
Abstract
Bovine mastitis is a worldwide disease affecting dairy cattle and causes major economic losses in the dairy industry. Recently, the emergence of microbial resistance to the current antibiotics complicates the treatment protocol which necessitates antibiotic stewardship and further research to find new active compounds. Recently, phytobiotics have gained interest in being used as an alternative to antibiotics in the poultry industry as an antibiotic stewardship intervention. This study evaluated the in vitro antibacterial activity of 16 flavonoids against bovine mastitis pathogens. Two flavones: 2-(4-methoxyphenyl)chromen-4-one (1) and 2-(3-hydroxyphenyl)chromen-4-one (4) showed inhibition of the growth of Klebsiella oxytoca with MIC values range (25–50 µg mL− 1) followed by a structure-activity relationship (SAR) study indicating that the presence of a hydroxyl group at C-3` or methoxy at C-4` increases the activity against Klebsiella oxytoca while the presence of hydroxyl group at C-7 decreases the activity. Furthermore, a structure-based drug development approach was applied using several in silico tools to understand the interactions of active flavones at the active site of the DNA gyrase protein. Compound (4) showed a higher docking score than quercetin (standard) which is known to have antibacterial activity by inhibiting the DNA gyrase. In addition, the structure-based pharmacophores of compound (4) and quercetin showed similar pharmacophoric features and interactions with DNA gyrase. Based on our findings, compounds (1) and (4) are promising for further study as potential anti-microbial phytochemicals that can have a role in controlling bovine mastitis as well as to investigate their mechanism of action further.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Bovine mastitis is a worldwide disease affecting dairy cattle health and milk production and causes major economic losses in the dairy industry (Schabauer et al. 2018). Bovine mastitis is an inflammation of the bovine mammary glands caused by a defence response against bacterial infection or tissue damage. In general, the inflammatory response against harmful stimuli such as tissue damage or invading microorganisms is a normal immunological response to eliminate the harmful stimuli and start the healing process, but abnormal inflammatory response in the mammary gland may cause severe acute inflammation or chronic mastitis which negatively affects the quantity and quality of the milk and increases the mortality rate of cattle (Aitken et al. 2011; Yang et al. 2014). These consequences make bovine mastitis the most costly cattle disease (Azooz et al. 2020; Hogeveen and Van Der Voort. 2017; Hogeveen et al. 2011; Romero et al. 2018). Annual economic loss due to mastitis has been estimated to be €2 Bn in the European Union and the same figure is estimated for the USA (Wang et al. 2020). The causes of bovine mastitis are multifactorial and complex, but microbial infection is the most common cause. Several bacterial pathogens may cause bovine mastitis, and Staphylococcus aureus is the most common. Other common microbial causes include Streptococcus agalactiae, Streptococcus dysgalactiae, Streptococcus uberis, Escherichia coli, Pseudomonas spp., and Klebsiella spp. (Barreiros et al. 2022; De los Santos et al. 2022; Hillerton and Berry. 2005).
Antibiotics are the recommended treatment for bovine mastitis, with b-lactams being the most used. However, several studies have reported high levels of antimicrobial resistance to antibiotics in the clinical bacterial isolates exceeding 85% such as penicillin, ampicillin, and sulphonamide in coagulase-negative Staphylococci (Purgato et al. 2021). Due to some pathogens` ability to resist the available antibiotics, the treatment options have become limited, and the treatment failure rate has increased (Purgato et al. 2021). The cure rate of Staphylococcus aureus has recently decreased due to the emergence of resistance mechanisms such as biofilm production and efflux-mediated resistance (Kamaruzzaman et al. 2017; Silva et al. 2023).
Considering the economic burden of bovine mastitis, this suggests that more productive research is needed to find new active compounds against bovine mastitis pathogens as well as antibiotic stewardship to enhance proper antibiotic use. Phytobiotics are secondary metabolites produced by plants such as flavonoids, alkaloids, and terpenes, and they are known to have several biological activities such as antimicrobial and antioxidant activities (Kikusato. 2021; Morrissey and Osbourn. 1999). Recently, phytobiotics have gained interest in being used as an alternative to antibiotics in the poultry industry as an antibiotic stewardship intervention (Abd El-Hack et al. 2016). This helps restrict the therapeutic use of antibiotics and eliminates their sub-therapeutic use (Olagaray and Bradford. 2019).
It has been reported that the presence of flavonoid in large quantities in some plant extracts such as Schinus molle (known as Peruvian peppertree) and Brickellia veronicaefolia (known as hierba dorada) leads to the inhibitory activity of these plant extracts against bovine mastitis-causing pathogens (Macías Alonso et al. 2019) as well as Eucalyptus globulus (known as blue gum), Juglans regia (known as English walnut) extracts (Gomes et al. 2019), and Bauhinia variegate (known as orchid tree) (Mishra et al. 2013). Flavonoids are polyphenolic natural compounds found in abundance in plants and they have essential roles in several biological processes such as growth, development, and protection against several environmental factors such as microorganisms and ultraviolet light (Shen et al. 2022). Hence, flavonoids are promising antibiotic candidates against resistant microbial pathogens (Dixon. 2001). The basic skeletal structure of flavonoids is composed of three rings and based on the degree of oxidation of the central ring, flavonoids are classified into seven subgroups (Shen et al. 2022). Flavones and flavonols are subgroups of flavonoids and distinct from other flavonoids by the presence of a double bond between C-2 and C-3 and oxidized at C-4, no substitution at C-3 position in flavone while flavonols have OH group at C-3 position (Hostetler et al. 2017; Martens and Mithöfer. 2005; Nguyen and Bhattacharya. 2022). It was found that the positions of the hydroxy and methoxy substituents in the structure play a vital role in the bioactivity of flavonoids (Chirumbolo. 2010; Preet et al. 2023). Flavones and flavonols gained much interest due to their proven bioactivity in vitro and in vivo studies (Hostetler et al. 2017; Lee et al. 1993; Martens and Mithöfer. 2005; Shen et al. 2022). A clinical study carried out on dairy cows diagnosed with mastitis, showed that the intramammary administration of micronized purified flavonoid fraction (MPFF) is effective in the treatment of bovine mastitis caused by coagulase-negative Staphylococci (Gutiérrez-Reinoso et al. 2023).
With the emergence of antimicrobial resistance and the increased mortality and morbidity associated with it, flavonoids have gained renewed interest as potential antimicrobial agents, especially since flavonoids are very common in plants, and have safety profiles (Amawi et al. 2017). However, their mechanism of action is still unclear (Salmanli et al. 2021). The proposed antibacterial mechanisms of flavonoids are diverse, including inhibition of nucleic acid synthesis, inhibition of energy metabolism, inhibition of biofilm formation, alteration of the permeability of cell membranes, and inhibition of the function of the cytoplasmic membrane (Xie et al. 2015; Cushnie and Lamb. 2005; Takó et al. 2020). It has been shown that flavonoids have shown a significant topoisomerase inhibition effect, leading to their antibacterial activity. For example, quercetin, apigenin, and 3,6,7,3′,4′-pentahydroxyflavone displayed a significant inhibition against DNA gyrase (topoisomerase 2), a protein responsible for DNA replication in bacteria. (Nguyen et al. 2022; Ohemeng et al. 1993; Plaper et al. 2003).
Computer-aided drug design (CADD) methods make a significant contribution to drug development research (Sadybekov and Katritch. 2023; Sliwoski et al. 2013). Structure-based methods use the information on the target and ligand structures using molecular docking, structure-based pharmacophore modelling, and molecular dynamics simulation to identify promising compounds and understand their interactions with target proteins (Palermo and De Vivo. 2015). These computational tools have significantly accelerated the drug development process (Ou-Yang et al. 2012).
In this study, the in vitro antibacterial activity of 16 flavones and flavonols against five bovine mastitis-causing pathogens was evaluated, and the minimum inhibitory concentration and the minimum bactericidal concentration were also determined. The structure-activity relationships (SAR) were analysed to determine fundamental structural properties contributing to their antibacterial activity against bovine mastitis pathogens. To understand the interactions of the tested flavonoids at the active site of the protein receptor, a molecular docking study was performed using topoisomerase II (DNA gyrase) enzyme (Bax et al. 2010; Patel et al. 2014; Shamsudin et al. 2022), and structure-based pharmacophores were generated to identify the main pharmacophoric features responsible for the antibacterial activity. Furthermore, molecular dynamics simulation was performed to assess the binding stability of the target protein with the active flavonoids.
Methodology
Source of compounds (1–16)
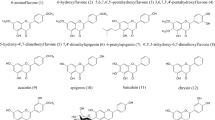
Compounds (1–16) were taken from the Marine Biodiscovery Centre Compound Library at the University of Aberdeen, UK (donated by the late Professor R.H. Thomson). The IUPAC names of these flavonoids are as follows: (1) 2-(4-methoxyphenyl)chromen-4-one; (2) 2-(3-hydroxyphenyl)-7-methoxychromen-4-one; (3) 7-methoxy-2-(2-methoxyphenyl)chromen-4-one; (4) 2-(3-hydroxyphenyl)chromen-4-one; (5) 8-methoxy-2-phenylchromen-4-one ; (6) 7-methoxy-2-phenylchromen-4-one ; (7) 7-hydroxy-5-methoxy-2-phenylchromen-4-one; (8) 7-hydroxy-2-(4-methoxyphenyl)chromen-4-one; (9) (4-oxo-2-phenylchromen-5-yl) acetate; (10) 2-(2,5-dihydroxyphenyl)-5,7-dihydroxychromen-4-one; (11) 2-(4-hydroxyphenyl)-3-methoxychromen-4-one; (12) 2-(3-ethoxy-4-methoxy-phenyl)-3,5,7-trimethoxy-chromen-4-one; (13) 2-(3-ethoxy-4-methoxy-phenyl)-3,5,7-trimethoxy-chromen-4-one; (14) 3-acetoxy-5,7-dimethoxy-2-(4-methoxy-phenyl)-chromen-4-one; (15) [3-acetyloxy-7-methoxy-2-(4-methoxyphenyl)-4-oxochromen-5-yl] acetate; (16) 2-(4-hydroxyphenyl)-3,5,7-trimethoxychromen-4-one.
Antimicrobial assays
The antimicrobial activity of 16 flavones and flavonols was initially assessed using the Kirby-Bauer disc-diffusion assay method against five bovine mastitis strains procured from NCIMB Ltd., Aberdeen, United Kingdom. The tested pathogens are gram-positive bacteria including Staphylococcus aureus subsp. aureus (NCIMB 701494), Streptococcus bovis (NCIMB 702087), and Enterococcus pseudoavium (NCIMB 13084) and gram-negative bacteria included Klebsiella oxytoca (NCIMB 701361) and Escherichia coli (NCIMB 701266). Flavonoids were dissolved in dimethyl sulphoxide (DMSO), ThermoFisher Scientific Ltd., United Kingdom at a concentration of 70 µg mL− 1. Sterile filter paper discs were impregnated with 20 µL of flavonoid solutions and left to dry overnight. The pathogens were inoculated in Mueller-Hinton broth, Sigma Aldrich Ltd., United Kingdom for one hour in the incubator at 28 °C then spread uniformly in the form of bacterial lawn on top of Mueller-Hinton agar plate. The previously loaded discs with flavonoid solutions were placed on the inoculated plates. Sterile filter paper discs loaded with DMSO were used as the negative control and 10 µg gentamicin discs, ThermoFisher Scientific Ltd., United Kingdom as the positive control. The plates were incubated at 28 °C for 24 h and the inhibition zone was observed. The antimicrobial assay was repeated twice to confirm the results.
The minimum inhibitory concentration against the test pathogens was evaluated by the broth macro-dilution method. The broth macro-dilution method is one of the most popular antimicrobial test methods which includes preparing serial dilution (two-fold dilutions) of the tested compound in a liquid growth medium in tubes with a minimum final volume of 2 mL (Balouiri et al. 2016). The pathogens were cultured in sterile Mueller Hinton broth in test tubes with serially diluted compounds (1) and (4) in DMSO. The final concentrations of the tested compounds were: 200, 100, 50, 25,12.5, 6.25, and 3.125 µg mL− 1. The test tubes were placed in the incubator at 28 °C for 24 h. The minimum inhibitory concentration was determined as the lowest concentration of the compound which showed no visually observed bacterial growth (i.e., no turbidity). A test tube containing sterile Mueller-Hinton broth and another tube containing the pathogen culture were used as a negative and positive control, respectively, to confirm the media sterility and adequate microbial growth over the incubation period. An aliquot of the positive control was plated to determine the baseline concentration of the pathogens used. The minimum bactericidal concentration against the tested pathogens was performed by plating the dilution, which represents the minimum inhibitory concentration, as well as the higher concentrated dilutions on Mueller-Hinton agar plates. The minimum bactericidal concentration was determined as the lowest concentration that kills 99.9% of the final bacterial inoculum after 24 h of incubation (i.e., demonstrated no bacterial growth in the plate) (Faller et al. 2004).
Molecular docking
A molecular docking study was carried out using AutoDock Vina version 1.2.0. (Eberhardt et al. 2021; Trott and Olson. 2010) using Samson by OneAngstrom, 2022 (https://www.samson-connect.net/). The crystal structure of the topoisomerase II (DNA gyrase) enzyme was downloaded from the Protein Data Bank (PDB ID: 2XCT) (http://www.rcsb.org/) and the active site (i.e., interaction site) of the protein was predicted by Ligand Scout 4.4.8 software (Wolber and Thierry. 2005) which generates a box around the active site with the following box centre and size dimensions: -16.2, 43.0, 88.0 and 25.7, 52.4, 22.5 Å (Fig. 1). The binding mode number was set as 10 and the maximum energy difference was set as 3 Kcal/mol. The molecular docking was carried out using the same settings at several exhaustiveness levels for convergence. The molecular docking process involved two main steps: prediction of possible ligand conformations and their orientations inside the active site (known together as a pose) followed by evaluation of their binding energy where each pose is given a docking score. The pose which showed the highest docking score was selected in this study.

Crystal structure of DNA gyrase (PDB ID: 2XCT), the box shows the binding site
Validation of molecular docking method
The docking method and parameters used in this study were validated by re-docking the co-crystallized ligand (Ciprofloxacin, a DNA gyrase inhibitor). This validation process involved the separation of the ligand and then re-docking the ligand again in the active site. The re-docked ligand was superimposed on the co-crystallized ligand and the Root Mean Square Deviation (RMSD) value was calculated. The low RMSD value of 0.785 Å between the experimental and the reference positions of the ligand indicated the same binding orientation which validated the method (Fig. S1).
Structure-based pharmacophore modelling
A structure-based pharmacophore was constructed based on docked complexes of DNA gyrase protein (PDB ID: 2XCT) with compounds (1) and (4) as well as the standard flavonoid (quercetin) to further understand the interaction between the active flavonoids with the active site of the target protein at the atomic level. Docked complexes were prepared using Autodock Vina 1.2.0. (Eberhardt et al. 2021; Trott et al. 2010) which is then used to create the pharmacophores using Ligand Scout 4.4.8 software (Wolber and Thierry. 2005).
Molecular dynamics (MD) simulation
To evaluate the conformational changes inside the active site and to assess the stability of the DNA gyrase complexes with compounds (1) and (4) over a specified time, molecular dynamics simulations were performed over 50 ns using Flare version 7.0.0 based on the OpenMM package (Eastman et al. 2017). The calculation method used was Open Force Field Explicit Water. The solvent model was Explicit TIP3P water, and the AM1-BCC method was used to compute partial charges. The following system settings were used: the solvent box shape was Truncated Octahedron, the solvent box buffer was kept at 10.0 Å, and the temperature and pressure were 298.0 K and 1.0 bar, respectively. The system was equilibrated for 200 ps. The time step 4.00 fs. The remaining settings are kept as default.
Results and discussion
Bioassay tests
The antimicrobial activity of 16 flavones and flavonols was tested initially using the disc diffusion assay against the following pathogens that cause bovine mastitis: Staphylococcus aureus subsp. aureus, Streptococcus bovis, Enterococcus pseudoavium, Klebsiella oxytoca, and Escherichia coli. The disc diffusion method is commonly used to evaluate the antimicrobial activity of pure compounds as well as microbial and plant extracts in laboratories and other institutions because of its low cost and ease of performance and interpretation (Balouiri et al. 2016). Only Compounds (1) and (4) showed a zone of inhibition at a concentration of 70 µg mL-1 against Klebsiella oxytoca. While other tested flavones and flavonols didn`t show a zone of inhibition against any of the tested pathogens. Therefore, the minimum inhibitory concentration and the minimum bactericidal concentration were determined only for compounds (1) and (4) against Klebsiella oxytoca. Bioassay data is summarized in (Table 1).
Structure-activity relationship (SAR)
Structure-Activity Relationship (SAR) is an important key to many aspects of drug discovery research, starting from the initial screening and ending up with lead optimisation (Guha. 2013). Thus, having a good understanding of the chemical properties of the flavonoids which are responsible for their bioactivity is essential in the design of new active compounds (Echeverría et al. 2017).
Compounds (1) and (4) showed activity against Klebsiella oxytoca while no flavonols showed activity against this strain. Thus, the presence of substituents at the C-3 position in the studied flavonols is not essential for the activity and might therefore decrease the antibacterial activity against this bacterial strain. It has been reported previously that hydroxylation at the C-3 position decreases antibacterial activity against gram-negative bacteria through inhibition of DNA gyrase while it is essential for antibacterial activity effect acting via the cell membrane (Shamsudin et al. 2022). Therefore, this increases the probability that the antibacterial activity of compounds (1) and (4) against Klebsiella oxytoca might be due to their inhibitory effect on DNA gyrase. In addition, we concluded that the presence of a hydroxyl group at C-3` or methoxy at C-4` increases the activity against Klebsiella oxytoca while the presence of a hydroxyl group at the C-7 position decreases the activity (Fig. 2). Although compound (8) has a methoxy group at C-4`, it also has a hydroxyl group at the C-7 position, which might reduce the activity.

Structure-activity relationship (SAR) of the flavones and flavonols shows the possible structural features for their antibacterial activity against Klebsiella oxytoca
In the literature, the antibacterial properties of flavonoids have been analysed by structure-activity relationship (SAR) studies; however, published data is inconsistent as to whether the presence of hydroxy or methoxy groups at different positions enhances or lowers the antibacterial activity. This inconsistency may be due to the amphiphilic balance (i.e., the balance between hydrophobic and hydrophilic characteristics) of the flavonoids, which is considered one of the most important aspects of antibacterial activity as it affects the ability of flavonoids to penetrate bacterial cells to exert their effect (Echeverría et al. 2017; Shamsudin et al. 2022). As mentioned above, the chemical properties of the flavonoids are essential for their antibacterial activity, however, the physical properties are also important as they affect the permeation of flavonoids through the cell wall and cell membrane of the bacteria to reach the site of action. The requirement of having phenolic groups in flavonoids for their antibacterial activity decreases the hydrophobicity and, in some cases, could affect the cell wall permeation. Hence, maintaining the balance between hydrophobicity and hydrophilicity in flavonoids is important to be taken into consideration in drug design (Echeverría et al. 2017).
Molecular docking
To further understand types of interaction at the molecular level between flavonoids and the active site of the target protein, the tested flavonoids were subjected to molecular docking against the DNA gyrase enzyme which is essential for DNA replication and transcription by catalysing the ATP-dependent negative supercoiling of DNA. This enzyme is essential in all bacteria, and it is absent in human cells, thus considered an attractive target for antibiotics (Collin et al. 2011). Flavonoids are known to have antibacterial activity through their inhibitory effect on the DNA gyrase enzyme (Shamsudin et al. 2022). The tested flavonoids (1–16) were docked against DNA gyrase protein (PDB ID: 2XCT) as well as quercetin which was chosen as standard because it is one of the flavonoids that is known to exhibit antibacterial activity by binding to DNA gyrase (Hossion et al. 2011; Plaper et al. 2003). Also, ciprofloxacin was docked to validate the docking method. Generally, molecular docking uses a scoring function that ranks the ligands based on their binding energy which reflects their binding affinity to the target protein as well as proposes structural hypotheses on the interactions between ligands and the target protein (Morris and Lim-Wilby. 2008). In this study, the docking scores of quercetin and ciprofloxacin were found to be similar (-7.1 Kcal mol− 1). The chemical structures of the tested flavones and flavonols and docking scores are shown below (Table 2). It is interesting to note that compound (4) which showed antimicrobial activity against Klebsiella oxytoca exhibited the highest binding affinity (-7.7 Kcal mol− 1) to the active site of DNA gyrase protein among the tested flavones and flavonols which further increases the probability that antibacterial activity of compounds (4) against Klebsiella oxytoca might be due to its inhibitory effect on DNA gyrase protein. The predicted binding poses of compounds (1) and (4) with DNA gyrase are shown below (Fig. 3). The interactions of compounds (1) and (4) with the target protein are discussed in detail in the structure-based pharmacophore modelling section.

The predicted binding poses of (a) compound (1) and (b) compound (4) inside the pocket of DNA gyrase
Structure-based pharmacophore modelling
Pharmacophore modelling is an important part of several computer-aided drug design (CADD) research, and it has been successfully applied in many computational methods such as virtual screening and lead optimization (Seidel et al. 2019). The definition of pharmacophore was provided by the International Union of Pure and Applied Chemistry (IUPAC) as “the ensemble of steric and electronic features that is necessary to ensure the optimal supra-molecular interactions with a specific biological target structure and to trigger (or to block) its biological response” (Wermuth et al. 1988). In this study, a structure-based pharmacophore was generated for compounds (1) and (4) using low-energy conformers where three main pharmacophoric features were identified: hydrogen bond acceptors (HBAs), hydrogen bond donors (HBDs), and hydrophobic features (H). In compound (1), ring A and ring B showed hydrophobic interactions with Tyr 1150D and Val 511D residues respectively, and the methoxy group at C-4` acts as a hydrogen bond acceptor for Ser1028D. In compound (4), Phe 1123B made a hydrophobic interaction with ring B, and the hydroxy group at C-3` acts as a hydrogen bond donor with Asp508D and Glu435D. It is worth noting that the standard (quercetin) showed the same pharmacophoric features as compound (4), ring B showed hydrophobic interaction with Phe 1123B and the hydroxy group at C-3` position acts as a hydrogen bond donor with Asp 510D (Fig. 4). This emphasizes the importance of Phe1123B toward the ligand-protein interaction as it interacts with compound (4) and quercetin. In compound (1), the interaction of the methoxy group at C-4` with the active site of DNA gyrase is consistent with our SAR findings about the importance of this methoxy group in the antibacterial activity of compound (1). Also, the interaction of the hydroxy group at C-3` in compound (4) with the active site of DNA gyrase confirms our SAR findings regarding the importance of this hydroxy substituent in the antibacterial activity of compound (4).

Structure-based pharmacophore of (a) compound (1), (b) compound (4), and (c) quercetin with DNA gyrase protein (2XCT)
Molecular dynamics (MD) simulation of DNA gyrase complexes with compounds (1) and (4)
A molecular dynamics simulation was carried out to assess the stability of the target protein complex with ligand over a specified time and to evaluate the conformational changes inside the active site. The stability of DNA gyrase complex with compounds (1) and (4) was simulated for 50 ns using Flare version 7.0.0 using the OpenMM package version. Root Mean Square Deviation (RMSD) reflects the stability of the protein complex with the ligand over time, while the Root Mean Square Fluctuation (RMSF) reflects the flexibility of each amino acid residue of the target protein inside the complex. The RMSD of the α carbons of amino acid residues of DNA gyrase complexes with compounds (1) and (4) were found to be 8.06 ± 2.54 Å and 7.25 ± 1.36 Å respectively (Fig. 5). The large increase in RMSD values of the protein-compound (1) complex during the first 15 ns of the simulation reflects significant conformational changes which indicated instability. High RMSD fluctuation was also observed in the time range between 15 ns and 27 ns. The second half of the simulation showed minimal RMSD fluctuation, and the complex attained stability. RMSD values of protein-compound (4) complex showed an increase at the beginning of the simulation (i.e., 0–7 ns) then minimal fluctuation was observed until the end of the simulation. Therefore, the DNA gyrase complex with compound (4) is predicted to be relatively more stable than its complex with compound (1). The RMSF of DNA gyrase in the complexes with the compounds (1) and (4) were found to be 3.73 ± 1.44 Å and 2.73 ± 1.00 Å respectively (Fig. 6). Both RMSF plots were like each other which indicated that amino acids of the binding site showed similar flexibility in both complexes.

RMSD plots of DNA gyrase complexes with compound (1) (Blue) and compound (4) (Green) as a function of time

RMSF plots of compound (1) (Blue) and compound (4) (Green) in complex with DNA gyrase as a function of amino acid residue number
The Radius of gyration (Rg) was also calculated to check the stability of the protein-ligand complex throughout the simulation time. Analysing the MD simulation of compounds (1) and (4) complexes with DNA gyrase indicated that the Rg of both fluctuated within a small range reflecting the stability of protein-ligand complexes (Fig. 7). The Rg of DNA gyrase complexes with compounds (1) and (4) were found to be 32.94 ± 0.22 Å and 33.04 ± 0.23 Å respectively.

The radius of gyration (Rg) values of DNA gyrase complexes with (a) compound (1) and (b) compound (4) during 50 ns of MD simulation
Conclusion
As a result of in vitro antimicrobial tests, compounds (1) and (4) showed activity against Klebsiella oxytoca with minimum inhibitory concentration values of 25 and 50 µg mL− 1, respectively. Based on the in vitro bioactivity shown by compounds (1) and (4), a structure-activity relationship (SAR) study indicates that the presence of a hydroxyl group at C-3` or methoxy at C-4` increases the activity against Klebsiella oxytoca while the presence of hydroxyl group at C-7 position decreases the activity. Also, a structure-based drug development approach was applied using several in silico tools including molecular docking, structure-based pharmacophore modelling, and molecular dynamics simulation. A molecular docking study showed a high affinity of these two compounds for the DNA gyrase protein, which is considered one of the target proteins that flavones act on to exhibit antibacterial activity. It is worth noting that compound (4) showed a higher binding affinity than the standard (quercetin) which is known to have antibacterial activity by inhibiting the DNA gyrase. In addition to that, the structure-based pharmacophores of compound (4) and quercetin showed similar pharmacophoric features and the same types of interactions. Based on our findings, compounds (1) and (4) are promising for further study as potential antimicrobial phytochemicals that can have a role in controlling bovine mastitis alone or in combination with other antimicrobial phytochemicals, as well as to investigate their mechanism of action further.
Data availability
No datasets were generated or analysed during the current study.
References
Abd El-Hack M, Alagawany M, Farag MR et al (2016) Nutritional; healthical and therapeutic efficacy of black cumin (Nigella sativa) in animals; poultry and humans. Int J Pharmacol. https://doi.org/10.3923/ijp.2016.232.248
Aitken SL, Corl CM, Sordillo LM (2011) Immunopathology of Mastitis: insights into Disease Recognition and Resolution. J Mammary Gland Biol Neoplasia. https://doi.org/10.1007/s10911-011-9230-4
Amawi H, Ashby CR, Tiwari AK (2017) Cancer chemoprevention through dietary flavonoids: what’s limiting? Chin J Cancer. https://doi.org/10.1186/s40880-017-0217-4
Azooz MF, El-Wakeel SA, Yousef HM (2020) Financial and economic analyses of the impact of cattle Mastitis on the profitability of Egyptian dairy farms. Vet World. https://doi.org/10.14202/vetworld.2020.1750-1759
Balouiri M, Sadiki M, Ibnsouda SK (2016) Methods for in vitro evaluating antimicrobial activity: a review. J Pharm Anal https://doi.org/10.1016%2Fj.jpha.2015.11.005
Barreiros Y, de Meneses AC, Alves JLF et al (2022) Xanthan gum-based film-forming suspension containing essential oils: production and in vitro antimicrobial activity evaluation against mastitis-causing microorganisms. https://doi.org/10.1016/j.lwt.2021.112470. LWT
Bax B, Chan P, Eggleston D et al (2010) Type IIA topoisomerase inhibition by a new class of antibacterial agents. Nat. https://doi.org/10.1038/nature09197
Chirumbolo S (2010) The role of Quercetin, Flavonols and flavones in modulating inflammatory cell function. Inflamm Allergy Drug Targets. https://doi.org/10.2174/187152810793358741
Collin F, Karkare S, Maxwell A (2011) Exploiting bacterial DNA gyrase as a drug target: current state and perspectives. Appl Microbiol Biotechnol. https://doi.org/10.1007/s00253-011-3557-z
Cushnie TP, Lamb AJ (2005) Antimicrobial activity of flavonoids. Int J Antimicrob Agents https://doi.org/10.1016%2Fj.ijantimicag.2005.09.002
De los Santos R, González-Revello Á, Majul L et al (2022) Sub-clinical Bovine Mastitis Associated with Staphylococcus spp. in Eleven Uruguayan dairy farms. J Infect Dev Ctries. https://doi.org/10.3855/jidc.12960
Dixon RA (2001) Natural products and plant disease resistance. Nat. https://doi.org/10.1038/35081178
Eastman P, Swails J, Chodera JD et al (2017) OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comp Biol. https://doi.org/10.1371/journal.pcbi.1005659
Eberhardt J, Santos-Martins D, Tillack AF et al (2021) AutoDock Vina 1.2.0: new docking methods, expanded force field, and Python Binding. Chem Inf Model. https://doi.org/10.1021/acs.jcim.1c00203
Echeverría J, Opazo J, Mendoza L et al (2017 structure-activity and Lipophilicity relationships of selected Antibacterial Natural flavones and flavanones of Chilean Flora. Mol. https://doi.org/10.3390/molecules22040608
Faller MA, Sheehan DJ, Rex JH (2004) Determination of fungicidal activities against yeasts and molds: lessons learned from bactericidal testing and the need for standardization. Clin Microbiol Rev. https://doi.org/10.1128/cmr.17.2.268-280.2004
Gomes F, Martins N, Ferreira ICFR et al (2019) Anti-biofilm activity of hydromethanolic plant extracts against Staphylococcus aureus isolates from bovine mastitis. Heliyon. https://doi.org/10.1016/j.heliyon.2019.e01728
Guha R (2013) On exploring structure-activity relationships. Methods Mol Biol. https://doi.org/10.1007/978-1-62703-342-8_6
Gutiérrez-Reinoso MA, Uquilla JB, Guamaní JL et al (2023) Intramammary infusion of Micronised purified Flavonoid Fraction (MPFF) in Mastitis-diagnosed dairy cows naturally infected by Staphylococcus spp. in the late lactation. Vet Sci. https://doi.org/10.3390/vetsci10050335
Hillerton JE, Berry EA (2005) Treating mastitis in the cow- a tradition or an archaism. J Appl Microbiol. https://doi.org/10.1111/j.1365-2672.2005.02649.x
Hogeveen H, Van Der Voort M (2017) Assessing the economic impact of an endemic disease: the case of Mastitis. Rev Sci Tech. https://doi.org/10.20506/rst.36.1.2623
Hogeveen H, Huijps K, Lam TJ (2011) Economic aspect of mastitis: new developments. N Z Vet J. https://doi.org/10.1080/00480169.2011.547165
Hossion AM, Zamami Y, Kandahary RK et al (2011) Quercetin diacylglycoside analogues showing dual inhibition of DNA gyrase and topoisomerase IV as novel antibacterial agents. J Med Chem. https://doi.org/10.1021/jm200010x
Hostetler GL, Ralston RA, Schwartz SL (2017) Flavones: food sources, bioavailability, metabolism, and Bioactivity. Adv Nutr https://doi.org/10.3945%2Fan.116.012948
Kamaruzzaman NF, Chong SQY, Edmondson-Brown KM (2017) Bactericidal and Anti-biofilm effects of Polyhexamethylene Biguanide in models of Intracellular and Biofilm of Staphylococcus aureus isolated from bovine mastitis. Front Microbiol. https://doi.org/10.3389/fmicb.2017.01518
Kikusato M (2021) Phytobiotics to improve health and production of broiler chickens: functions beyond the antioxidant activity. Anim Biosci. https://doi.org/10.5713/ab.20.0842
Lee SJ, Son KH, Chang HW et al (1993) Antiinflammatory activity of naturally occurring flavone and flavonol glycosides. Arch Pharm Res. https://doi.org/10.1007/BF02974123
Macías Alonso M, Salazar J, Osegueda S et al (2019) In vitro antimicrobial activity of Mexican plants on bovine mastitis bacteria: preliminary studies. https://doi.org/10.14393/BJ-v36n1a2020-42137. Biosci
Martens S, Mithöfer A (2005) Flavones and flavone synthases. https://doi.org/10.1016/j.phytochem.2005.07.013. Phytochem
Mishra A, Sharma AK, Kumar S et al (2013) Bauhinia variegata leaf extracts exhibit considerable anti-bacterial, antioxidant, and anticancer activities. Biomed Res Int. https://doi.org/10.1155/2013/915436
Morris GM, Lim-Wilby M (2008) Molecular docking. Methods Mol Biol. https://doi.org/10.1007/978-1-59745-177-2_19
Morrissey JP, Osbourn AE (1999) Fungal resistance to plant antibiotics as a mechanism of pathogenesis. Microbiol Mol Biol Rev https://doi.org/10.1128%2Fmmbr.63.3.708-724.1999
Nguyen TLA, Bhattacharya D (2022) Antimicrobial Activity of Quercetin: An Approach to Its Mechanistic Principle. Mol. https://doi.org/10.3390/molecules27082494
Ohemeng KA, Schwender CF, Fu KP et al (1993) DNA gyrase inhibitory and antibacterial activity of some flavones. Bioorg Med Chem Lett. https://doi.org/10.1016/S0960-894X(01)80881-7
Olagaray KE, Bradford BJ (2019) Plant flavonoids to improve productivity of ruminants- A review. https://doi.org/10.1016/j.anifeedsci.2019.02.004. AFST
Ou-Yang S, Lu J, Kong X et al (2012) Computational drug discovery. Acta Pharmacol Sin. https://doi.org/10.1038/aps.2012.109
Palermo G, De Vivo M (2015) Computational Chemistry for Drug Discovery. Encyclopedia of Nanotechnology. Springer, Dordrecht. https://doi.org/10.1007/978-94-007-6178-0_100975-1
Patel MM, Patel LJ (2014) Design, synthesis, molecular docking, and antibacterial evaluation of some novel flouroquinolone derivatives as potent antibacterial agent. Sci World. https://doi.org/10.1155/2014/897187
Plaper A, Golob M, Hafner I et al (2003) Characterization of quercetin binding site on DNA gyrase. Biochem Biophys Res Commun. https://doi.org/10.1016/s0006-291x(03)01006-4
Preet G, Haj Hasan A, Ramlagan P et al (2023) Anti-Neurodegenerating Activity: Structure-Activity Relationship Analysis of Flavonoids. Mol. https://doi.org/10.3390/molecules28207188
Purgato GA, Lima S, Baeta JVPB et al (2021) Salvinia auriculata: chemical profile and biological activity against Staphylococcus aureus isolated from bovine mastitis. Braz J Microbiol. https://doi.org/10.1007/s42770-021-00595-z
Romero J, Benavides E, Meza C (2018) Assessing Financial impacts of subclinical mastitis on Colombian dairy farms. Front Vet Sci. https://doi.org/10.3389/fvets.2018.00273
Sadybekov AV, Katritch V (2023) Computational approaches streamlining drug discovery. Nat. https://doi.org/10.1038/s41586-023-05905-z
Salmanli M, Yilmaz GT, Tuzuner T (2021) Investigation of the antimicrobial activities of various antimicrobial agents on Streptococcus Mutans Sortase A through computer-aided drug design (CADD) approaches. Comput. https://doi.org/10.1016/j.cmpb.2021.106454. Methods Programs Biomed
Schabauer A, Zutz C, Lung B et al (2018) Gentisaldehyde and its derivative 2,3-Dihydroxybenzaldehyde show antimicrobial activities against bovine mastitis Staphylococcus aureus. Front Vet Sci. https://doi.org/10.3389/fvets.2018.00148
Seidel T, Schuetz DA, Garon A et al (2019) The Pharmacophore Concept and its applications in computer-aided Drug Design. https://doi.org/10.1007/978-3-030-14632-0_4. Prog Chem Org Nat Prod
Shamsudin NF, Ahmed QU, Mahmood S et al (2022) Antibacterial effects of flavonoids and their structure-activity relationship study: a comparative interpretation. https://doi.org/10.3390%2Fmolecules27041149. Mol
Shen N, Wang T, Gan Q et al (2022) Plant flavonoids: classification, distribution, biosynthesis, and antioxidant activity. Food Chem. https://doi.org/10.1016/j.foodchem.2022.132531
Silva ATF, Gonçalves JL, Dantas STA et al (2023) Genetic and phenotypic characterization of subclinical mastitis-causing Multidrug-Resistant Staphylococcus aureus. https://doi.org/10.3390/antibiotics12091353. Antibiot
Sliwoski G, Kothiwale S, Meiler J et al (2013) Computational methods in drug discovery. Pharmacol Rev https://doi.org/10.1124%2Fpr.112.007336
Takó M, Kerekes EB, Zambrano C et al (2020) Plant phenolics and phenolic-enriched extracts as Antimicrobial agents against Food-contaminating microorganisms. https://doi.org/10.3390/antiox9020165. Antioxidants
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimisation and multithreading. J Comput Chem https://doi.org/10.1002%2Fjcc.21334
Wang J, Qu Q, Liu X et al (2020) 1-Hydroxyanthraquinone exhibited antibacterial activity by regulating glutamine synthetase of Staphylococcus xylosus as a virulence factor. Biomed Pharmacother. https://doi.org/10.1016/j.biopha.2019.109779
Wermuth CG, Ganellin CR, Lindberg P (1988) Glossary of terms used in medicinal chemistry (IUPAC recommendations 1998). Pure Appl Chem. https://doi.org/10.1351/pac199870051129
Wolber G, Thierry L (2005) LigandScout: 3-D pharmacophores derived from protein-bound ligands and their use as virtual screening filters. J Chem Inf Model. https://doi.org/10.1021/ci049885e
Xie Y, Yang W, Tang F et al (2015) Antibacterial activities of flavonoids: structure-activity relationship and mechanism. Curr Med Chem. https://doi.org/10.2174/0929867321666140916113443
Yang Z, Zhou E, Wei D et al (2014) Emodin inhibits LPS-induced inflammatory response by activating PPAR-γ in mouse mammary epithelial cells. Int Immunopharmacol. https://doi.org/10.1016/j.intimp.2014.05.019
Acknowledgements
We give sincere thanks to the late Professor R. H. Thomson for donating the pure compounds used in the study to the compound library of the Marine Biodiscovery Centre. Also, we thank Toka Mahmoud, Yasmine Madi, and Julius Tibyangye (Marine Biodiscovery Centre, Department of Chemistry, University of Aberdeen, UK) for their constructive suggestions. A sincere thanks to P.S. Oberoi (I.C.A.R, N.D.R.I., India) for their support.
Funding
This research was funded by the Deanship of Research at Jordan University of Science and Technology (JUST).
Author information
Authors and Affiliations
Contributions
A.H.: project conceptualisation, project protocol, method validation, investigation, funding acquisition, conducting the antimicrobial tests, using software, data analysis, writing the original draft, manuscript revision and proofreading.G.P.: project conceptualisation, project protocol, method validation, manuscript revision and proofreading.R.V.A., H.A.: figures generation, manuscript revision and proofreading.E.O.: project protocol, manuscript revision and proofreading.R.E.: supervision, project administration, manuscript revision and proofreading.M.J.: method validation, supervision, project administration, manuscript revision and proofreading.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publish
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Haj Hasan, A., Preet, G., Astakala, R.V. et al. Antibacterial activity of natural flavones against bovine mastitis pathogens: in vitro, SAR analysis, and computational study. In Silico Pharmacol. 12, 78 (2024). https://doi.org/10.1007/s40203-024-00253-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-024-00253-w