x = 0.00–0.40) via sol–gel synthesis and its electrochemical behaviour for use in Li-ion-batteries. Mater. Res. Bull. 43, 2119–2129 (2008)
Li, R., Li, M.: Enhancement of the electrochemical properties of LiMn2O4 by glycolic acid-assisted sol-gel method. Ionics 15, 215–219 (2009)
Vivekanandhan, S., Venkateswarlu, M., Satyanarayana, N., Suresh, P., Nagaraju, D.H., Munichandraiah, N.: Effect of calcining temperature on the electrochemical performance of nanocrystalline LiMn2O4 powders prepared by polyethylene glycol (PEG-400) assisted Pechini process. Mater. Lett. 60, 3212–3216 (2006)
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Potapenko, A.V., Chernukhin, S.I. & Kirillov, S.A. A new method of pretreatment of lithium manganese spinels and high-rate electrochemical performance of Li[Li0.033Mn1.967]O4. Mater Renew Sustain Energy 4, 40 (2015). https://doi.org/10.1007/s40243-014-0040-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40243-014-0040-7