
Abstract
Development of resistance in the Plasmodium falciparum to Artemisinin, the most effective anti-malarial compound, threatens malaria elimination tactics. To gain more efficacious Artemisinin derivatives, QSAR modeling and docking was performed. In the present study, 2D-QSAR model and molecular docking were used to evaluate the Artemisinin compounds and to reveal their binding modes and structural basis of inhibitory activity. Moreover, ADMET-related descriptors have been calculated to predict the pharmacokinetic properties of the effective compounds. The correlation expressed as coefficient of determination (r2) and prediction accuracy expressed in the form of cross-validated r2 (q2) of QSAR model are found 0.9687 and 0.9586, respectively. Total 239 descriptors have been included in the study as independent variables. The four chemical descriptors, namely radius of gyration, mominertia Z, SssNH count and SK Average have been found to be well correlated with anti-malarial activities. The model was statistically robust and has good predictive power which could be employed for virtual screening of proposed anti-malarial compounds. QSAR and docking results revealed that studied compounds exhibit good anti-malarial activities and binding affinities. The outcomes could be useful for the design and development of the potent inhibitors which after optimization can be potential therapeutics for malaria.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Several millions of the people worldwide are infected by the Plasmodium falciparum, leading to death of around 1 million annually (World Malaria Report 2013). Most of the therapeutic approaches are Artemisinin based combination therapies (ACTs) and chloroquine (Fidock et al. 2004). Semi-synthetic derivatives of Artemisinin are more frequently used in malaria chemotherapy, due to their better pharmacokinetic properties and higher efficacies as compared to parent compound. ACT is fast acting, well tolerated and is nearly 95% effective in the treatment of malaria. However resistance in parasite to ACTs has been reported in some south-east Asian countries (Kar and Kar 2010). As the P. falciparum resistance to Artemisinin has emerged, development of novel effective anti-malarial drugs is an urgent priority. It prompted to explore further efficient drug like compounds with new mechanisms of action. Currently, quantitative structure activity relationship (QSAR) is useful to check time consumption and cost throughout the analysis of biological activities (Ibezim et al. 2012). Since last few years, QSAR modeling became an important tool for drug design and structural optimization (Bhhatarai and Garg 2008; Xiang et al. 2009; Basak et al. 2010) and is widely used for virtual screening of compounds.
In the current study, molecules with wide range of activities (activity range of 1.4–10,630 nano molar) were used to understand the distinct contributing features for their high potency. The present work describes the development of a QSAR model by using multiple linear regression analysis (MLRA) technique which successfully and accurately predicted activity modulating descriptors. The developed model was used to screen Artemisinin derivatives and to predict the activity. The 11 compounds were identified with very good anti-malarial activities (less than 0.5 nano molar log IC50). Also, the pharmacokinetic properties were predicted through calculation of the absorption, distribution, metabolism, excretion and toxicity (ADMET) related descriptors. Furthermore, through docking possible binding sites and conserved pockets were identified for active compounds against plasmepsin-2 and falcipain-2 of the P. falciparum. It will be useful for understanding the molecular mechanism and directing the molecular design of more potent Artemisinin based inhibitors.
Materials and methods
QSAR model development
The QSAR study was performed using a series of Artemisinin derived compounds reported in the literature (Qidwai et al. 2012; Yadav et al. 2014; Posner et al. 1999; O’Neill et al. 2001; Avery et al. 2003; Liu et al. 2011). The biological activities were represented by the log IC50. The data set of 47 Artemisinin derivatives which were randomly divided in training set (32 compounds) for model development and test set (15 compounds) for model validation. The seventy percent training and thirty percent test set compounds were maintained during the analysis. The chemical structures and their biological activities have been shown (Supp Table 1). The V-life MDS (Molecular Design Suite) TM 3.5 software, supplied by V-life Sciences Technologies was used to build QSAR model (V-life MDS software package, version 3.5 2008).
Ligand preparation and optimization
The molecular structures of all of the molecules were sketched using the ChemBioDraw-Ultra-v12.0 (2010) software. The chemical structures of known drugs were retrieved from the PubChem compound database, which is available at NCBI (http://www.pubchem.ncbi.nlm.nih.gov). Three dimensional geometry of the compounds was generated using Accelrys Discovery Studio (DS) v3.1 (Software Inc., San Diego, CA, USA).
Descriptor calculation
A large number (239) of theoretical two dimensional descriptors were selected for QSAR analysis. The descriptors that have been computed from chemical structures to identify structure/activity relationship of Artemisinin compounds are vdW surface area (van der Waals surface area of the molecule), ve potential surface area (total van der Waals surface area with negative electrostatic potential of the molecule), +ve potential surface area (total van der Waals surface area with positive electrostatic potential of the molecule) dipole moment, Y compDipole (y component of the dipole moment), element count, slogP, path count, cluster, distance based topological indices, connectivity index, hydrophobic and hydrophilic areas like SA most hydrophilic (most hydrophilic value on the vdW surface by Audry method using Slogp), SA most hydrophobic–hydrophilic distance (distance between most hydrophobic and hydrophilic point on the vdW surface by Audry method using Slogp), SA hydrophilic area (vdW surface descriptor showing hydrophilic surface area by Audry method using SlogP) and SK most hydrophilic (most hydrophilic value on the vdW surface by Kellog method using Slogp), radius of gyration, Wiener’s index, moment of inertia, semi-empirical descriptors, HOMO (Highest occupied molecular orbital), LUMO (lowest unoccupied molecular orbital), heat of formation and ionization potential, polarizability AHC, Chi1, Chi2, polar surface area, radius of gyration, Mom InertiaZ, SssNH count, SK average, MomInertia X, MomInertia Y, MomInertia Z.
Cross validation
The models generated by two dimensional QSAR studies were cross validated using a standard leave one out (LOO) procedure. The cross validated coefficient of determination (r2) i.e. q 2 value was calculated. The q 2 obtained is indicative of the predictive power of the current model. The q 2 was calculated using Eq. (1).
The external predictive power of the model is assessed by predicting the log IC50value of test set molecules, which were not included in the QSAR model development. The predictive ability of the selected model was confirmed by pred r 2. The selected descriptors were then used to build QSAR model.
Molecular docking studies
Molecular docking was carried out with the help of Glide module of Schrodinger (2016) and PyRx (Autodoc vina) (Trott and Olson 2010). Three dimensional structures of target proteins were retrieved from Protein Data Bank (PDB) (http://www.pdb.org). The Artemisinin derivatives were docked to the crystal structures of plasmepsin-2 (PDB code: 2IGY) and falcipain-2 (PDB code: 3BPF).
Protein preparation
The protein preparation wizard from Schrodinger was used to prepare protein structure retrieved from the PDB. Hydrogen atoms were added to the protein structures corresponding to pH value of 7.4. All-atom constrained energy minimizations were carried out using the OPLS-2005 force field. An energy minimization was terminated when the root mean square deviation (RMSD) was larger than 0.30 Å.
Ligand preparation for docking
All ligands for docking were prepared using LigPrep module of Schrodinger (2016) by generating low energy ionization and tautomeric states within the pH range of 7.4 ± 0. They were further minimized using the OPLS-2005 force field.
Receptor grid generation and docking
Docking is based on a grid represented by physical properties in the receptor volume that is searched for ligand-receptor interaction during docking process. Glide uses a grid file to describe the profile of a binding pocket, with the center of the grids defined by the co-crystallized ligand. Grid files were prepared with the “Receptor Grid Generation” panel of Glide. Grid points were calculated within a region or an enclosing box defined with the centroid of the bound ligand and the size of a docked ligand with length ≤20 Å. Docking was performed by Glide of Schrodinger (2016). The score function of Glide, or Glide score, was used for binding affinity prediction.
ADMET studies
ADMET profiling of compounds were performed by applying ADMET descriptors algorithm and toxicity prediction protocol of Qikprop of Schrodinger (2016) and ADMETSAR database (freely available at http://www.admetexp.org) (Cheng et al. 2012).
Results
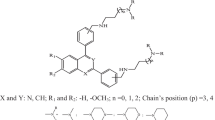
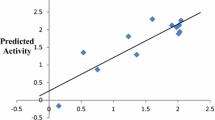
The best QSAR model was selected on the basis of predicted fitness plots and statistical values of the model. The developed model is found with coefficient of determination (r2), cross-validated r2 (q2) and the r2 for external test set (pred_r2) values of 0.9687, 0.9586 and 0.9247 respectively. Standard error of estimate (SEE) r2 (r2_se) and standard error of estimate (SEE) q2 (q2_se) are predicted 0.2206 and 0.2535 respectively. The statistical significance of the model (F-test) is 208.58 and degree of freedom (df) is found 32. The linear and radar plots of experimental versus predictive log IC50 values of compounds in training and test data set are depicted in Figs. 1, 2 respectively. The descriptors based on the model used in the present study are indicated (Fig. 3). Figure 4 depicts two dimensional structures of proposed Artemisinin compounds. QSAR model based predicted properties of Artemisinin compounds are illustrated in Table 1.

Multiple linear regression plot expressing the experimental and predicted log IC50 values for the training (a) and test (b) set compounds

The radar plots denoting the training (a) and the test (b) sets by the red (actual activity) and blue (predicted activity) lines

a Four descriptors, radius of gyration (geometrical descriptor), Mom inertia Z (topological descriptor), SssNH count (sum of ssNH-electrotopological-states), a topological descriptor and SK average (semi-empirical descriptors) have been shown correlation with anti-malarial activity. b Anti-malarial activity (log IC50) modulation by topological descriptor SssNH count

The above figure depicts two dimensional structures of proposed Artemisinin compounds
In this study, flexible docking was performed using the extra precision (XP) mode in Glide with the default settings. The improvement of XP over standard precision (SP) mode includes the addition of large desolvation penalties to both ligand and protein, assignment of specific structural motifs that give significantly to binding affinity, and expanded sampling algorithms required by scoring function improvement. The XP scoring function includes four constituents: Ecoul (Coulomb energy), Evdw (Van de Waals’s energy), Ebind (items favouring binding), and Epenalty (items hindering binding). To cross check weather docking protocol is working fine, deviation from crystal pose and docked pose was measured by RMSD. The measured deviation should not be more than 2 Å. Tables 2 and 3 illustrate the docking scores resulting from the docked poses of ligands with the anti-malarial targets, plasmepsin-2 and falcipain-2. Figure 5 shows the two and three dimensional ligand-interaction diagram illustrating the major interactions between the ligand and the active sites amino acid residues of plasmepsin-2.


The ligand-interaction diagram illustrates the major interactions between the ligand and the active sites amino acid residues of plasmepsin-2 (PDB: 2IGY). The residues forming the potential interactions are shown as colour
The predicted pharmacokinetic properties of the studied compounds are presented in the Tables 4 and 5.
Discussion
In an attempt to determine the role of structural features, which appears to influence the anti-malarial activity, QSAR study is important. The predicted QSAR model showed good predictivity as it satisfies the required parameters. For evaluation of the external predictive power of the model, it was applied for the prediction of log IC50 values of test set which was not part of training set during model development. The linear graphical representation of fitness plots illustrates the good overlap of observed and predicted activities of the data set (Fig. 1). The radar plot for training set shows a good r2 value as the two lines show a good overlap whereas a good overlap for the test set represents high pred_r2 value (Fig. 2). The statistical output of this model is presented as following:
The robustness and the predictive capacity of the QSAR model were predicted through the statistical parameters. QSAR studies are affected by several factors from which the most important are: (i) the selection of the best molecular descriptors that should include maximum information of molecular structures and a minimum overlap between them (ii) the optimal number of descriptors to be included in the model (Ibezim et al. 2012). The descriptors are commonly used to express different characteristics/attributes of the chemical structure in order to give information about the activity/property being studied. The developed model has identified four descriptors, radius of gyration (geometrical descriptor), Mom inertia Z (topological descriptor), SssNH count (sum of ssNH-Electrotopological-states), a topological descriptor and SK average (semi-empirical descriptors) that have been found to be well correlated with anti-malarial activity. The descriptor SssNH count is produced marked change in anti-malarial activity of the studied compounds. The log IC50 of the compounds radically increases with descriptor SssNH count (Fig. 3b). The compounds 4c, d, 5a–24, 5a, b, 6, 9b, DHA and Artemether are highly active compounds have SssNH count zero and lower radius of gyration values as compared to the compounds 10b, g, k, 12c, 12f. The N8 which is the most active compound has been predicted with zero SssNH count whereas compounds 12f and 12N1 (the least active) are predicted to have two SssNH count. In this context, it was of interest to estimate the effect of this descriptor on the anti-malarial potential of structurally diverse Artemisinin compounds. The results of virtual screening show that 11 compounds (N1, N2, N8, N30, N31, N33, N34, N35, N39, N47 and 10a-N6) are found to have potential anti-malarial activities. Further, compound N8 has great potential of being lead as it has highest predicted activity and also has good binding affinity to both targets (Table 3). Although previous studies were carried out to explore the activity modulating effect of various descriptors in the Artemisinin however in the present study, the predicted descriptors are new that allow a better understanding of the structure of molecules and their activity. Also, the designed and virtually screened compounds are new. The selected Artemisinin compounds have good anti-malarial activities and have greater potential of being drug after optimization.
Molecular docking studies
Although several studies have been done to unravel mode of action of Artemisinin, still exact mechanism is confusing (Golenser et al. 2006) and it is under investigation. Eckstein-Ludwig et al. (2003) assumed that Artemisinin act by inhibiting P. falciparum ATP6 outside the food vacuole after activation by iron. Artemisinin has structural similarities to thapsigargin, an immensely precise inhibitor of sarco/endoplasmic reticulum Ca2-ATPase (SERCA). Studies suggested that Artemisinin likely targets to the P. falciparum cysteine protease and aspartic protease (Qidwai et al. 2012; Eckstein-Ludwig et al. 2003; Ettari et al. 2010). In human erythrocytes, the malaria parasite hydrolyses host hemoglobin for its amino acid requirement. The hemoglobin degradation is carried out by three classes of enzymes named as plasmepsin, falcipain and amino-peptidases and their inhibition may block parasite protein biosynthesis and leading to harmful effect to the parasite (Rosenthal et al. 2002). Therefore, targeting of theses enzymes would be important for malaria treatment and can be utilized as potential anti-malarial drug targets. In order to understand the interaction of Artemisinin derivatives with parasite hemoglobinase (plasmepsin-2 and falcipain-2), docking was performed (Table 2). Docking protocol was checked by RMSD calculations. Deviation from crystal and docked pose was measured. RMS = 1.0111; maximum diff = 3.2469 between atoms 65 and 65 have been predicted. The result of study suggested that studied compounds have good binding affinities to plasmepsin-2 and falcipain-2 (Tables 2, 3). The docked compound N30 has been found with highest binding affinity as predicted by both Glide and PyRx. Similarly, the docked ligand N39 has good binding affinity. Both ligands also have good predicted anti-malarial activities. Comparison of binding affinities of Artemisinin compounds with two anti-malarial targets suggested that the binding affinities are higher for falcipain-2 as compared to plasmepsin-2. The results showed that, studied compounds having minimum binding energy and have good affinity toward the active pocket, thus, they may be considered as good inhibitor of P. falciparum proteases digesting host hemoglobin. Results showed that all the derivatives showed interaction in the active binding pocket of targets and have higher binding affinity as compared to the standard drugs DHA and Artemisinin. QSAR and docking work suggested that the compounds N30 and N39 have great potential of being drug like compounds after optimization against P. falciparum.
Artemisinin may not target to parasite hemoglobinase
The idea that Artemisinin may not acts via targeting to parasite proteases came through the docking of Deoxy-artemisinin. Deoxy-Artemisinin has no effect on P. falciparum and does not inhibit its growth. However, docking energies resulting from the interaction of Deoxy-artemisinin-plasmepsin-2 and Deoxy-artemisinin-falcipain-2 have been predicted nearly equal to that of interaction of Artemisinin-plasmepsin-2 and Artemisinin-falcipain-2. The IC50 values of compound 12f (the least active compound in the model) and Artemisinin are 10,630 and 3.6 nM respectively. The binding affinities of plasmepsin-2 and falcipain-2 with compound 12f are higher than their binding affinities with Artemisinin (Table 3). It seems that Artemisinin and its derivatives may exert their effect through either binding with another target or with multiple targets. Deoxy-artemisinin and 12f docking results may not support that Artemisinin specifically binds to protein and inhibits it. Artemisinin and other members of this class that encompass the peroxide bond, may interact with the electron transport chain of malaria parasites, make free radicals, which could damage mitochondrial functions and ultimately lead to cell death (Wang et al. 2010). Probably Artemisinin does not perform its functions through binding precisely to protein target site and then inhibiting its function. The anti-malarial effect of Artemisinin is due to its selective interaction with malarial mitochondria, which makes reactive oxygen species. The peroxide bond of the molecule is vital to the production of free radicals.
Absorption, distribution, metabolism, excretion, and toxicity studies
During the drug discovery effort many drugs are miscarried due to blood brain permeation failure, toxicity and poor efficacy. Since Artemisinin possess anti-malarial activity and it has poor pharmacokinetic properties, therefore virtual derivatives were designed and properties were investigated. Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties are key players in drug development. Lack of efficacy and unacceptable toxicity are mainly associated with failures of drug discovery. Suitable lipophilic property is important for the drugs to cross membrane and cytosol to reach the target. The ADMET-related properties for instance water solubility, human intestinal absorption, oral bioavailability, blood brain barrier (BBB) penetration, transporter, plasma protein binding, volume of distribution, CYP450, toxicity are used to refine the drug likeness properties (Vlife and software package, version 3.5 2008). In the present study, ADMET properties of the Artemisinin and its derivatives were predicted through ADMETSAR database which is freely available. Several ADMET associated properties, such as water solubility, human intestinal absorption, oral bioavailability, blood brain barrier penetration, P-glycoprotein substrate and inhibitor, renal organic cation transporter, plasma protein binding, volume of distribution, CYP450 substrates and inhibition (CYP1A2, 2C9, 2C19, 2D6 and 3A4), drug-induced liver injury, human Ether-a-go-go-Related gene (hERG) inhibition, rat acute toxicity, skin sensitivity, AMES mutagenicity, carcinogens, fish toxicity, the Tetrahymena pyriformis toxicity have been predicted. For Artemisinin probabilities of being human intestinal absorption, blood–brain barrier penetration, CYP450 substrates and inhibition, human Ether-a-go-go-Related gene (hERG) inhibition and AMES mutagenicity, carcinogenicity, fish toxicity, aqueous solubility (LogS), Caco-2 Permeability and rat acute toxicity have been found 0.9747, 0.9490, 0.8493, 0.9850, 0.9711, 0.7785, 0.9161, 0.8911, -4.3515, 1.3569 and 1.7351 respectively. The detailed predicted properties of other studied compounds with respect to Artemisinin artemether and DHA have been shown (Table 4). Blood–brain barrier penetration probabilities of compounds N2, N22, N35, N39, N40, N42, N45, N46 and N47 are higher than Artemisinin and artemether while such probabilities have been found lower for compounds 10a-N5 and 12N1. Probabilities of human intestinal absorption for N35, N39, N40, N42 and N45 are higher than Artemisinin, DHA and artemether while lower for compounds 10a-N5 and 12N1. Compounds N30, N35, N44 and N47 have been predicted with higher probabilities of Non AMES Toxicity as compared to A62, Artemisinin and artemether. Compounds N2, N8, N33, N35, N39, N40 and N42 have been found to be carcinogens with lower probabilities. Compound N8 has 90% probability of not possessing mutagenic properties and 74% chance of being non AMES toxic. Proposed compounds have been found with better ADME properties, therefore have greater potential of being lead compounds.
Further, pharmaceutically significant properties of Artemisinin analogs were analyzed through QikProp software (Table 5) and compared them with standard drugs, Artemisinin, artemether and DHA. The results predicted through QikProp suggested that compounds N2, N8, N33, N35, N39, N40 and N42 have been found with good ADME properties. The major descriptors reported in QikProp (Schrodinger) software are molecular weight (mol_MW) (150–650), aqueous solubility (QPlogS) (−6.5 to 0.5), apparent MDCK cell permeability (QPPMDCK) (500 great), brain/blood partition coefficient (QPlogBB) (−3.0 to 1.2) and percent human oral absorption (≥80% is high, ≤25% is poor). All the studied compounds have percent human oral absorption 80–100% (Table 5). The first three properties are based on Lipinski rule of five, molecular weight (mol_MW) less than 650, partition coefficient between octanol and water (logPo/w) between −2 and 6.5 and solubility (QPlogS) greater than −7. Brain/blood partition coefficient (QPlogBB) parameter specified about the capacity of the drug to pass through the blood–brain barrier which is important for ADME for investigating druggability. Lipinski’s rule of five pharmacokinetics filter is used as a drug likeness test. This rule is based on the observation that most orally administered drugs have a molecular weight (MW) of five hundred or less, a logP no higher than five, five or fewer hydrogen bond donor sites, and ten or fewer hydrogen bond acceptor sites (N and O atoms). In this study, all the compounds have been found with H donor sites lesser than five and H acceptor sites lesser than ten. Compounds 10a-N5, 12-N1 and 12f have been predicted with molecular weight more than 500. In addition, the bioavailability of all derivatives was assessed through topological polar surface area analysis. The polar surface area (PSA) was calculated by using topological PSA. This descriptor was shown to correlate well with passive molecular transport through membranes and therefore allows prediction of transport properties of drugs and has been linked to drug bioavailability. Generally, any molecule with PSA value of less 100 Å2 is expected to possess good absorption properties. Passively absorbed molecules with a PSA 140 Å2 are thought to have low oral bioavailability. The PSA value for compounds 12-N1, 12F, N31, N34 are more than 100 Å2 while other studied compounds have lower PSA values (Table 5). Artemisinin has PSA value of 64.326 Å2. Because of low oil and water solubility of Artemisinin its use in malaria treatment is limited. Derivatives of Artemisinin showed more potency than their parent compound. The parent compound showed CNS toxicity and limited plasma half-life also.
Conclusion
The present study was carried out to explore predictive QSAR model capable of revealing the structural requirements for potential anti-malarial inhibitors targeting P. falciparum. The developed model is significant, robust and has good reliability and predictive ability. The molecular descriptors found in QSAR equation have encoded information about radius of gyration, mominertia Z, SssNH count and SK Average. The compounds N1, N2, N8, N30, N33, and N39 have been predicted with good anti-malarial activities. The docked poses revealed that, all of the derivatives have high binding affinities against parasite plasmepsin-2 and falcipain-2 (hemoglobin digesting enzymes). QSAR model, oral bioavailability, ADME and toxicity risk assessments suggested that compounds N1, N2, N8, N30, N33, and N39 possesses good drug like properties than Artemisinin and DHA. Therefore, the current study advocates the use of combined methodology of QSAR molecular docking and ADME to search potential leads against the P. falciparum. The information can be useful for the identification, design and development of diverse, potent anti-malarial with great potential of being drug like compounds.
References
Avery MA, Muraleedharan KM, Desai PV, Bandyopadhyaya AK, Furtado MM, Tekwani BL (2003) Structure-activity relationships of the antimalarial agent artemisinin. 8. Design, synthesis, and CoMFA studies toward the development of artemisinin-based drugs against leishmaniasis and malaria. J Med Chem 46:4244–4258
Basak SC, Mills D, Garg R, Bhhatarai B (2010) A QSAR study of HIV protease inhibitors using theoretical descriptors. Curr Comput Aided Drug Des 6:269–282
Bhhatarai B, Garg R (2008) Comparative QSAR as a cheminformatics tool in the design of dihydro-pyranone based HIV-1 protease inhibitors. Curr Comput Aided Drug Des 4:283–310
Cheng F, Li W, Zhou Y et al (2012) AdmetSAR: a comprehensive source and free tool for assessment of chemical ADMET properties. J Chem Inf Model 52:3099–3105
Eckstein-Ludwig U, Webb RJ, van Goethem IDA et al (2003) Artemisinin target the SERCA of Plasmodium falciparum. Lett Nat 424:957–961
Ettari R, Bova F, Zappala M, Grasso S (2010) Micale N falcipain-2 inhibitors. Med Res Rev 30:136–137
Fidock DA, Rosenthal PJ, Croft SL, Brun R, Nwaka S (2004) Antimalarial drug discovery: efficacy models for compound screening. Nat Rev Drug Discov 6:509–520
Golenser J, Waknine JH, Krugliak M, Hunt NH, Grau GE (2006) Current perspectives on the mechanism of action of artemisinins. Int J Parasitol 36:1427–1441
Ibezim E, Duchowicz PR, Ortiz EV, Castro EA (2012) QSAR on aryl-piperazine derivatives with activity on malaria. Chemomet Intell Lab Syst 110:81–88
Kar S, Kar S (2010) Control of malaria. Nat Rev Drug Discov 7:511–512
Liu Y, Cui K, Lu W (2011) Synthesis and antimalarial activity of novel dihydro-artemisinin derivatives. Mole 16:4527–4538
O’Neill PM, Miller A, Bishop LPD et al (2001) Synthesis, antimalarial activity, biomimetic iron (II) chemistry, and in vivo metabolism of novel, potent C-10-phenoxy derivatives of dihydroartemisinin. J Med Chem 44:58–68
Vlife MDS software package, version 3.5 (2008) Supplied by Vlife science technologies Pvt. Ltd, Pune
Posner GH, Parker MH, Northrop J et al (1999) Orally active, hydrolytically stable, semisynthetic, antimalarial trioxanes in the artemisinin family. J Med Chem 42:300–304
Qidwai T, Yadav DK, Khan F et al (2012) QSAR, docking and ADMET studies of artemisinin derivatives for antimalarial activity targeting plasmepsin II, a hemoglobin-degrading enzyme from P. falciparum. Curr Pharm Des 37:6133–6154
Rosenthal PJ, Sijwali PS, Singh A, Shenai BR (2002) Cysteine proteases of malaria parasites: targets for chemotherapy. Curr Pharm Des 8:1659–1672
Schrodinger (2016) Schrodinger Release 2016-4. Schrödinger, LLC, New York
Trott O, Olson AJ (2010) AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization and multithreading. J Comput Chem 31:455–461
Wang J, Huang L, Li J et al (2010) Artemisinin directly targets malarial mitochondria through its specific mitochondrial activation. PLoS One 3:e9582
World Malaria Report (2013) Global Malaria Programme, WHO, Geneva. http://www.who.int/malaria/about_us/en/
Xiang Y, Zhang Z, Xiao A, Huo J (2009) Recent studies of QSAR on inhibitors of estrogen receptor and human eosinophil phosphodiesterase. Curr Comput Aided Drug Des 5:200–214
Yadav DK, Dhawn S, Chauhan A et al (2014) QSAR and docking based semi-synthesis and in vivo evaluation of artemisinin derivatives for antimalarial activity. Curr Drug Targets 8:753–761
Acknowledgement
The author wishes to express gratitude to Prof. B.N. Mishra, Dept. of Biotechnology, IET, Lucknow, for giving inspiring and innovative suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made.
The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder.
To view a copy of this licence, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Qidwai, T. QSAR modeling, docking and ADMET studies for exploration of potential anti-malarial compounds against Plasmodium falciparum . In Silico Pharmacol. 5, 6 (2017). https://doi.org/10.1007/s40203-017-0026-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40203-017-0026-0