
Abstract
A 336-base pair (bp) sized mRNA sequence encoding 111 amino acid size crustin isoform (MC-crustin) was obtained from the gill sample of the green mud crab, Scylla serrata. MC-crustin possessed an N-terminal signal peptide region comprising of 21 amino acid residues, followed by a 90 amino acid mature peptide region having a molecular weight of 10.164 kDa, charge + 4.25 and theoretical pI of 8.27. Sequence alignment and phylogenetic tree analyses revealed the peptide to be a Type I crustin, with four conserved cysteine residues forming the cysteine rich region, followed by WAP domain. MC-crustin was cationic with cysteine/proline rich structure and was predicted with antimicrobial, anti-inflammatory, anti-angiogenic and anti-hypertensive property making it a potential molecule for possible therapeutic applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epidemics and pandemics in recent years have proven that public health is strongly under global threat and therefore new and effective drugs in combating these emerging infectious diseases is an urgent requirement. World Health Organization (WHO) reports antibiotic resistance as a serious threat mounting worldwide and hence cautions a period in near future in which infections can no longer be mitigated with the antibiotics currently in use (Xie et al. 2017). The same scenario is being faced not only in human health sector, but also in animal husbandry and aquaculture (Schar et al. 2021). Drug-resistant Vibrio parahaemolyticus strains have been reported in China and South Korea (Loo et al. 2020). Over the last two decades, naturally evolved molecules, the Antimicrobial peptides (AMPs) have been recognized as promising antibiotic-alternatives capable of overcoming antibiotic resistance (Erdem Büyükkiraz and Kesmen 2022). About 40 antimicrobial peptides are presently undergoing clinical trials among which nisin, gramicidin, polymyxins, daptomycin and melittin are in clinical use (Dijksteel et al. 2021).
Marine invertebrates present a wide repertoire of different mechanisms and biomolecules, which enables them to survive in their natural environment as they are directly exposed to microorganisms. As these organisms lack immunological memory, they mainly depend on the innate immune system, comprising humoral and cellular responses (Tincu and Taylor 2004). Among marine invertebrates, crustaceans are the largest and most diverse animal group, abundant and spread in all aquatic habitats, from hypersaline to freshwater ecosystems. As they are constantly bathed in an environment with frequent exposure to both commensals as well as opportunistic pathogens, crustaceans defend themselves by a combination of nonadaptive antimicrobial and antiviral responses guided by haemocytes, the circulating immunocompetent cells (Kulkarni et al. 2021) as well as epithelial cells lining different organs, such as gills and intestine (Silveira et al. 2018; Alenton et al. 2019). It is well known that crustacean AMPs are constitutively synthesised and stored in cytoplasmic granules of specific haemocyte populations (Destoumieux et al. 2000; Vu et al. 2018). Since the discovery of the first crustacean AMPs (Schnapp et al. 1996; Destoumieux et al. 1997), several genetically-encoded AMP families and other antimicrobial-related molecules have been known and characterized at molecular level, especially in decapods due to their commercial importance. Till date, 12 gene-encoded AMP families are recognised in crustaceans i.e., crustins, anti-lipopolysaccharide factors (including crab scygonadins), penaeidins, stylicins, proline-rich AMPs (including the 6.5 kDa bac-like peptide), glycine-rich AMPs, arasins (including the callinectin peptide), hyastatins, astacidins, panusins (including the homarin peptide), paralithocins and armadillidins (Matos and Rosa 2022).
Chisholm and Smith (1992) identified certain antibacterial factors in granular haemocytes of shore crab, Carcinus maenas and Relf et al. (1999) isolated the bioactive peptide, crustin from the granular haemocytes of C. maenas, showing activity against Gram-positive marine bacteria. This was named ‘carcinin’ (Smith and Chisholm 2001) which became the prototype for the later identified group of similar peptides, ‘crustins’, from other crustaceans i.e., Litopenaeus vannamei and L. setiferus (Bartlett et al. 2002). Since then, several crustin like sequences have been reported from various crustaceans such as Penaeus subtilis (Rosa et al. 2007), Hyas araneus and Paralithodes camtschaticus (Sperstad et al. 2009), Fenneropenaeus indicus (Antony et al. 2010; Sruthy et al. 2017), P. monodon (Antony et al. 2011), Scylla serrata (Afsal et al. 2011), Macrobrachium rosenbergii (Huang et al. 2016) and Rimicaris sp. (Wang et al. 2021a, b). Crustins are 7–22 kDa antibacterial proteins in the crustacean hemolymph with the WAP domain containing protein superfamily (Smith et al. 2008; Wang et al. 2021a, b). They have a distinctive signature of 12 cysteine residues out of which four form the cysteine-rich region, while the remaining eight participate in four disulphide bonds, recognized as the 4-disulphide core domain/whey acidic protein domain (4DSC/WAP). The central cysteine of the 4DSC domain (C7–C11 of the crustin signature) seems to be important to maintain the structure (Vargas-Albores and Martínez-Porchas 2017). This three-dimensional arrangement is also found in other proteins displaying different biological functions i.e., antimicrobial activity, proteinase inhibition (Ota et al. 2002) and tissue differentiation (Ranganathan et al. 1999). Contradictory to the conservative C-terminal WAP domain, the N-terminal region of crustins is variable, based on which crustins were classified i.e., Types I–III (Smith et al. 2008). Currently, this classification system also includes proteins containing two WAP domains exhibiting antimicrobial activity (Type IV or double WAP domain-containing proteins) and crustins from hymenopteran insects (Type V) (Matos and Rosa 2022). The present study focuses on molecular and functional characterization of a novel crustin isoform (MC-Crustin) from the gill of the mud crab, Scylla serrata using in silico approach. Scylla serrata mainly inhabits estuaries, mangrove swamps and sheltered coastal habitats and are usually found in muddy bottom. This species is considered as one of the most popular seafood item in South-East Asian countries due to its delicious nature and high demand in international markets (Chandra 2012; Pripanapong and Tongdee 1998). Mud crab farming/fattening has been very attractive and thereby the health management becomes important, necessitating an understanding of their defence potential. In this scenario, a preliminary prediction of properties and potential of MC-crustin was carried out to unravel one of the most important component of its innate immunity, the antimicrobial peptides that confer protection to the animals.
Materials and methods
Sample collection
Mud-crab Scylla serrata (Fig. 1) was collected from a local fish market and transported to lab live. Gills of the crab were excised and macerated thoroughly in TRI Reagent® (Sigma-Aldrich) and preserved at − 20 °C in a Freezer (Sanyo, Japan) prior to RNA isolation.

Green mud-crab, Scylla serrata used in the study
Total RNA isolation and cDNA synthesis
Total RNA was isolated using TRI Reagent® (Sigma-Aldrich) as per manufacturer’s protocol and dissolved in DEPC treated water. After confirming purity and quality of RNA in a 0.8% agarose gel, single stranded cDNA was synthesized. For this, only RNA that showed A260:A280 ≥ 1.8 was used. First strand cDNA was generated using 5 μg total RNA, 1 × RT buffer, 2 mM dNTP, 2 mM oligo d(T20), 20U of RNase inhibitor, and 100U of MMLV reverse transcriptase. cDNA synthesis was performed at 42 °C for 1 h followed by an inactivation step at 85 °C for 15 min. Quality of cDNA was tested by PCR amplification of internal control β-actin, primed by crustacean specific β-actin primers (F—5′ CTTGTGGTTGACAATGGCTCCG 3′ and R—5′ TGGTGAAGGAGTAGCCACGCTC 3′).
PCR amplification, TA cloning and sequencing
Crustin was amplified from the S. serrata gill cDNA using crustinF and crustinR primers (crustinF—5′ GAGAGCAGAATTAGACACTGT 3′, crustinR—5′ ATATAGTATAACATAACCATACTTC 3′) designed by Afsal et al. (2013) based on consensus sequence regions of crustins from GenBank. The reaction mixture which included EmeraldAmp® PCR Master Mix, template, primers and water was subjected to 95 °C for 2 min followed by 35 cycles of 94 °C for 15 s, 57 °C for 30 s, and 72 °C for 30 s and a final extension at 72 °C for 10 min. The PCR amplicons were then ligated to pGEM®T Easy vector (Promega) and transformed to DH5α E. coli competent cells, using manufacturer’s protocol. Transformed clones were cultured in LB broth (500 μl), at 37 °C with continuous shaking at 150 rpm for 1 h 30 min and subsequently plated onto LB agar plates containing Ampicillin (100 μg ml−1), X-gal (80 μg ml−1) and IPTG (100 mM). Positive colonies were identified by blue/white screening and the colonies that remained white due to insertional inactivation of the alpha-subunit of beta-galactosidase were selected and cultured overnight in LB broth containing antibiotic (Ampicillin 100 μg ml−1) with continuous shaking at 200 rpm and 37 °C. Plasmids were isolated with GenElute™ Plasmid Miniprep Kit (Sigma), followed by quality confirmation on a 0.8% agarose gel. The recombinant plasmids were subjected to PCR amplification using vector-specific and gene-specific primers. The plasmid was sequenced using T7F and SP6R primers on an ABI 3730XL DNA sequencer (Applied Biosystem, USA) at AgriGenome Sequencing Facility, India.
Sequence analysis and molecular property prediction
The sequence was subjected to BLAST search at NCBI (https://blast.ncbi.nlm.nih.gov/) and the peptide was identified based on similarity score and the sequence was submitted to NCBI BankIt (https://www.ncbi.nlm.nih.gov/WebSub/). ExPASy translate tool (https://web.expasy.org/translate/) was used to deduce amino acid sequence from the nucleotide mRNA sequence. Signal peptide and propeptide sites were denoted by SignalP 5.0 (http://www.cbs. dtu.dk/services/SignalP/) and ProP 1.0 (http://www.cbs. dtu.dk/services/ProP/) (Armenteros et al. 2019; Duckert et al. 2004). Presence of specific peptide domain was predicted using SMART (http://smart.emblheidelberg.de/) (Letunic and Bork 2018). ScanProsite (https://prosite.expasy.org/scanprosite/) provided the cysteine disulphide bonds in the identified domain (Castro et al. 2006). APD3 (http://aps.unmc.edu/AP/main.php) and ProtPram tool (https://web.expasy.org/protparam/) of ExPASy were used to compute various physico-chemical characteristics of the peptide (Wang et al. 2016; Gasteiger et al. 2005). Dipeptide compositions of the peptide were analysed by Pfeature (https://webs.iiitd.edu.in/raghava/pfeature/) (Pande et al. 2019). Hydropathy plot was computed using Kyte and Doolittle by ExPASy ProtScale (https://web.expasy.org/protscale/). Jalview Version: 2.11.1.0 and Mega X applications were used for clustal alignment and phylogenetic analysis respectively (Waterhouse et al. 2009; Kumar et al. 2018). Secondary structure was analyzed in RaptorX (Wang et al. 2010). 3D structure for the peptide was predicted from SWISS-MODEL workspace, Expasy (https://www.expasy.org/resources/swiss-model) and the PDB data generated was visualized in DeepView/Swiss-PdbViewer 4.1.0 (http://www.expasy.org/spdbv/) to compute disulphide bonds, distribution of cationic and hydrophobic residues as well as to predict the electrostatic potential distribution in the 3D model of the peptide (Waterhouse et al. 2018; Guex and Peitsch 1997). PDBsum (http://www.ebi.ac.uk/thornton-srv/databases/pdbsum/Generate.html) provided further structural analyses and Ramachandran plot for the predicted model (Laskowski and Thornton 2022). TANGO (http://tango.crg.es/) and AGGRESCAN (http://bioinf.uab.es/aggrescan/) were used for in vitro and in vivo aggregation analyses (Rousseau et al. 2006; Fernandez-Escamilla et al. 2004; Linding et al. 2004; Conchillo-Solé et al 2007). FoldAmyloid (http://bioinfo.protres.ru/fold-amyloid/) predicted amyloidogenic regions in the peptide (Garbuzynskiy et al. 2010).
Bioactive potential prediction
Online platforms used for prediction of various bioactive potentials of MC-crustin is given in Table 1.
Results
A 500 base pair (bp) amplicon was obtained using crustinF and crustinR primers from the cDNA of gill sample of the green mud crab, Scylla serrata (Fig. 2.).

500 bp amplicon using crustinF and crustinR primers. Lane 1–100 bp ladder, Lane 2–500 bp amplification
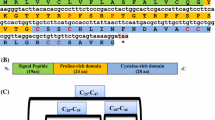
The sequence analysis of the cloned amplicon revealed it to contain an open reading frame of 336 base pair (bp), encoding 111 amino acid (aa) residue crustin peptide (NCBI Accession number ON513450). With 100% query coverage, the peptide displayed varying range of identities (98.2–73.87%) with previously reported crustins from crabs viz., Scylla olivacea (QJW82625.1) 98.20%, Scylla tranquebarica (AFI56572.1) 98.20%, Scylla serrata (ADW11096.1) 94.59%, Portunus pelagicus (AFN37210.1) 89.19%, Scylla paramamosain (ABY20727.1, ABY20728.1) 90.9% and Portunus trituberculatus (ACM89167.2) 73.87%. A signal peptide cleavage site processed by signal peptidase 1 (Sec/SPI) was predicted between positions Ala21 and Ser22 with N-region Met1–Lys4, H-region Ile5–Val16 and C-region Ala17–Ala21. There were no propeptide cleavage sites identified in the peptide. A transmembrane helix region was predicted from region Ile5–Tyr27. SMART analysis annotated a 4DSC (4-disulphide core)/WAP (Whey Acidic Protein SM000217) domain from His60–Glu109 (Fig. 3).

cDNA sequence (336 bp) (GenBank Accession number-ON513450) encoding crustin antimicrobial peptide (111aa) from the green mud crab Scylla serrata. The translated peptide sequence is shown in colour. The Italicised amino acids form the signal peptide comprising the N-region (yellow), H-region (blue) and C-region (green) with signal peptidase cleavage site predicted at A21–S22 (arrow mark). The remaining sequence comprise of the mature peptide (grey) of MC-crustin within which the SMART annotated WAP/4DSC domain is shown in bold. ‘*’ denotes stop codon. Region forecasted as transmembrane helix is underlined
Physico-chemical properties and domain annotation
Molecular weight of the 90 aa, cysteine rich (13%), mature peptide region (MC-crustin) was 10.164 kDa with a theoretical pI of 8.27. Net charge of the peptide accounted to be + 4.25 with 12 (Arginine, R + Lysine, K) positively charged residues and 9 (Aspartate, D) + Glutamate, E) negatively charged residues. Amino acid composition is given in Fig. 4, showing the peptide to be cysteine (C) rich (13%) followed by proline (P) (11%). GRAVY (Grand average hydropathy value) was predicted as 0.45, Wimley–White whole-residue hydrophobicity of the peptide, 17.18 kcal/mol. and the APD defined total hydrophobic ratio as 36%. The ExPasy hydropathy plot also denoted major portion of the peptide falling in the hydrophilic region (Fig. 5). Boman index of the mature peptide was 1.79 kcal/mol. The estimated half-life of peptide was 1.9 h (mammalian reticulocytes, in vitro), > 20 h (yeast, in vivo) and > 10 h (Escherichia coli, in vivo). The instability index (II) of MC-crustin mature peptide was 56.10% which was higher than the instability index of the full-length peptide including the signal peptide, which had the II value of 48%.

Amino acid composition of MC-Crustin from Scylla serrata (calculated by APD3 server http://aps.unmc.edu/AP/main.php)

ExPasy plotted hydropathy plot for mature peptide MC-crustin using the Kyte and Doolittle
MC-crustin is a Type I crustin, observed with four conserved cysteine residues in the region between the signal peptide and SMART annotated C-terminus WAP domain. MC-crustin has a higher percentage of proline (P) residues (16%) within this region. The WAP motif alone comprises of 50 amino acids with eight cysteine residues, following the conserved arrangement across different types of crustins as well as other WAP domain containing proteins, displaying four disulphide bridges, referred to as the four-disulphide core domain (4DSC). ScanProsite predicted disulphide bonding pattern in WAP domain as C1–C6, C2–C7, C3–C5 and C4–C8. The arrangement of eight cysteines of the WAP domain and the other four cysteine residues outside to it displayed the ‘crustin signature domain’ for MC-crustin as in other crustins (except SWD Type III crustins) which have only WAP domain.
Sequence alignment and phylogenetic analysis
Alignment of crustins revealed the signal sequence of MC-crustin to be Valine (V) rich. However, the signal peptide sequences vary across species and types of crustins, with lengths varying from 16 to 24 aa. The region between the WAP domain and signal peptide marks the types of crustins. Type I crustins including MC-crustin in the present study has a cysteine (11%) rich region with four conserved cysteine residues. Type II crustins comprise of an additional glycine (G) rich region prior to the cysteine rich region. Type III crustins do not contain any sequences between the signal and WAP domain. However, the WAP domains, even though variable, displayed certain conserved patterns. Generally, in WAP proteins, the WAP domain is marked by the beginning of KXGXCP motif. However, MC-crustin has HXGXCP motif instead. A Proline residue was found to be conserved adjacent to C1 residue of WAP domain. The location of aspartic acid and lysine residues across all crustins and WAP domain containing proteins was highly conserved, following the pattern of C3XXDXXC4XXXXKC5C6. Another observation was that among the crustins, adjacent to the second cysteine residue, no methionine residue was observed as was seen for the WAP domains of Elafin and SLPI (Fig. 6).

MUSCLE alignment of crustin peptides using default parameters of the Jalview version 2 workbench. MC-crustin was aligned against crustin sequences of crustaceans from the NCBI database. The WAP domain of Elafin and SLPI was also included in the alignment. Percentage identity colour scheme of Jalview was applied in the alignment
Phylogenetic analysis revealed that Type I, Type II and Type V crustins are forming a monophyletic group and the Type III and Type IV crustins another group. MC-crustin was well nested within the Type I crustins and was grouped with the Scylla crustins. The Type I crustins were observed to have a common ancestor that included the carcinins (from Carcinus maenas). The monophyletic group consisting of carcinins were observed to contain Type I crustin sequences from crabs, lobsters and crayfish (Fig. 7).

Evolutionary tree of Crustins inferred using the Neighbor-Joining method. Percentage of replicate trees in which the associated taxa clustered together in the bootstrap test (1000 replicates) are shown next to the branches. All ambiguous positions were removed for each sequence pair (pairwise deletion option)
Secondary structure analysis
Secondary structure prediction by RaptorX for MC-crustin showed coil regions predominating (98%) and presence of strand (1%) within the sequence (Fig. 8). TANGO did not predict any beta aggregation regions. However, AGGRESCAN predicted aggregation prone segments for the peptide (Fig. 9). Similarly, amyloidogenic regions of 21–25 and 73–77 were predicted by amyloid tool (Fig. 10).

Distribution of the three state secondary structure class of MC-Crustin predicted by RaptorX

a Graph demonstrating the aggregation tendency profile of the sequence in the MC-crustin mature peptide. b Graph portraying exclusively the area employed in a hot spot (normalized by the peptide’s length). Regions 23–26 and 84–90 of the mature peptide are foreseen as potential aggregation regions

FoldAmyloid predicted amyloidogenic regions in MC-crustin: A21L22Y23C24C25 and C73C74Y75D76A77
Tertiary structure analysis
SWISS-MODEL user template-based prediction provided a 3D structure for the WAP domain of MC-crustin which was modelled based on Elafin [PDB ID:1FLE (chain B)] (Fig. 11a). Pdbsum analysis revealed the structure with coil, helix and strand conformations (Fig. 11b). Ramachandran plot analysis showed more than 90% residues within the most favoured and additional allowed regions (Fig. 11c). Even though the mature peptide full length was provided as sequence input, the output predicted structure was only available for the WAP domain region of Histidine 39 (H39)–Valine 87 (V87). PDBsum structural details revealed that four strands formed the polypeptide chain fold with external two strands (strand 1 and strand 2) and internal core strands (β1 and β2) forming beta-sheets. Strand 1 is connected to strand 2 via a larger loop and strand 2 is connected to the internal beta-strand 1 (β1) by a helical region (D64, G65, A66) (Fig. 11d) and a γ-turn like segment (F69, R70, S71) (Fig. 11f) leading to the beta-strand 1. β1 then takes a γ-turn (D76, A77, C78) and a β-hairpin (D76, A77, C78, V79, E80, H81) (Fig. 11e) to position the second beta strand forming the anti-parallel beta sheet. The overall structure is further stabilized by the disulphide bonds of cysteines C1–C6, C2–C7 and C3–C5 connecting all the four strands with one another and the fourth disulphide bond C4–C8 that connects the first and second loops. The disulphide bonds, distribution of cationic and hydrophobic amino acid residues and the electrostatic potential distribution were predicted by Swiss-PDB viewer (Figs. 11 and 12). The loop region prior to WAP domain was observed to have a greater positive charge distribution (Fig. 12b).

a Tertiary structure of WAP domain of MC-crustin based on Elafin chain B (1FLE) template predicted by SWISS-model. The structure was viewed and disulphide bonds were predicted in Swiss-Pdb viewer version 4. b Ramachandran plot for predicted structure. c Wiring diagram predicted by PDBsum showing MC-crustin’s secondary structural elements (alpha-helices and beta-sheets) together with various structural motifs such as d helices, e beta-hairpins and f gamma-turns

a Swiss-PDB viewer prediction of the cationic (blue) and hydrophobic residues (green). b Electrostatic distribution denoting positive charged regions (blue) and the negatively charged regions (red)
Therapeutic potential
The therapeutic potentials of MC-crustin were predicted using several online prediction tools and the prediction scores above the threshold were considered as positive and below as negative (Table 2). MC-crustin was predicted to be a bioactive peptide by Peptide Ranker with a score of 0.986. iAMP was employed to predict three classes of AMPs as anti-bacterial, anti-fungal and anti-viral. MC-crustin was predicted to be anti-bacterial, anti-fungal as well as anti-viral. The anti-viral property was further predicted by FIRM-AVP to be positive. Pro-inflammatory peptides are a class of peptides that can be used as therapeutic candidates to treat and alleviate several disorders. PIP-EL predicted MC-crustin to have pro-inflammatory property. Chronic inflammation and auto-immune diseases develop when inflammatory responses continue after they have completed their normal function. MC-crustin was predicted as anti-inflammatory by Pre-AIP as well as AIP-Pred tools. PredAPP predicted the peptide to have anti-parasitic potentials with a prediction potential of 0.62. Anti-angiogenic peptides regulate angiogenesis and tumour growth. MC-crustin was predicted to have anti-angiogenic property with a prediction value of 1.48. The peptide was also predicted with anti-hypertensive property by the AntiangioPred tool, and the WAP domain seemed to have higher prediction score of 0.79. The peptide was predicted as non- cell penetrating type and non-haemolytic.
Discussion
For decades, the unregulated use of antibiotics for therapeutic purposes and as prophylactics in agriculture and animal husbandry has led to the emergence of antimicrobial resistance (AMR) in an alarming rate across the globe. Search for alternatives to antibiotics are on the run to tackle this crisis (Iskandar et al. 2022). In this context, the potentials of naturally evolved antimicrobial peptides, present in all groups of organisms, right from bacteria to the plants, humans, etc. are being explored widely as lead molecules for management of various diseases (Erdem Büyükkiraz and Kesmen 2022). Marine organisms are exposed to a plethora of pathogens and hence have an efficient first line of defence against the invading organisms (Wu et al. 2021). Marine invertebrates defend themselves against pathogens mainly through innate immunity. With respect to crustacean immunity, there is a well-played orchestra of immunity factors such as agglutination, prophenoloxidase system, encapsulation, melanization and production of antimicrobial peptides to deal with the invaders (Cerenius et al. 2010).
Crustin in the present study (MC-crustin) was obtained from the gill transcripts of Scylla serrata, and was predicted to have 10.164 kDa with a theoretical pI of 8.27. In general, crustins range from 6 to 22 kDa, with pI values in the range 4–8.5 (Smith et al. 2008). A medium value of predicted Boman index denotes that MC-crustin may not be involved in protein binding like the receptor binding activity (Boman 2003). The negative GRAVY value denotes the peptide to have relatively lower hydrophobicity. This is further supported by the lower hydrophobicity ratio for MC-crustin. Charge of the peptide is positive with R and K contributing to the greater positive charge. Thus, the cationicity and electrostatic potential may be a driving force compared to the hydrophobicity for the peptide’s initial interaction with bacterial membranes (Moravej et al. 2018; López Cascales et al. 2018). The primary contact of peptides with pathogenic surfaces involves biochemical or biophysical affinity carried out via hydrophobic or electrostatic interactions (Moravej et al. 2018). Higher proline composition (11%) of MC-crustin is a notable feature as the importance of proline residues in antimicrobial activities have been well documented. Introduction of positive charges and proline residues in F5W-magainin resulted in increased therapeutic potential (Azuma et al. 2020). The proline residue in maculatin 1.1 facilitated membrane insertion via lipid disordering (Fernandez et al. 2013). A higher instability index of the MC-crustin mature peptide than the full-length peptide denotes the signal cleaved mature peptide as less stable than the precursor. The limited stability and short half-life of few hours of the active mature peptide may limit damage to the ‘self’, once their purpose of antimicrobial activity has been served (Brockton et al. 2007). The signal peptide cleavage site is predicted to be recognized by the Sec/SP1 system, with N-region, H-region and C-regions. The transmembrane helix region predicted from positions 5–27 of the full-length MC-crustin may be the region responsible for anchoring the nascent peptide to membrane of endoplasmic reticulum (Valore and Ganz 1992; Ganz 2003).
Classification and formula annotations for different types of crustins have been attempted by several authors, and it is still an area under perusal (Bartlett et al 2002; Smith et al 2008; Tassanakajon et al. 2015, 2011; Vargas-Albores and Martínez-Porchas 2017). Type1 crustins or the carcinin like peptides (Zhao and Wang 2008) have cysteine rich region, with four conserved cysteine residues between the signal peptide and WAP domain (Smith et al 2008; Matos and Rosa 2022). Crustin signature domain is characterized by 12 conserved cysteine residues among which the last eight cysteine residues are present in C-terminal WAP domain, also observed in MC-crustin (Ranganathan et al 1999; Bartlett et al. 2002; Smith et al. 2008). Other consensus patterns recognized in the C-terminal WAP domain for MC-crustin and others are: (i) HXGXCP containing C1, similar to the KXGXCP recognized as WAP motif (Ranganathan et al. 1999) and (ii) aspartic acid (D) and lysine (K) residues conserved as C3XXDXXC4XXXXKC5C6XDXC7 (Ranganathan et al. 1999; Bartlett et al. 2002; Smith et al. 2008). The region between C-terminal WAP domain and N-terminal signal peptide contains proline (P) and cysteine (C) rich region that follows the Type I crustin consensus pattern of C-X3-C-X (8–12)-C-C-X (16–17)-C-X6-C-X (9–10) (Smith et al. 2008). In mammals, the WAP domain is recognized to have serine protease inhibition, which is believed to be due to the presence of a methionine (M) residue adjacent to the second cysteine in the 4DSC (Ota et al. 2002; Francart et al. 1997). However, in WAP domain-containing proteins with antibacterial properties, this is replaced by a cationic or hydrophobic amino acid (Smith et al. 2008). MC-crustin and other crustins do not contain methionine residue at the respective site, indicating antibacterial activity.
Type II crustins hold a long glycine-rich hydrophobic region at the N-terminus followed by the 12 conserved cysteine containing ‘crustin signature domain’ which includes the C-terminal single WAP domain. The Type III/single WAP domain (SWD) crustins are defined by the presence/absence of a short proline and arginine-rich region at the N-terminus and a single WAP domain at the C-terminal of mature peptide. They do not contain the complete ‘crustin signature domain’ (Smith et al. 2008; Smith 2011; Tassanakajon et al. 2011). Type IV crustins/DWD crustins, as their name suggest, have two WAP domains (Smith 2011). Finally, Type V crustins from insect order Hymenoptera, are similar to Type I crustins; nevertheless, with an aromatic amino acid-rich domain between the cysteine-rich domain and WAP domain (Zhang and Zhu 2012). Zhao and Wang (2008) had proposed a divergent evolution for WAP domains in crustaceans, exhibiting different function such as antimicrobial activity and proteinase inhibitor activity (Du et al. 2010; Jiang et al. 2013; Zhang et al. 2017). Neighbour joining analysis in present study revealed two main branches of WAP domain containing proteins in crustaceans, one group containing the ‘crustin signature domain’ with WAP domain peptides (Type I and Type II crustins) and the other group which do not possess the signature crustin domain, yet containing the WAP domains (SWDs and DWDs), similar to previous studies such as Zhao and Wang, (2008) and Mu et al., (2010). In the first group of crustins, Type I and Type II crustins formed two distinct clades. Similarly, in second group, Type II and Type III also formed two distinct clades.
WAP domain structural analysis revealed antiparallel beta-sheet structure in the core region besides the major loop regions, stabilized by disulphide bonds (Ranganathan et al. 1999; Koizumi et al. 2008). Recent MD simulation studies in a crustin (Aacrus1) from the barnacle, Amphibalanus amphitrite revealed that the loop region is formed by 17 amino acid residues, extending from a serine (S) to phenyl alanine (F), adjacent to the first and second cysteine residues of its WAP domain, responsible for the deep anchoring of the peptide into bacterial membrane (Zhang et al. 2022a, b). The corresponding region in MC-crustin is from R37 to V53 of the mature peptide WAP domain region. However, the residues specifically involved in the binding needs to be assessed. Nevertheless, the loop region of MC-crustin from H39 to R54 was observed to have greater cationicity in the electrostatic potential distribution. It is likely that this region may be involved in strong binding to the negatively charged bacterial membranes and the stabilization of the structure due to the C1–C6 cysteine bond could be responsible for a stable binding and penetration into bacterial membranes, as observed in Aacrus1.
It is well known that AMPs show less ‘in solution’ aggregation and higher aggregation tendencies when in hydrophobic environments like bacterial membranes. Hydrophobicity influences aggregation of peptides. Increasing hydrophobicity in peptides beyond a critical level resulted in larger aggregates of peptides either ‘in solution’ itself or on bacterial membranes, consequently reducing its bioavailability. Hence, higher hydrophobicity and ‘in solution’ aggregation is generally considered to reduce the activity of the peptide. Nevertheless, optimum aggregation on membrane is essential for antimicrobial action (Torrent et al. 2011; Yin et al. 2012; Clark et al. 2021). MC- crustin did not show in solution aggregation (TANGO prediction) and only fewer aggregation prone residues as per AGGRESCAN aggregation tendency profile. The predicted aggregation hotspot regions (Y23–G26 and C84–Y90) and amyloidogenic regions (A21–C25 and C73–A77) confirm probability for peptide aggregations in bacterial membranes to execute their membrane disruption mechanisms (Yadav 2021; Kurpe et al. 2020; Zhang et al. 2021).
The potentials of MC-crustin were further supported by various in silico potential predictors, classifying the peptide as antimicrobial peptide. Search for biologically active peptide is increasing on account of the increased resistance by pathogens against the antibiotics in use. Bioactive peptides can be classified into short (< 20 aa) and long (> 20 aa) peptides (Mooney et al. 2012). In general, leucine (L), glycine (G) and lysine (K) are preferred for long bioactive antimicrobial peptides. Moreover, long bioactive peptides have certain general distribution of amino acid composition when compared to short bioactive peptides (3.9% of F, 7.5% of G, 5.8% of E, 4.8% of T, 4.5% of N, 5% of R). Ninety aminoacid sized MC-crustin was classified by Peptide Ranker as a bioactive peptide, falling in the long bioactive peptide class (Mooney et al. 2012). Antibacterial peptides have greater distribution of positively charged residues at the N and C-terminus of the peptides. Moreover, it has been observed that residues W, I, L, F are more frequent at the 2nd position of N-terminus of antibacterial peptides in comparison to non-antibacterial peptides. Residues such as G, R, K, L and C are in greater proportion in antibacterial peptides (Lata et al. 2007). MC-crustin had 7% each of G, R and K, 13% C and 6% G and greater distribution of positively charged amino acid residues—K, R and H in the C-terminus WAP domain, making it a suitable candidate as antibacterial peptide supplemented by prediction of iAMPpred (Meheret al. 2017). It has been postulated that higher frequency of the positively charged residues at C-terminus may help to interact with the negatively charged bacterial membranes (Lata et al. 2007).
Instability index greater than 40, slightly negative GRAVY score and abundance of aliphatic residues like I, V and L (total 15% for MC-crustin) are properties associated to be observed with antiviral peptides (Chang and Yang 2013). Crustins are known to show antiviral properties (Du et al. 2019; Wang et al. 2021a, b). iAMP pred supported the anti-viral potential for MC-crustin in the present study. Residues like C, G, H, K, R, and S are prominent in antifungal peptides. MC-crustin being a cysteine rich peptide (13% C) and equal distribution of lysine and arginine (7% each) as well as histidine and serine (6% each) could be the possible amino acid composition features suitable for antifungal peptide, supported by prediction from iAMPpred.
Inflammation is a biological response of the immune system that can be triggered by a variety of factors, including pathogens, damaged cells and toxic compounds (Chen et al. 2017). Proinflammatory peptides (or PIPs) are therapeutic candidates to cure various diseases that help in vaccine development. In general, arginine and leucine are preferred amino acids in proinflammatory peptides. The most abundant dipeptide compositions in PIPs are aliphatic-aliphatic, positively charged-aliphatic and hydroxyl group aliphatic or aromatic amino acids (FF, SL, SR, SF, SV, LL, LI, RT, RA) (Manavalan et al. 2018a, b). MC-crustin had equal distribution (1.12% each) of the dipeptide compositions SL, SF and LI in the sequence. Inflammatory responses are tightly controlled under normal conditions and are essential for the initiation of protective immunity. However, when they are not balanced after their routine functions, it can result in auto-immune disorders and chronic inflammation. For in silico predictions, arginine, leucine and lysine are observed to be dominant in anti-inflammatory peptides and the preferred dipeptide compositions are LL, SL, LE, LI, LS, LK, YL, IK and RI (Manavalan et al. 2018a, b). MC-crustin had equal distribution of SL, LI and YL dipeptide compositions (1.2% each). Moreover, the expression of crustin genes is reported to be mainly regulated by NF-κB transcription factors activated by both Toll and IMD signalling pathways, which are also a part of inflammatory signalling (Wang et al. 2013; Huang et al. 2016).
Angiogenesis promotes blood flow to injured tissues for healing. However, it is kept under regulation by several endogenous anti-angiogenic factors like anti-angiogenic peptides, thereby preventing tumour genesis (Rosca et al 2011). In MC-crustin, amino acids viz., C, P, S, R and W contributed to the anti-angiogenic property. MC-crustin was abundant with cysteines (13%) and prolines (11%), followed by arginine (7%) and serine (6%). Moreover, the N-terminal had S, P, W and C residues, a pattern observed in anti-angiogenic peptides (Ettayapuram Ramaprasad et al. 2015).
Anti-hypertensive peptides are known to target angiotensin-1 converting enzyme (ACE). Generally, anti-hypertensive peptides, which are inert inside the original protein, which become active once cleaved, have been reported to contain usually 2–20 amino acids (Himaya et al. 2012). All classes of anti-hypertensive peptides are known to have proline abundance, which was also observed for MC-crustin. Moreover, residues like aspartic acid and serine are known to be less frequent in these peptides (6% each in MC-crustin) (Kumar et al. 2015). A notable prediction is the WAP domain as anti-hypertensive, when compared to other regions of the mature peptide. Furthermore, the higher proline residue and reduced hydrophobicity may be responsible for the non-hemolytic property of the MC-crustin.
Conclusion
Marine derived AMPs have potential biomedical importance, making them anticipated alternatives for rational drug strategies and pharmaceuticals with a large potential in biomedical and aquaculture applications. MC-crustin with its cationic, cysteine and proline rich structure, was predicted with antimicrobial, anti-viral, anti-inflammatory, anti-angiogenic and anti-hypertensive property. Presence of aggregation prone segments with positive charge on WAP domain loop region are supportive of membrane binding and disruption property of the peptide. Being non-haemolytic with notable bioactive property, MC-crustin would be a promising drug molecule for therapeutic applications as an alternative to the antibiotics.
Data availability
The data generated and/or analysed during the current study are not publicly available (exception is GenBank accessions) but are available from the corresponding author on reasonable request.
References
Afsal VV, Antony SP, Sathyan N, Philip R (2011) Molecular characterization and phylogenetic analysis of two antimicrobial peptides: anti-lipopolysaccharide factor and crustin from the brown mud crab, Scylla serrata. Results Immunol 1(1):6–10. https://doi.org/10.1016/j.rinim.2011.06.001
Afsal VV, Antony SP, Bright AR, Philip R (2013) Molecular identification and characterization of Type I crustin isoforms from the hemocytes of portunid crabs, Scylla tranquebarica and Portunus pelagicus. Cell Immunol 284(1–2):45–50. https://doi.org/10.1016/j.cellimm.2013.07.003
Alenton RRR, Koiwai K, Nakamura R, Thawonsuwan J, Kondo H, Hirono I (2019) A hint of primitive mucosal immunity in shrimp through Marsupenaeus japonicus Gill C-Type lectin. J Immunol 203(8):2310–2318. https://doi.org/10.4049/jimmunol.1900156
Antony SP, Bright Singh IS, Philip R (2010) Molecular characterization of a crustin-like, putative antimicrobial peptide, Fi-crustin, from the Indian white shrimp. Fenneropenaeus indicus. Fish Shellfish Immunol 28(1):216–220. https://doi.org/10.1016/j.fsi.2009.10.013
Antony SP, Singh IS, Sudheer NS, Vrinda S, Priyaja P, Philip R (2011) Molecular characterization of a crustin-like antimicrobial peptide in the giant tiger shrimp, Penaeus monodon, and its expression profile in response to various immunostimulants and challenge with WSSV. Immunobiology 216(1–2):184–194. https://doi.org/10.1016/j.imbio.2010.05.030
Armenteros JJA, Tsirigos KD, Sønderby CK, Petersen TN, Winther O, Brunak S et al (2019) SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37(4):420–423. https://doi.org/10.1038/s41587-019-0036-z
Azuma E, Choda N, Odaki M, Yano Y, Matsuzaki K (2020) Improvement of therapeutic index by the combination of enhanced peptide cationicity and proline introduction. ACS Infect Dis 6(8):2271–2278. https://doi.org/10.1021/acsinfecdis.0c00387
Bartlett TC, Cuthbertson BJ, Shepard EF, Chapman RW, Gross PS, Warr GW (2002) Crustins, homologues of an 1.15-kDa antibacterial peptide, from two species of penaeid shrimp, Litopenaeus vannamei and Litopenaeus setiferus. Mar Biotechnol (New York, N.Y.) 4(3):278–293. https://doi.org/10.1007/s10126-002-0020-2
Boman HG (2003) Antibacterial peptides: basic facts and emerging concepts. J Intern Med 254(3):197–215. https://doi.org/10.1046/j.1365-2796.2003.01228.x
Brockton V, Hammond JA, Smith VJ (2007) Gene characterisation, isoforms and recombinant expression of carcinin, an antibacterial protein from the shore crab, Carcinus maenas. Mol Immunol 44(5):943–949. https://doi.org/10.1016/j.molimm.2006.03.017
Cerenius L, Jiravanichpaisal P, Liu HP, Söderhill I (2010) Crustacean immunity. Adv Exp Med Biol 708:239–259. https://doi.org/10.1007/978-1-4419-8059-5_13
Chandra KJ (2012) A survey on the production and marketing of mud crab, Scylla serrata (forskal, 1755) in the south-west part of Bangladesh. Int Res J Appl Life Sci 1(3)
Chang KY, Yang JR (2013) Analysis and prediction of highly effective antiviral peptides based on random forests. PLoS One 8(8):e70166
Chen L, Deng H, Cui H, Fang J, Zuo Z, Deng J, Li Y, Wang X, Zhao L (2017) Inflammatory responses and inflammation-associated diseases in organs. Oncotarget 9(6):7204–7218. https://doi.org/10.18632/oncotarget.23208
Chisholm JR, Smith VJ (1992) Antibacterial activity in the haemocytes of the shore crab, Carcinus maenas. J Mar Biol Assoc UK 72(3):529–542
Chowdhury AS, Reehl SM, Kehn-Hall K, Bishop B, Webb-Robertson BM (2020) Better understanding and prediction of antiviral peptides through primary and secondary structure feature importance. Sci Rep 10(1):19260. https://doi.org/10.1038/s41598-020-76161-8
Clark S, Jowitt TA, Harris LK, Knight CG, Dobson CB (2021) The lexicon of antimicrobial peptides: a complete set of arginine and tryptophan sequences. Commun Biol 4(1):1–14
Conchillo-Solé O, de Groot NS, Avilés FX, Vendrell J, Daura X, Ventura S (2007) AGGRESCAN: a server for the prediction and evaluation of “hot spots” of aggregation in polypeptides. BMC Bioinform 8(1):65. https://doi.org/10.1186/1471-2105-8-65
de Castro E, Sigrist CJ, Gattiker A, Bulliard V, Langendijk-Genevaux PS, Gasteiger E, Bairoch A, Hulo N (2006) ScanProsite: detection of PROSITE signature matches and ProRule-associated functional and structural residues in proteins. Nucl Acids Res 34(Web Server issue):W362–W365. https://doi.org/10.1093/nar/gkl124
Destoumieux D, Bulet P, Loew D, Van Dorsselaer A, Rodriguez J, Bachère E (1997) Penaeidins, a new family of antimicrobial peptides isolated from the shrimp Penaeus vannamei (Decapoda). J Biol Chem 272(45):28398–28406. https://doi.org/10.1074/jbc.272.45.28398
Destoumieux D, Munoz M, Bulet P, Bachère E (2000) Penaeidins, a family of antimicrobial peptides from penaeid shrimp (Crustacea, Decapoda). Cell Mol Life Sc CMLS 57(8–9):1260–1271. https://doi.org/10.1007/pl00000764
Dijksteel GS, Ulrich M, Middelkoop E, Boekema B (2021) Review: lessons learned from clinical trials using antimicrobial peptides (AMPs). Front Microbiol 12:616979. https://doi.org/10.3389/fmicb.2021.616979
Du ZQ, Li XC, Wang ZH, Zhao XF, Wang JX (2010) A single WAP domain (SWD)-containing protein with antipathogenic relevance in red swamp crayfish, Procambarus clarkii. Fish Shellfish Immunol 28(1):134–142. https://doi.org/10.1016/j.fsi.2009.10.009
Du ZQ, Wang Y, Ma HY, Shen XL, Wang K, Du J, Yu XD, Fang WH, Li XC (2019) A new crustin homologue (SpCrus6) involved in the antimicrobial and antiviral innate immunity in mud crab, Scylla paramamosain. Fish Shellfish Immunol 84:733–743. https://doi.org/10.1016/j.fsi.2018.10.072
Duckert P, Brunak S, Blom N (2004) Prediction of proprotein convertase cleavage sites. Protein Eng Des Sel 17(1):107–112. https://doi.org/10.1093/protein/gzh013
Erdem Büyükkiraz M, Kesmen Z (2022) Antimicrobial peptides (AMPs): a promising class of antimicrobial compounds. J Appl Microbiol 132(3):1573–1596. https://doi.org/10.1111/jam.15314
Ettayapuram Ramaprasad AS, Singh S, Gajendra PSR, Venkatesan S (2015) AntiAngioPred: a server for prediction of anti-angiogenic peptides. PLoS One 10(9):e0136990. https://doi.org/10.1371/journal.pone.0136990
Fernandez DI, Lee TH, Sani MA, Aguilar MI, Separovic F (2013) Proline facilitates membrane insertion of the antimicrobial peptide maculatin 1.1 via surface indentation and subsequent lipid disordering. Biophys J 104(7):1495–1507. https://doi.org/10.1016/j.bpj.2013.01.059
Fernandez-Escamilla AM, Rousseau F, Schymkowitz J, Serrano L (2004) Prediction of sequence-dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 22(10):1302–1306. https://doi.org/10.1038/nbt1012
Francart C, Dauchez M, Alix AJ, Lippens G (1997) Solution structure of R-elafin, a specific inhibitor of elastase. J Mol Biol 268(3):666–677. https://doi.org/10.1006/jmbi.1997.0983
Ganz T (2003) Defensins: antimicrobial peptides of innate immunity. Nat Rev Immunol 3(9):710–720. https://doi.org/10.1038/nri1180
Garbuzynskiy SO, Lobanov MY, Galzitskaya OV (2010) FoldAmyloid: a method of prediction of amyloidogenic regions from protein sequence. Bioinformatics 26(3):326–332
Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. The proteomics protocols handbook. Humana Press, Totowa, pp 571–607
Guex N, Peitsch MC (1997) SWISS-MODEL and the Swiss-Pdb Viewer: an environment for comparative protein modeling. Electrophoresis 18(15):2714–2723
Himaya SWA, Ngo DH, Ryu B, Kim SK (2012) An active peptide purified from gastrointestinal enzyme hydrolysate of Pacific cod skin gelatin attenuates angiotensin-1 converting enzyme (ACE) activity and cellular oxidative stress. Food Chem 132(4):1872–1882
Holton TA, Pollastri G, Shields DC, Mooney C (2013) CPPpred: prediction of cell penetrating peptides. Bioinformatics (oxford, England) 29(23):3094–3096. https://doi.org/10.1093/bioinformatics/btt518
Huang X, Wang W, Ren Q (2016) Dorsal transcription factor is involved in regulating expression of crustin genes during white spot syndrome virus infection. Dev Comp Immunol 63:18–26. https://doi.org/10.1016/j.dci.2016.05.006
Iskandar K, Murugaiyan J, Hammoudi Halat D, Hage SE, Chibabhai V, Adukkadukkam S, Roques C, Molinier L, Salameh P, Van Dongen M (2022) Antibiotic discovery and resistance: the chase and the race. Antibiotics (basel, Switzerland) 11(2):182. https://doi.org/10.3390/antibiotics11020182
Jiang HS, Sun C, Wang T, Zhao XF, Wang JX (2013) A single whey acidic protein domain containing protein (SWD) inhibits bacteria invasion and dissemination in shrimp Marsupenaeus japonicus. Fish Shellfish Immunol 35(2):310–318. https://doi.org/10.1016/j.fsi.2013.04.035
Khatun MS, Hasan MM, Kurata H (2019) PreAIP: computational prediction of anti-inflammatory peptides by integrating multiple complementary features. Front Genet 10:129. https://doi.org/10.3389/fgene.2019.00129 (Published 2019 Mar 5)
Koizumi M, Fujino A, Fukushima K, Kamimura T, Takimoto-Kamimura M (2008) Complex of human neutrophil elastase with 1/2SLPI. J Synchrotron Radiat 15(3):308–311
Kulkarni A, Krishnan S, Anand D, Kokkattunivarthil Uthaman S, Otta SK, Karunasagar I, Kooloth Valappil R (2021) Immune responses and immunoprotection in crustaceans with special reference to shrimp. Rev Aquac 13(1):431–459
Kumar R, Chaudhary K, Sharma M, Nagpal G, Chauhan JS, Singh S, Gautam A, Raghava GP (2015) AHTPDB: a comprehensive platform for analysis and presentation of antihypertensive peptides. Nucleic Acids Res 43(Database issue):D956–D962. https://doi.org/10.1093/nar/gku1141
Kumar S, Stecher G, Li M, Knyaz C, Tamura K (2018) MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35(6):1547–1549. https://doi.org/10.1093/molbev/msy096
Kurpe SR, Grishin SY, Surin AK, Panfilov AV, Slizen MV, Chowdhury SD, Galzitskaya OV (2020) Antimicrobial and amyloidogenic activity of peptides. Can antimicrobial peptides be used against SARS-CoV-2? Int J Mol Sci 21(24):9552. https://doi.org/10.3390/ijms21249552
Laskowski RA, Thornton JM (2022) PDBsum extras: SARS-CoV-2 and AlphaFold models. Protein Sci Publ Protein Soc 31(1):283–289. https://doi.org/10.1002/pro.4238
Lata S, Sharma BK, Raghava GP (2007) Analysis and prediction of antibacterial peptides. BMC Bioinform 8:263. https://doi.org/10.1186/1471-2105-8-263
Letunic I, Bork P (2018) 20 years of the SMART protein domain annotation resource. Nucleic Acids Res 46(D1):D493–D496. https://doi.org/10.1093/nar/gkx922
Linding R, Schymkowitz J, Rousseau F, Diella F, Serrano L (2004) A comparative study of the relationship between protein structure and betaaggregation in globular and intrinsically disordered proteins. J Mol Biol 342(1):345–353. https://doi.org/10.1016/j.jmb.2004.06.088
Loo K-Y, Letchumanan V, Law JW-F, Pusparajah P, Goh B-H, Abmutalib N-S, He Y-W, Lee L-H (2020) Incidence of antibiotic resistance in Vibrio spp. Rev Aquac 12:2590–2608. https://doi.org/10.1111/raq.12460
López Cascales JJ, Zenak S, García de la Torre J, Lezama OG, Garro A, Enriz RD (2018) Small cationic peptides: influence of charge on their antimicrobial activity. ACS Omega 3(5):5390–5398. https://doi.org/10.1021/acsomega.8b00293
Manavalan B, Shin TH, Kim MO, Lee G (2018a) AIPpred: sequence-based prediction of anti-inflammatory peptides using random forest. Front Pharmacol 9:276. https://doi.org/10.3389/fphar.2018.00276
Manavalan B, Shin TH, Kim MO, Lee G (2018b) PIP-EL: a new ensemble learning method for improved proinflammatory peptide predictions. Front Immunol 9:1783. https://doi.org/10.3389/fimmu.2018.01783
Manavalan B, Basith S, Shin TH, Wei L, Lee G (2019) mAHTPred: a sequence-based meta-predictor for improving the prediction of anti-hypertensive peptides using effective feature representation. Bioinformatics (oxford, england) 35(16):2757–2765. https://doi.org/10.1093/bioinformatics/bty1047
Matos GM, Rosa RD (2022) On the silver jubilee of crustacean antimicrobial peptides. Rev Aquac. https://doi.org/10.1111/raq.12614
Meher PK, Sahu TK, Saini V, Rao AR (2017) Predicting antimicrobial peptides with improved accuracy by incorporating the compositional, physico-chemical and structural features into Chou’s general PseAAC. Sci Rep 7(1):1–12
Mooney C, Haslam NJ, Pollastri G, Shields DC (2012) Towards the improved discovery and design of functional peptides: common features of diverse classes permit generalized prediction of bioactivity. PloS One 7(10):e45012. https://doi.org/10.1371/journal.pone.0045012
Moravej H, Moravej Z, Yazdanparast M, Heiat M, Mirhosseini A, Moosazadeh Moghaddam M, Mirnejad R (2018) Antimicrobial peptides: features, action, and their resistance mechanisms in bacteria. Microb Drug Resis (larchmont, N.y.) 24(6):747–767. https://doi.org/10.1089/mdr.2017.0392
Mu C, Zheng P, Zhao J, Wang L, Zhang H, Qiu L, Gai Y, Song L (2010) Molecular characterization and expression of a crustin-like gene from Chinese mitten crab, Eriocheir sinensis. Dev Comp Immunol 34(7):734–740. https://doi.org/10.1016/j.dci.2010.02.001
Ota Y, Shimoya K, Zhang Q, Moriyama A, Chin R, Tenma K, Kimura T, Koyama M, Azuma C, Murata Y (2002) The expression of secretory leukocyte protease inhibitor (SLPI) in the fallopian tube: SLPI protects the acrosome reaction of sperm from inhibitory effects of elastase. Hum Reprod (oxford, england) 17(10):2517–2522. https://doi.org/10.1093/humrep/17.10.2517
Pande A, Patiyal S, Lathwal A, Arora C, Kaur D, Dhall A et al (2019) Computing wide range of protein/peptide features from their sequence and structure. BioRxiv 9:599126
Pripanapong S, Tongdee NA (1998) Review of the mud crab (Scylla sp.) fisheries and culture in Thailand. Newsletter of Danish-SE Asian Collaboration in Tropical Coastal Ecosystems Research and Training Project, Denmark, Thailand and Malaysia, vol 2, no 2, pp 7–10
Ranganathan S, Simpson KJ, Shaw DC, Nicholas KR (1999) The whey acidic protein family: a new signature motif and three-dimensional structure by comparative modeling. J Mol Graph Model 17(2):106–136. https://doi.org/10.1016/s1093-3263(99)00023-6
Relf JM, Chisholm JR, Kemp GD, Smith VJ (1999) Purification and characterization of a cysteine-rich 1.15-kDa antibacterial protein from the granular haemocytes of the shore crab, Carcinus maenas. Eur J Biochem 264(2):350–357. https://doi.org/10.1046/j.1432-1327.1999.00607.x
Rosa RD, Bandeira PT, Barracco MA (2007) Molecular cloning of crustins from the hemocytes of Brazilian penaeid shrimps. FEMS Microbiol Lett 274(2):287–290. https://doi.org/10.1111/j.1574-6968.2007.00866.x
Rosca EV, Koskimaki JE, Rivera CG, Pandey NB, Tamiz AP, Popel AS (2011) Anti-angiogenic peptides for cancer therapeutics. Curr Pharm Biotechnol 12(8):1101–1116. https://doi.org/10.2174/138920111796117300
Rousseau F, Schymkowitz J, Serrano L (2006) Protein aggregation and amyloidosis: confusion of the kinds? Curr Opin Struct Biol 16(1):118–126. https://doi.org/10.1016/j.sbi.2006.01.011
Schar D, Zhao C, Wang Y, Larsson D, Gilbert M, Van Boeckel TP (2021) Twenty-year trends in antimicrobial resistance from aquaculture and fisheries in Asia. Nat Commun 12(1):5384. https://doi.org/10.1038/s41467-021-25655-8
Schnapp D, Kemp GD, Smith VJ (1996) Purification and characterization of a proline-rich antibacterial peptide, with sequence similarity to bactenecin-7, from the haemocytes of the shore crab, Carcinus maenas. Eur J Biochem 240(3):532–539. https://doi.org/10.1111/j.1432-1033.1996.0532h.x
Silveira AS, Matos GM, Falchetti M, Ribeiro FS, Bressan A, Bachère E, Perazzolo LM, Rosa RD (2018) An immune-related gene expression atlas of the shrimp digestive system in response to two major pathogens brings insights into the involvement of hemocytes in gut immunity. Dev Comp Immunol 79:44–50. https://doi.org/10.1016/j.dci.2017.10.005
Smith VJ (2011) Phylogeny of whey acidic protein (WAP) four-disulfide core proteins and their role in lower vertebrates and invertebrates. Biochem Soc Trans 39(5):1403–1408. https://doi.org/10.1042/BST0391403
Smith VJ, Chisholm JR (2001) Antimicrobial proteins in crustaceans. Adv Exp Med Biol 484:95–112. https://doi.org/10.1007/978-1-4615-1291-2_10
Smith VJ, Fernandes JM, Kemp GD, Hauton C (2008) Crustins: enigmatic WAP domain-containing antibacterial proteins from crustaceans. Dev Comp Immunol 32(7):758–772. https://doi.org/10.1016/j.dci.2007.12.002
Sperstad SV, Haug T, Paulsen V, Rode TM, Strandskog G, Solem ST, Styrvold OB, Stensvåg K (2009) Characterization of crustins from the hemocytes of the spider crab, Hyas araneus, and the red king crab, Paralithodes camtschaticus. Dev Comp Immunol 33(4):583–591. https://doi.org/10.1016/j.dci.2008.10.010
Sruthy KS, Nair A, Puthumana J, Antony SP, Singh I, Philip R (2017) Molecular cloning, recombinant expression and functional characterization of an antimicrobial peptide, Crustin from the Indian white shrimp, Fenneropenaeus indicus. Fish Shellfish Immunol 71:83–94. https://doi.org/10.1016/j.fsi.2017.09.071
Tassanakajon A, Amparyup P, Somboonwiwat K, Supungul P (2011) Cationic antimicrobial peptides in penaeid shrimp. Mar Biotechnol (New York, N.Y.) 13(4):639–657. https://doi.org/10.1007/s10126-011-9381-8
Tassanakajon A, Somboonwiwat K, Amparyup P (2015) Sequence diversity and evolution of antimicrobial peptides in invertebrates. Dev Comp Immunol 48(2):324–341. https://doi.org/10.1016/j.dci.2014.05.020
Tincu JA, Taylor SW (2004) Antimicrobial peptides from marine invertebrates. Antimicrob Agents Chemother 48(10):3645–3654. https://doi.org/10.1128/AAC.48.10.3645-3654.2004
Torrent M, Andreu D, Nogués VM, Boix E (2011) Connecting peptide physicochemical and antimicrobial properties by a rational prediction model. PLoS One 6(2):e16968. https://doi.org/10.1371/journal.pone.0016968
Valore EV, Ganz T (1992) Posttranslational processing of defensins in immature human myeloid cells. Blood 79(6):1538–1544
Vargas-Albores F, Martínez-Porchas M (2017) Crustins are distinctive members of the WAP-containing protein superfamily: an improved classification approach. Dev Comp Immunol 76:9–17. https://doi.org/10.1016/j.dci.2017.05.012
Vu GH, Do D, Rivera CD, Dickinson PS, Christie AE, Stemmler EA (2018) Characterization of the mature form of a β-defensin-like peptide, Hoa-D1, in the lobster Homarus americanus. Mol Immunol 101:329–343. https://doi.org/10.1016/j.molimm.2018.07.004
Wang PH, Gu ZH, Wan DH, Liu BD, Huang XD, Weng SP, Yu XQ, He JG (2013) The shrimp IKK-NF-κB signaling pathway regulates antimicrobial peptide expression and may be subverted by white spot syndrome virus to facilitate viral gene expression. Cell Mol Immunol 10(5):423–436. https://doi.org/10.1038/cmi.2013.30
Wang G, Li X, Wang Z (2016) APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res 44(D1):D1087–D1093. https://doi.org/10.1093/nar/gkv1278
Wang Y, Zhang C, Fang WH, Ma HY, Li XC (2021a) SpCrus2 glycine-rich region contributes largely to the antiviral activity of the whole-protein molecule by interacting with vp26, a wssv structural protein. Mar Drugs 19(10):544. https://doi.org/10.3390/md19100544
Wang Y, Zhang J, Sun Y, Sun L (2021b) A Crustin from Hydrothermal Vent Shrimp: Antimicrobial Activity and Mechanism. Mar Drugs 19(3):176. https://doi.org/10.3390/md19030176
Wang Z, Zhao F, Peng J, Xu J (2010) Protein 8-class secondary structure prediction using conditional neural fields. In: 2010 IEEE international conference on bioinformatics and biomedicine (BIBM). IEEE, pp 109–114
Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ (2009) Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics (oxford, England) 25(9):1189–1191. https://doi.org/10.1093/bioinformatics/btp033
Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer T, Rempfer C, Bordoli L, Lepore R, Schwede T (2018) SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46(W1):W296–W303. https://doi.org/10.1093/nar/gky427
Win TS, Malik AA, Prachayasittikul V, Wikberg JES, Nantasenamat C, Shoombuatong W (2017) HemoPred: a web server for predicting the hemolytic activity of peptides. Future Med Chem 9(3):275–291. https://doi.org/10.4155/fmc-2016-0188
Wu R, Patocka J, Nepovimova E, Oleksak P, Valis M, Wu W, Kuca K (2021) Marine invertebrate peptides: antimicrobial peptides. Front Microbiol 12:785085. https://doi.org/10.3389/fmicb.2021.785085
Xie J, Zhao Q, Li S, Yan Z, Li J, Li Y et al (2017) Novel anti-microbial peptide CPF-C1 analogs with superior stabilities and activities against multidrug- resistant bacteria. Chem Biol Drug Des 90:690–702
Yadav JK (2021) Structural and functional swapping of amyloidogenic and antimicrobial peptides: redefining the role of amyloidogenic propensity in disease and host defense. J Pept Sci off Publ Eur Pept Soc 28:e3378. https://doi.org/10.1002/psc.3378. (advance online publication)
Yin LM, Edwards MA, Li J, Yip CM, Deber CM (2012) Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J Biol Chem 287(10):7738–7745
Zhang Z, Zhu S (2012) Comparative genomics analysis of five families of antimicrobial peptide-like genes in seven ant species. Dev Comp Immunol 38(2):262–274. https://doi.org/10.1016/j.dci.2012.05.003
Zhang HW, Man X, Wang Y, Song QS, Stanley D, Hui KM, Zhang XW (2017) Characterization of a double WAP domain-containing protein from the red swamp crayfish Procambarus clarkii. Fish Shellfish Immunol 71:329–337. https://doi.org/10.1016/j.fsi.2017.10.019
Zhang QY, Yan ZB, Meng YM, Hong XY, Shao G, Ma JJ, Cheng XR, Liu J, Kang J, Fu CY (2021) Antimicrobial peptides: mechanism of action, activity and clinical potential. Mil Med Res 8(1):48. https://doi.org/10.1186/s40779-021-00343-2
Zhang W, Xia E, Dai R, Tang W, Bin Y, Xia J (2022a) PredAPP: predicting anti-parasitic peptides with undersampling and ensemble approaches. Interdiscip Sci Comput Life Sci 14(1):258–268. https://doi.org/10.1007/s12539-021-00484-x
Zhang W, Xu X, Zhang J, Ye T, Zhou Q, Xu Y, Li W, Hu Z, Shang C (2022b) Discovery and characterization of a new crustin antimicrobial peptide from Amphibalanus amphitrite. Pharmaceutics 14(2):413. https://doi.org/10.3390/pharmaceutics14020413
Zhao XF, Wang JX (2008) The antimicrobial peptides of the immune response of shrimp. Invertebr Surviv J 5(2):162–179
Acknowledgements
The authors are thankful to the Director, Centre for Marine Living Resources and Ecology (CMLRE) and Ministry of Earth Sciences (MoES), Government of India, for the research grant (MoES/10-MLR/01/2012) and scientific support for the work. The first author is grateful to the Council of Scientific and Industrial Research (CSIR) for the award of the fellowship (09/239(0536)/2018-EMR-I) and the corresponding author to UGC, Government of India for the BSR Faculty Grant (F.18-1/2011(BSR) dt. 16 May 2019). The authors wish to thank the Department of Marine Biology, Microbiology and Biochemistry, School of Marine Sciences, Cochin University of Science and Technology and National Centre for Aquatic and Animal Health (NCAAH), and Cochin University of Science and Technology for the facilities provided for the successful completion of the work.
Author information
Authors and Affiliations
Contributions
NS carried out the experiment of the present work with support from AMV, AVV, APP, AK and MMS. RP supervised the work and corrected the manuscript. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that there is no conflict of interest.
Research involving human and animal participants
This article does not contain any study that requires ethical approval.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Neelima, S., Anju, M.V., Anooja, V.V. et al. Characterisation of a novel crustin isoform from mud crab, Scylla serrata (Forsskål, 1775) and its functional analysis in silico. In Silico Pharmacol. 11, 2 (2023). https://doi.org/10.1007/s40203-022-00138-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-022-00138-w