
Abstract
Purpose of Review
Amyotrophic lateral sclerosis and frontotemporal dementia (ALS-FTD) spectrum disorder is a rare fatal disease with strong genetic influences. The implementation of short-read sequencing methodologies in increasingly large patient cohorts has rapidly expanded our knowledge of the complex genetic architecture of the disease. We aim to convey the broad history of ALS gene discovery as context for a focused review of 11 ALS gene associations reported over the last 5 years. We also summarize the current level of genetic evidence for all previously reported genes.
Recent Findings
The history of ALS gene discovery has occurred in at least four identifiable phases, each powered by different technologies and scale of investigation. The most recent epoch, benefitting from population-scale genome data, large international consortia, and low-cost sequencing, has yielded 11 new gene associations. We summarize the current level of genetic evidence supporting these ALS genes, highlighting any genotype-phenotype or genotype-pathology correlations, and discussing preliminary understanding of molecular pathogenesis. This era has also raised uncertainty around prior ALS-associated genes and clarified the role of others.
Summary
Our understanding of the genetic underpinning of ALS has expanded rapidly over the last 25 years and has led directly to the clinical application of molecularly driven therapies. Ongoing sequencing efforts in ALS will identify new causative and risk factor genes while clarifying the status of genes reported in prior eras of research.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The ALS-FTD Spectrum of Disease Has Complex Genetic Architecture
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease clinically recognized for progressive paralysis due to the degeneration of spinal cord and cortical motor neurons [1]. Even with improvements in the multi-disciplinary care of patients and two medications approved by the FDA, ALS is invariably fatal due to respiratory failure, with patients surviving a median of 3 years after symptom onset. The clinical picture of a typical ALS patient is so dominated by mounting motor disability that ALS historically been considered a “motor neuron disease.” This nomenclature likely delayed recognition that ALS is a multisystem disease in which a range of non-motor neuronal populations can also be affected [2, 3]. The most commonly involved non-motor neurons are those regulating fronto-executive cognitive function; more than half of ALS patients develop fronto-executive dysfunction during the course of the disease with 5–10% meeting criteria for fronto-temporal dementia (FTD) [4,5,6,7]. In addition to frequent clinical co-occurrence, the ALS-FTD spectrum shares an increasing number of genetic causes and underlying neuropathology (mislocalized and aggregated TDP-43 protein) [8, 9]. This pathological hallmark makes the ALS-FTD spectrum of disease one among many neurodegenerative diseases that are characterized by pathological protein aggregates, the so-called proteinopathies.
Knowledge of key biological pathways important to ALS-FTD pathogenesis has been greatly advanced by progress in uncovering causative genes and genetic risk factors for the disease. From the very first published case series, familial ALS (fALS) has been recognized, with 7–10% of individuals having a family history of either ALS or FTD [10]. It is within these families that the bulk of monogenic causes have been found. However, 85–90% of ALS patients, so-called sporadic ALS (sALS), have no known family history and typically do not carry rare and high penetrant mutations in known genes. Genetic factors influence disease propensity in this group as well, with studies showing polygenic risk from many common variants [11, 12] or, as suggested by preliminary data, for smaller numbers of larger effect variants (so-called oligogenic inheritance) [13, 14]. Thus, the genetic structure of ALS is proving to be complex, with a broad range of allele frequencies and effect sizes (reviewed in [15]).
Timeline of Gene Discovery in ALS
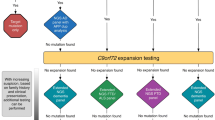
Since Charcot described motor neuron disease in the late 1800s, over 40 ALS genes have now been associated with ALS, explaining 25–35% of FALS and 5–10% of apparent SALS. Genetic discoveries in ALS have occurred in four distinguishable epochs, each leveraging important methodological advances (Fig. 1).

Strategies for the identification of ALS genes. Strategies have evolved from individual gene mapping to increasingly powered technological and statistical methodologies. Since the implication of SOD1, 42 additional genes have been implicated in ALS to date with variable genetic support in replication and functional studies. GWAS, genome-wide association studies; WES, whole-exome sequencing; WGS, whole-genome sequencing
In the first era (c.1990–1999), gene discovery required large ALS families powered for high-resolution linkage analysis followed by the arduous task of positional cloning and sequencing. Accordingly, discoveries came slowly in this era for all human diseases and yielded only a single gene for typical ALS, SOD1 [16].
Publication of the human genome inaugurated the second era (c.2000–2008). As an exhaustive compendium of genes and their genomic location, the draft genome allowed a simple database query to replace time-consuming positional cloning (ALS2, [17]). Genes within a linkage peak could be quickly prioritized for sequencing based on predicted biological roles. Large families were still required to identify ALS-associated loci but hypothesis-driven candidate gene sequencing could now be pursued. Candidate genes could be those causing other human diseases with phenotypic overlap (DCTN1, GRN, CHMP2B, FIG 4), implicated by neuropathological analysis of human tissues (TARDBP, PRPH), biomarker investigations (ANG), or insights from mouse models of ALS. During this era, the availability of increasingly cheap genome-wide genotyping of common variants made unbiased genome-wide association studies (GWAS) possible [18]. Instead of depending on large families, the success of candidate gene and GWAS methods hinge on large numbers of DNA samples and replication in additional cohorts. This fact prompted organized efforts to collect and bank samples from patients, family members, and controls (predominantly of European ancestry). Some of these efforts (e.g., the NINDS MND collection housed at Coriell) recognized the importance of consenting participants for broad sample and data sharing, ensuring some cohorts would follow an open-access model rather than remaining in the hands of single institutions or consortia. During this era, nearly a dozen genes were reported to cause ALS and common variations at a dozen loci were implicated by GWAS. Only two of these associations have been repeatedly replicated (the region around C9ORF72 and at UNC13A, with OR ~ 1.2 each [12, 19••]), but the evidence for others has either rejected an association or been inconclusive.
The explosion in sequencing speed and reduction in costs brought by “next-generation” short-read sequencing methods ushered in the third era (c.2009–2014) by permitting the rapid and simultaneous sequencing of larger numbers of genes (targeted panels and whole exomes) in increasing numbers of ALS patients. Studies could rapidly identify mutated genes within a locus implicated by linkage in larger ALS families, if they could be found—families with DNA from multiple affected individuals were increasingly scarce [20]. Alternatively, families too small for linkage could be analyzed for recurrently mutated genes. Because whole-exome sequencing returned genome-wide mutational data, it was learned that several other known non-ALS disease genes had ALS presentations within their phenotypic spectrum (VCP, MATR3). These methods also showed that portions of the ALS population carried potentially pathogenic variants in more than one ALS gene and possibly represented oligogenic inheritance [14, 21]. This era also saw the first use of whole-genome sequencing (WGS) and repeat-primed PCR to discover the most common cause of ALS to date, expansions of a GGGGCC hexanucleotide repeat in the C9orf72 gene [22, 23], and even larger-scale and increasingly prospective DNA sample collections (e.g., Project MinE), including in non-European populations. More than 15 genes and additional GWAS loci were reported in this 5-year period, but approximately one-third of these associations remain uncertain pending additional replication and/or functional studies.
As panels of known ALS genes became inexpensive and diverse geoancestry cohorts assembled, a fuller characterization of gene mutation frequencies in both familial and “sporadic” patients have revealed important differences between ancestries [24]. For example, ~ 60% of European families are explained by known genes (C9ORF72>>SOD1>>TARDBP>FUS), while in Asia, the 40% of families that are currently explained have a different causative gene distribution (SOD1>>>FUS>TARDBP>C9ORF72). Mutations in these same genes can be found in ~ 8% of “sporadic” European patients (C9ORF72>>>>SOD1>>TARDBP>FUS) and ~ 3% in Asian populations (SOD1>>FUS>TARDBP=C9ORF72) [25].
The fourth and current era (c.2015 onward) has been enabled by the large and well-annotated sample repositories focused on simplex/sporadic cases, broader data sharing/consortia efforts, and the falling costs of whole-genome sequencing. Ever larger GWA studies have validated known loci and provided strong evidence for new regions [12, 19••], while comprehensive ALS gene ascertainment has continued uncovering individuals with possible oligogenic ALS [21, 26]. By far the biggest advance in ALS gene discovery has been the application of improved statistical frameworks for genome-wide rare variant case-control studies (e.g., collapsing analysis, rare-variant burden testing [27, 28, 29••]. The ability to identify genes or other genomic regions enriched for mutations in ALS has played a role in implicating at least 11 new ALS-associated genes including TBK1, TUBA4A, KIF5A, ANXA11, TIA1, CCNF, DNAJC7, NEK1, C21orf2, LGALSL, and GLT8D1. These genes are summarized below, focusing on the strength of genetic and functional evidence, genotype-phenotype correlations, proposed mechanisms by which these genes could lead to neurodegeneration, and gaps in our understanding that need filling by future investigation.
Many ALS Genes Highlighting Key Shared Pathways
By overlapping the known functions of causative ALS genes, the field has gained valuable insight into key cellular pathways underlying the pathogenesis of the disease. Although many pathways are highlighted by the genetics of ALS, several are overwhelmingly implicated by the convergence of multiple genes. These include RNA processing, proteostasis, neuroinflammation, vesicle trafficking, and axonal transport.
RNA Processing and Metabolism
TARDBP (Tar DNA binding protein 43 kDa; encoding TDP-43 protein) and FUS encode “prion-like domain” (PLD)–containing proteins with increased aggregation propensity that carry key functions in RNA processing. Predominantly localized to the nucleus, their ability to shuttle between the nucleus and cytosol is compromised in ALS, resulting in pathological TDP-43 and FUS inclusions in the cytosol (reviewed in [30]). Mutations in the genes themselves (TDP-43 C-terminal domain) remain rare [31], yet TDP-43 protein deposition within insoluble cytoplasmic inclusions is seen in almost all SALS cases, while a minority of FALS cases present instead FUS and SOD1 deposition, with SOD1/FUS and TDP-43 deposition being mutually exclusive. TDP-43 aggregation and pathomechanisms are therefore more widely applicable to our understanding of the pathogenesis of ALS [32, 33]. Misfolding and accumulation into cytoplasmic non-functional aggregates of RNA-binding proteins involved in all steps of mRNA transcription, processing, storage, and degradation have severe consequences in multiple cell types. The loss of TDP-43 and FUS from the nucleus (“loss-of-function”), the formation of protein aggregates (“gain-of-function”), and a combination of both have been implicated in ALS. Loss-of-function mechanisms include dysregulation in RNA metabolism, splicing, and mRNA transport (via hnRNPs for TDP-43 and ELAV-4 for FUS) [34, 35]. Mutations in TARDBP can exhibit disease-causing effects in a toxic gain-of-function manner too: misfolding and accumulation of TDP-43 in the cytoplasm sequester mRNA transport proteins and even directly sequester mRNAs into aggregates, impeding efficient mRNA transport in the long processes of astrocytes and neurons, thereby disrupting essential functions away from the nucleus [36]. In addition to TDP-43 or FUS ubiquitylation, aging, or cellular stress factors, genetic factors related to cellular architecture have emerged as central underlying contributors to ALS pathology. A role for nuclear import (Nup proteins) and dysfunction in stress granule (SG) dynamics in seeding TDP-43 aggregation has crystallized recently with mutations in TIA1 and CCNF [37], see below). Tankyrase-1/2 inhibitor Veliparib was found to mitigate cytoplasmic accumulation of TDP-43 in SGs in mammalian systems, likely by inhibiting PARylation of SGs and allowing TDP-43 to shuttle back to the nucleus offering a potential therapeutic target [38]. The specificity of the mechanism of action will be interesting to test as hnRNPA1, another RBP and ALS gene, undergoes PARylation for nucleocytoplasmic shuttling [39]. The interplay between TDP-43 aggregation, SGs, and the nuclear membrane is not restricted to ALS and develops as a common theme in neurodegeneration with similar involvements of SGs and the nuclear pore in Tau and TDP-43 pathology in Alzheimer disease [40, 41].
Proteostasis
Cellular stress and subsequent pathological protein misfolding are a common pattern in all neurodegenerative diseases, including ALS-FTD where aggregated TDP-43 and other misfolded proteins are pathological hallmarks [33]. Many established ALS genes play key roles in regulating autophagy or the ubiquitin-mediated pathways for degrading misfolded proteins. These include OPTN, SQSTM1, UBQLN2, and VCP. Several of the newly reported genes also act in these pathways. TBK1 is a clear regulator of autophagy and directly interacts with OPTN [42]. DNAJC7 is a heat shock protein whose mutations may disrupt its ability to act as an effective intracellular chaperone, possibly resulting in increased burden of protein misfolding in ALS-FTD [28]. TIA1 plays a key role in the nucleation of stress granules [43], which are emerging as a potential nidus for the aggregation of TDP-43 and other ALS-related proteins FUS and hnRNPs [44,45,46]. CCNF encodes a protein called cyclin F, a binding partner of another ALS-associated protein VCP. A recent study demonstrated that, in cell culture, CCNF mutations may contribute to ALS pathogenesis by increasing the ATPase activity of VCP in the cytoplasm, which in turn increases TDP-43 aggregation [47].
Neuroinflammation
The role of microglia and other effector cells of the immune system has been well recognized from human neuropathological and animal modeling data. Perhaps the recent genetic validation of its importance comes from the discovery of mutations in TANK-binding Kinase (TBK1). A primary function of TBK1 is to activate autophagy and inflammatory pathways in response to pathogen exposure (mediated through activation of both NF-kB and interferon signaling). The complex role of neuroinflammation in ALS is illustrated by mouse studies of the interaction between TBK1 and SOD1: loss of one copy of TBK1 precipitated earlier disease onset with impaired autophagy but counter-intuitively extended survival by reducing inflammatory activation in later stages of disease [48, 49]. These studies appear to provide evidence for the deleterious effects of neuroinflammation in late disease, highlighting the therapeutic potential of targeted anti-inflammatory therapies as disease-modifying agents.
Impaired Axonal Transport
Maintaining adequate axonal transport is crucial in large cells such as motor neurons and is known to be dysregulated in ALS [50]. Axonal transport requires the assembly and maintenance of microtubule networks that act as transport highways, facilitating the anterograde and retrograde movement of cargo, including mRNAs, organelles, proteins, and much more. NEK1, through its interactions with FEZ1 and FEZ2, has been shown to be involved in pathways crucial to the maintenance, growth, and repair of these networks with mutations demonstrating impaired microtubule stability [51]. Furthermore, mutations in TUBA4A examined in cell culture demonstrated an impairment in microtubule network assembly and reduction in microtubule stability [52]. Mutations in KIF5A are also thought to result in impaired axonal transport as KIF5A is a kinesin, a microtubule-associated motor protein, responsible for organelle transport, including lysosomes [19••]. Cells also rely on local translation of mRNAs to respond to local stimuli without the need for global changes in the cellular environment [32]. Local translation of mRNAs is reliant on efficient mRNA transport along axons. A recently identified ALS-associated gene, ANXA11 [53], is responsible for tethering mRNAs as part of an RNA granule complex to actively transported lysosomes, enabling them to hitchhike to their terminal destination where they facilitate local neuronal translation.
Challenges in the Interpretation of ALS Genetics
The number of sequenced ALS cases and controls has grown exponentially over the last decade such that the number of reported ALS genes has been doubling every 4 years [15]. This pace coincides with equally rapid advances in molecular therapies like viral gene delivery or gene-silencing and anti-sense oligonucleotides (ASOs) for gene downregulation or splicing modifications. ASOs targeting SOD1, C9orf72, and FUS are already in clinical trials, and several pharmaceutical or biotech companies are expected to launch trials of AAV vectors targeting SOD1 in the next year. As companies deliberate on which genes to target next, it is essential that the genetic and functional evidence for reported genes be continuously re-evaluated.
It is worth explicitly stating that our certainty that a gene strongly influences ALS is correlated with the route by which the ALS association was made and the length of time since the gene was first reported (Fig. 1). Those genes that were identified in large family linkage studies are clearly causative even on the strength of a single reported mutation (e.g., SOD1, C9orf72, VAPB, SETX). Other genes have been securely implicated when smaller unrelated families show segregation of the same mutation (TARDBP, FUS), when multiple different mutations produce the same molecular defect (KIF5A, TBK1), or when the mutations reported in smaller families cluster regionally within protein domains (UBQLN2). With rare-variant burden testing and GWA studies, the strong evidence comes from the unbiased nature of the approach, the statistical power of genome-wide association, and replication.
For many ALS genes reported from candidate gene sequencing efforts or found in small families, the evidence is not as definitive. In many cases, the literature consists of variants unique to ALS patients identified by sequencing many simplex/sporadic patients. If variants have been found in familial cases, only rarely has DNA been available from other individuals for segregation testing. Even when a second affected individual is available for testing, it is typically a first-degree relative who has a 50% chance of sharing any variant, ALS-related or not. In the absence of clear-cut evidence from the genetic data, neuropathological signatures can strengthen the case for gene pathogenicity (e.g., aggregates of the ANXA11 gene product exclusive to mutation carriers [53] or the remarkable number of Lewy body–like inclusions found in TIA1 carriers [54]). Unfortunately, the number of genes where even a single mutation carrier has had neuropathological assessment is small. Finally, experimental data from functional studies or animal modeling can be useful in interpreting the importance of these genes. In ALS, even for genes with long-standing and incontrovertible evidence for monogenic causation, recapitulating disease features in mice or finding reproducible cellular phenotypes in patient-derived stem cell models have been challenging. For many of the uncertain ALS genes, there is some experimental data showing functional effects of some ALS-associated mutations, sometimes even in the robustly implicated pathways outlined earlier. However, the specificity of the demonstrated defects is almost never investigated for the gene variants that were also found in controls. Thus, there is uncertainty on the status of data for individual genes which is represented in Fig. 1 by color coding. These assignments are based on interpretation of the current literature, but other experts may feel more or less confident in particular individual genes based on their own weighting of the available evidence. The important point of the color-coding is to emphasize that not every gene reported to cause ALS and appearing on a timeline shares the same level of certainty.
Recently Discovered ALS Genes
As many of the newly discovered genes account for smaller proportions of the ALS population and have only recently been discovered, most have yet to have their neuropathological phenotypes studied or been studied in model systems. Further studies are clearly warranted to fully understand the functional consequences of these gene mutations.
TBK1: TBK1 (TANK-binding kinase 1) plays important roles in autophagy and activation of innate immunity and is a direct regulator of optineurin, another ALS-associated gene [42, 55]. It was the first gene for ALS identified using rare variant burden collapsing [56] in a predominantly sporadic cohort and was subsequently validated by a similar approach in familial ALS [57]. The strongest signal in both studies came from loss of function mutations, several of which have shown segregation in large families or been found in multiple studies. Thus, loss-of-function mutations in TBK1 are a clearly established cause of ALS. Several single amino acid deletions have been reported and may be acting by reducing its phosphorylation [58]. Individual studies and meta-analysis [59] have confirmed increased risk with missense variants but implicating specific mutations has been challenging: only a single missense mutation (p.Arg573Gly) has shown segregation in familial disease (primary lateral sclerosis or FTD, [60]). Therefore, at this time, the pathogenicity of specific missense changes in TBK1 is impossible to ascertain in the absence of segregation or clear functional data [61]. Indeed, some missense variants from ALS patients impair TBK1 signaling [57, 58]. Given the challenges of interpreting the pathogenicity of missense changes, it is difficult to estimate how common TBK1-induced ALS is. A German cohort reported around 1% of LOF mutations in FALS [62].
TBK1 mutations carriers show typical ages at onset, sites at onset, and rates of progression but appear to have higher rates of cognitive involvement and frank FTD [10]. Mutation carriers with FTD alone have been reported [63]. In fact, TBK1 may be the second only to C9orf72 in frequency among patients with ALS and FTD [64]. Atypical phenotypes, including primary lateral sclerosis [60], progressive supranuclear palsy (PSP) [65], cerebellar ataxia [66], and corticobasal degeneration [67] have also been described. PSP and ataxia in individuals carrying a single amino acid deletion also reported in ALS-FTD and for which functional loss-of-function data exists [58].
TUBA4A: TUBA4A is a major constituent protein of microtubules, essential for cytoskeletal integrity and axonal transport. TUBA4A was first implicated in ALS by burden testing methods in a large familial ALS cohort with confirmation in a second cohort [52]. Unfortunately, all pedigrees with potential mutations were too small or lacked DNA for definitive segregation of any single mutation. Among the other reported rare TUBA4A variants is p.A383T, found in both an Italian FALS patient and a sporadic patient of Chinese ancestry [67]. Other investigations in familial ALS or FTD have found few or no additional TUBA4A mutations [68, 69], and burden testing in large cohorts have not demonstrated an association in sporadic patients (ALSdb and Project Mine). The functional effects of only a few mutations in TUBA4A have been examined in cell culture systems. Not surprisingly, some variants demonstrate impaired microtubule network assembly and reductions in microtubule stability [52]. Neuropathological analysis of mutation carriers has not been reported, but tubulin A4A is not clearly mislocalized in spinal cord from sporadic ALS patients [52]. To date, patients with putative mutations in TUBA4A have shown spinal onset with typical upper and lower motor neuron involvement and only a small number have had cognitive impairment or FTD [52, 67, 68].
KIF5A: KIF5A is a kinesin, a microtubule-associated motor protein, responsible for organelle transport, including lysosomes and mitochondria, and missense mutations in the N-terminal motor domain of KIF5A have long been known to cause spastic paraplegia type 10/CMT2 (SPG10/CMT2) [70,71,72,73]. Recent evidence links loss-of-function mutations in the C-terminal cargo binding domain to ALS. This evidence came from both the largest GWAS in ALS to date and two rare variant burden analyses in familial ALS cohorts [19••, 49]. ALS-associated mutations were found to cluster at the 3′ end of the gene where they disrupt splicing of exon 27 and disrupt the C-terminal cargo binding domain. Loss of the cargo-binding domain or haploinsufficiency presumably disrupts axonal transport in some fashion, though this has yet to be demonstrated. The neuropathology of KIF5A loss-of-function carriers has not been reported. ALS patients with loss-of-function mutations in KIF5A show a lower age at onset (46 vs 65 years) and a remarkably long survival (10 vs. 3 years) [19••]. It should be noted that while the strongest association is ALS with C-terminal loss-of-function mutations and SPG10/CMT2 with N-terminal missense mutations, there have been some individuals within SPG10/CMT2 families presenting with ALS phenotypes, raising the possibility that even missense KIF5A mutations in the N-terminal motor domain can present with ALS [74].
ANXA11: ANXA11 is involved in the calcium-dependent formation of vesicles and potentially mRNA tethering for transport along axons. Its association with ALS was discovered when whole exome data from 50 family ALS cases were overlapped for recurrently mutated genes or variants [53]. This analysis “rediscovered” p.M337V in TARDBP and also revealed a second mutation—p.D40G in ANXA11. This mutation was identified in additional patients with ALS and represents a shared European founder. Post-mortem analysis of a single p.D40G mutation carrier showed large cytoplasmic inclusions of insoluble ANXA11 protein [53]. However, the most definitively causative mutations do not seem to enhance aggregation of ANXA11, but instead disrupt calcyclin binding. There is also evidence that some mutations impair the ability of ANXA11 to tether RNA granules to actively transported lysosomes [75]. ANXA11 mutations are rare in ALS, the handful of reported patients showing typical ALS features. Only a single individual has had clinically apparent dementia [75].
TIA1: TIA1 is an RNA-binding protein like TARDBP and FUS, with a key role in the formation of stress granules. Mutations in TIA were already associated with a distal myopathy with vacuoles (just like other ALS genes VCP, MATR3, HNRNPA2B1). Whole-exome sequencing in a small ALS-FTD family identified a segregating TIA1 variant in the low-complexity domain and subsequent analysis of this specific region in cases and controls showed a statistical burden of rare variants in ALS cases [43]. Targeted sequencing in several Chinese ALS cohorts have identified other rare mutations in this region of TIA1, but no segregating mutations have been reported [76, 77]. Attempts to replicate this genetic association within three larger cohorts have been unsuccessful [29••, 78, 79••], emphasizing that more investigation is needed. However, there are strong indications from neuropathologic evaluation that TIA1 mutations contribute to ALS pathogenesis. Nine individuals with seven different TIA1 mutations (P362L; M334I; A381T; G355R; V294M; V360M; A381T) in the TIA1 gene showed a remarkably consistent pathological phenotype characterized by frequent round eosinophilic, Lewy body–like inclusions [54] in addition to widespread TDP-43 aggregation. In addition to this shared unique pathology, almost all reported carriers have had clinical FTD in addition to ALS.
CCNF: CCNF encodes cyclin F, a member of the FBOX family of proteins and serves as the substrate-binding module of the SKP1-CUL1-F-box protein (SCF) ubiquitin ligase complex. In this role, it binds substrates and directs their ubiquitylation for subsequent degradation by the ubiquitin proteasomal pathway. CCNF was linked to ALS when single-nucleotide polymorphism (SNP) and microsatellite linkage analyses in a large Australian family pointed to Chromosome 16p13.3 and whole-exome sequencing identified p.S621G in CCNF [80]. Other rare or even novel variants have been identified in other cohorts, but none of them have been recurrent or shown to segregate with disease. Mutations reported in ALS do not alter cyclin F stability or disrupt formation of the SCF complex [47], but in vitro can increase the ATPase activity of VCP (another ALS-causative gene) while others impair ubiquitin-mediated proteasomal degradation [47]. Either mechanism disrupts the normal processing of TDP-43, leading to its abnormal cytoplasmic accumulation [47, 81]. The strongest effect occurred with the clearly segregating mutation (p.S621G), which also disrupts Lys48-specific ubiquitylation and impairs autophagy [81]. When expressed in zebrafish, this mutation also induced increased spinal cord neuronal cell death, impaired motor axonal length, and produced a motor deficit [82]. In sum, the functional analysis of cyclin F supports pathogenicity of the only segregating mutation (p.S621G), with work yet to be done in clarifying whether other identified mutations are causative. The entire spectrum of ALS-FTD has been reported in individuals with rare variants in CCNF with no standout features to date, and neuropathological evaluation has yet to be reported [80].
DNAJC7: DNAJC7 encodes a heat-shock protein co-chaperone known to regulate key protein-folding chaperones Hsp70 and Hsp90 with roles in neuroprotection. Understanding of DNAJC7’s role in protein folding and quality control has largely been in the context of steroid hormone regulation, but there is emerging evidence that it may suppress cellular responses to innate immunity [83]. Gene-based burden methodologies demonstrated enrichment of premature truncation variants in DNJAC7 [28]. The discovery of DNAJC7 was enabled by the aggregation of three previous ALS whole-exome datasets into a much larger collaborative cohort with unprecedented power. This illustrates the important fact that further gene discovery in ALS will require increasing cooperation and harmonization across the many genetic projects in the field and makes the case for open-access models of data production and sharing. Premature truncating mutations in DNAJC7 are presumed to act via haploinsufficiency, but functional investigations have not been reported. Similarly, the neuropathological and clinical phenotypes of mutation carriers are yet to be described.
NEK1: NEK1 (NIMA related kinase 1) is a serine/threonine kinase involved in cell-cycle regulation, axonal development, and implicated in axonal polarity and axon guidance as well as DNA damage repair [84, 85]. Loss-of-function mutations were first implicated in ALS by gene burden testing in a mostly sporadic cohort [56] but then validated in two independent familial cohorts of European ancestry [84, 86] and in a Chinese population [87]. The weight of evidence, as with TBK1 and OPTN, favors loss-of-function mutations as causative, but rare missense variation also modestly increases risk [86], especially the p.Arg261His variant [84, 88]. How loss of NEK1 function or missense mutations lead to ALS is not yet known, but may lead to the accumulation of DNA damage that contributes directly to cell damage and cell death in neurons that are already vulnerable due to widespread protein misfolding, as demonstrated in cell culture [89]. It is also possible that NEK1 mutations directly contribute to protein misfolding. Indeed, insoluble SOD1 aggregates were recently reported in an ALS case with a NEK1 R812X mutation at post-mortem [90]. Detailed phenotypic information for either loss-of-function or missense variant carriers has not been published.
C21orf2: The biological role of C21orf2 (or CFAP410) is poorly understood, but limited evidence suggests that it functions in DNA damage control and repair similar to NEK1 and plays a role in cilia formation [91]. Its drosophila homolog is implicated in actin structure [92], with possible relevance to ALS genes like TUBA4A, PFN1, KIF5A, and ANXA11. C21orf2 was identified in a large genome-wide association study of common variation [19••] with subsequent replication in a large cohort study [12]. The same study also used gene burden testing to demonstrate an excess of rare non-synonymous C21orf2 variants in patients with ALS. To date, segregating or recurrent mutations have not been reported, making it difficult to interpret the pathogenicity of individual mutations. Investigations into the functional implications of ALS-associated variants have only recently begun, with structural modeling suggesting that a fraction of them are likely to disrupt the protein structure [93]. The clinical and neuropathological phenotypes of ALS patients carrying C21orf2 mutations have not yet been reported.
LGALSL: LGALSL encodes a galectin-related protein whose function is largely unknown. As with most of the new genes highlighted in this review, LGALSL was identified using gene burden methods on whole-exome sequencing. Unlike other discoveries however, this study used burden testing methods capable of implicating mutational hotspots or domains rather than entire genes [29••]. As has been the case for most other gene burden methods, loss-of-function mutations were the strongest association with ALS. This very recent report has not yet been replicated, and no mutations segregating in familial ALS or ALS-FTD have yet been reported. A subsequent paper demonstrated LGALSL variants in 0.382% of cases and 0.068% of controls, hence identifying it as a non-significant candidate [56]. In the initial paper, carriers of LGALSL mutations had significantly early age at symptom onset (13 years earlier than average for the cohort) but were otherwise typical of ALS. Neuropathology of mutation carriers has not been reported. If future studies confirm an association between LGALSL and ALS, this gene could implicate an entirely new biological pathway in ALS pathogenesis.
GLT8D1: GLT8D1 is a ubiquitously expressed glycosyltransferase of unknown function implicated in ALS by family-based whole-exome analysis and candidate gene sequencing [94]. Study of a small ALS family identified a disease-associated haplotype containing rare missense mutations in two genes, ARPP21 and GLT8D1 [94]. The same haplotype was found in additional ALS patients, along with a much smaller number of patients who carried one or the other mutation, but not both. The identification of two ALS patients with other rare variants in exon 4 of GLT8D1 made this gene statistically more likely to be causative, but a role for ARPP21 has not been excluded. The authors hypothesized there could be a synergistic effect between the two mutations. Interestingly, a study in Chinese patients found a non-significant enrichment of mutations in ARPP21 but not GLT8D1 [95], making further study of these two genes essential for clarifying their role in ALS. ARPP21 is an RNA-binding protein with known roles in neuronal dendrite elaboration [96], while GLT8D1 is a ubiquitously expressed glycosyltransferase of unknown function. Missense variants demonstrate a mild deficit in enzyme activity and, in an overexpression model, induce a mild increase in cytotoxicity in vitro and impaired locomotion in zebrafish [95]. As with LGALSL, conclusive implication of GLT8D1 would highlight a previously unexplored pathway in ALS pathogenesis. The small number of cases reported thus far makes phenotypic patterns tentative, but plausible as mutation carriers showed earlier onset of disease and those carrying both mutations on the haplotype (GLT8D1 and ARPP21) trended toward shorter survival.
Conclusions and Future Directions
Recent advances in understanding the complex genetic architecture of ALS have been the direct consequence of global collaborative approaches to gather large cohorts with harmonized, well-curated clinical information from a large number of sites. It is therefore crucial that this continues and that there is a standardized approach to such collaborative efforts with the adoption of comparable methodologies, open access to data, and an emphasis on high-quality genetic and biological validation. Even with these concerted efforts, the genes identified will be rare in the ALS population and the mutations difficult to interpret on the bases of genetic data alone. This argues that concerted efforts for neuropathological evaluation and model systems to assess functional effects of mutations will be increasingly important in the interpretation of ALS genetic variants.
This review highlights the importance of ongoing aggregation of datasets through data sharing and collaboration at the very inception of large-scale genomic investigation to improve ascertainment of structural variants, somatic variation, copy number variation, and novel repeats contributing to disease. Future studies exploring these large datasets, including the potential application of artificial intelligence, will need to focus on the development of capabilities to interpret non-coding variation with robust and reproducible effect sizes. Also, the collection of longitudinal data may facilitate the identification of key associations with disease progression and other phenotypic traits, which would generate promising targets for molecularly directed personalized therapies.
References
Papers of particular interest, published recently, have been highlighted as: •• Of major importance
Geser F, Brandmeir NJ, Kwong LK, Martinez-Lage M, Elman L, McCluskey L, et al. Evidence of multisystem disorder in whole-brain map of pathological TDP-43 in amyotrophic lateral sclerosis. Arch Neurol. 2008;65(5):636–41.
Fang T, Jozsa F, Al-Chalabi A. Nonmotor symptoms in amyotrophic lateral sclerosis: a systematic review. Int Rev Neurobiol. 2017;134:1409–41.
Silani V, Ludolph A, Fornai F. The emerging picture of ALS: a multisystem, not only a “motor neuron disease”. Arch Ital Biol. 2017;155(4):99–109.
Goldstein LH, Abrahams S. Changes in cognition and behaviour in amyotrophic lateral sclerosis: nature of impairment and implications for assessment. Lancet Neurol. 2013;12(4):368–80.
Strong MJ, Abrahams S, Goldstein LH, Woolley S, Mclaughlin P, Snowden J, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18(3–4):153–74.
Chiò A, Moglia C, Canosa A, Manera U, Vasta R, Brunetti M, et al. Cognitive impairment across ALS clinical stages in a population-based cohort. Neurology. 2019; [Internet]. Available from;93:e984–94. https://doi.org/10.1212/WNL.0000000000008063.
Crockford C, Newton J, Lonergan K, Chiwera T, Booth T, Chandran S, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018;91(15):e1370–80.
Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, et al. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351(3):602–11.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–3.
Byrne S, Walsh C, Lynch C, Bede P, Elamin M, Kenna K, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82(6):623–7.
Ryan M, Heverin M, McLaughlin RL, Hardiman O. Lifetime risk and heritability of amyotrophic lateral sclerosis [published online ahead of print, 2019 Jul 22]. JAMA Neurol. 2019;76(11):1367–74.
van Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48(9):1043–8.
Lattante S, Doronzio PN, Marangi G, et al. Coexistence of variants in TBK1 and in other ALS-related genes elucidates an oligogenic model of pathogenesis in sporadic ALS. Neurobiol Aging. 2019;84:239.e9–239.e14.
Cady J, Allred P, Bali T, Pestronk A, Goate A, Miller TM, et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. 2015;77(1):100–13.
Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13(2):96–104.
Rosen DR, Siddique T, Patterson D, Figlewicz DA, Sapp P, Hentati A, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis [published correction appears in nature. 1993 Jul 22;364(6435):362]. Nature. 1993;362(6415):59–62.
Hadano S, Hand CK, Osuga H, Yanagisawa Y, Otomo A, Devon RS, et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat Genet. 2001;29(2):166–73.
Schymick JC, Scholz SW, Fung HC, Britton A, Arepalli S, Gibbs JR, et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2007;6(4):322–8.
•• Nicolas A, Kenna KP, Renton AE, et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron. 2018;97(6):1268–1283.e6. This is the largest ALS GWAS to date, validating several risk loci and finding a new Mendelian cause.
Conte A, Lattante S, Luigetti M, del Grande A, Romano A, Marcaccio A, et al. Classification of familial amyotrophic lateral sclerosis by family history: effects on frequency of genes mutation. J Neurol Neurosurg Psychiatry. 2012;83(12):1201–3.
van Blitterswijk M, van Es MA, Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21(17):3776–84.
Renton AE, Majounie E, Waite A, Simón-Sánchez J, Rollinson S, Gibbs JR, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72(2):257–68.
DeJesus-Hernandez M, Mackenzie IR, Boeve BF, Boxer AL, Baker M, Rutherford NJ, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72(2):245–56.
Zou ZY, Zhou ZR, Che CH, Liu CY, He RL, Huang HP. Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2017;88(7):540–9.
Mejzini R, Flynn LL, Pitout IL, Fletcher S, Wilton SD, Akkari PA. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. 2019;13:1310 Published 2019 Dec 6.
Kuuluvainen L, Kaivola K, Mönkäre S, et al. Oligogenic basis of sporadic ALS: the example of SOD1 p.Ala90Val mutation. Neurol Genet. 2019;5(3):e335 Published 2019 Apr 23.
Povysil G, Petrovski S, Hostyk J, Aggarwal V, Allen AS, Goldstein DB. Rare-variant collapsing analyses for complex traits: guidelines and applications. Nat Rev Genet. 2019;20(12):747–59.
Farhan SMK, Howrigan DP, Abbott LE, et al. Exome sequencing in amyotrophic lateral sclerosis implicates a novel gene, DNAJC7, encoding a heat-shock protein [published correction appears in Nat Neurosci. 2019 Dec 19]. Nat Neurosci. 2019;22(12):1966–74.
•• Gelfman S, Dugger S, de Araujo Martins Moreno C, et al. A new approach for rare variation collapsing on functional protein domains implicates specific genic regions in ALS. Genome Res. 2019;29(5):809–18. Illustrates new bioinformatic methods for rare variant collapsing, a technique driving new gene discovery in large genome sequencing studies.
Dormann D, Haass C. TDP-43 and FUS: a nuclear affair. Trends Neurosci. 2011;34(7):339–48.
Johnson BS, Snead D, Lee JJ, McCaffery JM, Shorter J, Gitler AD. TDP-43 is intrinsically aggregation-prone, and amyotrophic lateral sclerosis-linked mutations accelerate aggregation and increase toxicity [published correction appears in J Biol Chem. 2009 Sep 11;284(37):25459]. J Biol Chem. 2009;284(30):20329–39.
Barton SK, Gregory JM, Chandran S, Turner BJ. Could an impairment in local translation of mRNAs in glia be contributing to pathogenesis in ALS? Front Mol Neurosci. 2019;12:124 Published 2019 May 21.
Wells C, Brennan SE, Keon M, Saksena NK. Prionoid proteins in the pathogenesis of neurodegenerative diseases. Front Mol Neurosci. 2019;12:271 Published 2019 Nov 12.
Fahrenkrog B, Harel A. Perturbations in traffic: aberrant nucleocytoplasmic transport at the heart of neurodegeneration. Cells. 2018;7(12):232 Published 2018 Nov 26.
Štalekar M, Yin X, Rebolj K, Darovic S, Troakes C, Mayr M, et al. Proteomic analyses reveal that loss of TDP-43 affects RNA processing and intracellular transport. Neuroscience. 2015;293:157–70.
Kattuah W, Rogelj B, King A, Shaw CE, Hortobágyi T, Troakes C. Heterogeneous nuclear ribonucleoprotein E2 (hnRNP E2) is a component of TDP-43 aggregates specifically in the A and C pathological subtypes of frontotemporal lobar degeneration. Front Neurosci. 2019;13:551 Published 2019 Jun 4.
McGurk L, Gomes E, Guo L, et al. Poly (ADP-ribose) prevents pathological phase separation of TDP-43 by promoting liquid demixing and stress granule localization. Mol Cell. 2018;71(5):703–717.e9.
McGurk L, Mojsilovic-Petrovic J, Van Deerlin VM, et al. Nuclear poly (ADP-ribose) activity is a therapeutic target in amyotrophic lateral sclerosis. Acta Neuropathol Commun. 2018;6(1):84 Published 2018 Aug 29.
Duan Y, Du A, Gu J, et al. PARylation regulates stress granule dynamics, phase separation, and neurotoxicity of disease-related RNA-binding proteins. Cell Res. 2019;29(3):233–47.
Wolozin B, Ivanov P. Stress granules and neurodegeneration. Nat Rev Neurosci. 2019;20(11):649–66.
Eftekharzadeh B, Daigle JG, Kapinos LE, et al. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease [published correction appears in Neuron. 2019 Jan 16;101(2):349]. Neuron. 2018;99(5):925–940.e7.
Li F, Xu D, Wang Y, Zhou Z, Liu J, Hu S, et al. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy. 2018;14(1):66–79.
Mackenzie IR, Nicholson AM, Sarkar M, et al. TIA1 mutations in amyotrophic lateral sclerosis and frontotemporal dementia promote phase separation and alter stress granule dynamics. Neuron. 2017;95(4):808–816.e9.
Mateju D, Franzmann TM, Patel A, Kopach A, Boczek EE, Maharana S, et al. An aberrant phase transition of stress granules triggered by misfolded protein and prevented by chaperone function. EMBO J. 2017;36(12):1669–87.
Alberti S, Mateju D, Mediani L, Carra S. Granulostasis: protein quality control of RNP granules. Front Mol Neurosci. 2017;10:84 Published 2017 Mar 27.
Maziuk B, Ballance HI, Wolozin B. Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Front Mol Neurosci. 2017;10:89 Published 2017 Apr 4.
Yu Y, Nakagawa T, Morohoshi A, Nakagawa M, Ishida N, Suzuki N, et al. Pathogenic mutations in the ALS gene CCNF cause cytoplasmic mislocalization of Cyclin F and elevated VCP ATPase activity. Hum Mol Genet. 2019;28(20):3486–97.
Brenner D, Sieverding K, Bruno C, Lüningschrör P, Buck E, Mungwa S, et al. Heterozygous Tbk1 loss has opposing effects in early and late stages of ALS in mice. J Exp Med. 2019;216(2):267–78.
Gerbino V, Kaunga E, Ye J, et al. The loss of TBK1 kinase activity in motor neurons or in all cell types differentially impacts ALS disease progression in SOD1 mice. Neuron. 2020;106(5):789–805.e5.
Millecamps S, Julien JP. Axonal transport deficits and neurodegenerative diseases. Nat Rev Neurosci. 2013;14(3):161–76.
Meirelles GV, Perez AM, de Souza EE, Basei FL, Papa PF, Melo Hanchuk TD, et al. “stop ne(c) king around”: how interactomics contributes to functionally characterize Nek family kinases. World J Biol Chem. 2014;5(2):141–60.
Smith BN, Ticozzi N, Fallini C, Gkazi AS, Topp S, Kenna KP, et al. Exome-wide rare variant analysis identifies TUBA4A mutations associated with familial ALS. Neuron. 2014;84(2):324–31.
Smith BN, Topp SD, Fallini C, et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med. 2017;9(388):eaad9157.
Hirsch-Reinshagen V, Pottier C, Nicholson AM, et al. Clinical and neuropathological features of ALS/FTD with TIA1 mutations. Acta Neuropathol Commun. 2017;5(1):96 Published 2017 Dec 7.
Oakes JA, Davies MC, Collins MO. TBK1: a new player in ALS linking autophagy and neuroinflammation. Mol Brain. 2017;10(1):5.
Cirulli ET, Lasseigne BN, Petrovski S, Sapp PC, Dion PA, Leblond CS, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347(6229):1436–41.
Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015;18(5):631–6.
van der Zee J, Gijselinck I, Van Mossevelde S, Perrone F, Dillen L, Heeman B, et al. TBK1 mutation spectrum in an extended European patient cohort with frontotemporal dementia and amyotrophic lateral sclerosis. Hum Mutat. 2017;38(3):297–309.
Cui R, Tuo M, Li P, Zhou C. Association between TBK1 mutations and risk of amyotrophic lateral sclerosis/frontotemporal dementia spectrum: a meta-analysis. Neurol Sci. 2018;39(5):811–20.
Gómez-Tortosa E, Van der Zee J, Ruggiero M, Gijselinck I, Esteban-Pérez J, García-Redondo A, et al. Familial primary lateral sclerosis or dementia associated with Arg573Gly TBK1 mutation. J Neurol Neurosurg Psychiatry. 2017;88(11):996–7.
Freischmidt A, Müller K, Ludolph AC, Weishaupt JH, Andersen PM. Association of mutations in TBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol. 2017;74(1):110–3.
Müller K, Brenner D, Weydt P, Meyer T, Grehl T, Petri S, et al. Weishaupt JH; German ALS network MND-NET. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. 2018;89(8):817–27.
Pottier C, Bieniek KF, Finch N, van de Vorst M, Baker M, Perkersen R, et al. Whole-genome sequencing reveals important role for TBK1 and OPTN mutations in frontotemporal lobar degeneration without motor neuron disease. Acta Neuropathol. 2015;130(1):77–92.
Le Ber I, De Septenville A, Millecamps S, Camuzat A, Caroppo P, Couratier P, et al. TBK1 mutation frequencies in French frontotemporal dementia and amyotrophic lateral sclerosis cohorts. Neurobiol Aging. 2015;36(11):3116.e5–8.
Wilke C, Baets J, De Bleecker JL, Deconinck T, Biskup S, Hayer SN, et al. Beyond ALS and FTD: the phenotypic spectrum of TBK1 mutations includes PSP-like and cerebellar phenotypes. Neurobiol Aging. 2018;62:244.e9–244.e13.
Lamb R, Rohrer JD, Real R, Lubbe SJ, Waite AJ, Blake DJ, et al. A novel TBK1 mutation in a family with diverse frontotemporal dementia spectrum disorders. Cold Spring Harb Mol Case Stud. 2019;5(3):a003913.
Li J, He J, Tang L, Chen L, Ma Y, Fan D. Screening for TUBA4A mutations in a large Chinese cohort of patients with ALS: re-evaluating the pathogenesis of TUBA4A in ALS. J Neurol Neurosurg Psychiatry. 2018;89(12):1350–2.
Perrone F, Nguyen HP, Van Mossevelde S, Moisse M, Sieben A, Santens P, et al. Investigating the role of ALS genes CHCHD10 and TUBA4A in Belgian FTD-ALS spectrum patients. Neurobiol Aging. 2017;51:177.e9–177.e16.
Li J, He J, Tang L, Chen L, Xu L, Ma Y, et al. TUBA4A may not be a significant genetic factor in Chinese ALS patients. Amyotroph Lateral Scler Frontotemporal Degener. 2015;17(1–2):148–50.
Reid E, Kloos M, Ashley-Koch A, Hughes L, Bevan S, Svenson IK, et al. A kinesin heavy chain (KIF5A) mutation in hereditary spastic paraplegia (SPG10). Am J Hum Genet. 2002;71(5):1189–94.
Crimella C, Baschirotto C, Arnoldi A, Tonelli A, Tenderini E, Airoldi G, et al. Mutations in the motor and stalk domains of KIF5A in spastic paraplegia type 10 and in axonal Charcot-Marie-Tooth type 2. Clin Genet. 2012;82(2):157–64.
Liu X, Jian X, Boerwinkle E. dbNSFP v2.0: a database of human non-synonymous SNVs and their functional predictions and annotations. Hum Mutat. 2013;34(9):E2393–402.
Jennings S, Chenevert M, Liu L, Mottamal M, Wojcik EJ, Huckaba TM. Characterization of kinesin switch I mutations that cause hereditary spastic paraplegia. PLoS One. 2017;12:e0180353.
Filosto M, Piccinelli SC, Palmieri I, Necchini N, Valente M, Zanella I, et al. A novel mutation in the stalk domain of KIF5A causes a slowly progressive atypical motor syndrome. J Clin Med. 2018;8(1):E17.
Liao YC, Fernandopulle MS, Wang G, Choi H, Hao L, Drerup CM, et al. RNA granules hitchhike on lysosomes for long-distance transport, using annexin A11 as a molecular tether. Cell. 2019;179(1):147–164.e20.
Gu X, Chen Y, Wei Q, Cao B, Ou R, Yuan X, et al. Mutation screening of the TIA1 gene in Chinese patients with amyotrophic lateral sclerosis/frontotemporal dementia. Neurobiol Aging. 2018;68:161.e1–3. https://doi.org/10.1016/j.neurobiolaging.2018.04.010.
Zhang K, Liu Q, Shen D, Tai H, Fu H, Liu S, et al. Genetic analysis of TIA1 gene in Chinese patients with amyotrophic lateral sclerosis. Neurobiol Aging. 2018;67:201.e9–201.e10. https://doi.org/10.1016/j.neurobiolaging.2018.03.020.
Baradaran-Heravi Y, Dillen L, Nguyen HP, Van Mossevelde S, Baets J, De Jonghe P, et al. No supportive evidence for TIA1 gene mutations in a European cohort of ALS-FTD spectrum patients. Neurobiol Aging. 2018;69:293.e9–293.e11.
•• van der Spek RA, van Rheenen W, Pulit SL, Kenna KP, Ticozzi N, Kooyman M, et al. Reconsidering the causality of TIA1 mutations in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2018;19(1–2):1–3. This paper uses the largest whole genome cohort in ALS to challenge the association of a previously associated gene and illustrates the power of large scale genomics to refine the genetic landscape.
Williams KL, Topp S, Yang S, Smith B, Fifita JA, Warraich ST, et al. CCNF mutations in amyotrophic lateral sclerosis and frontotemporal dementia. Nat Commun. 2016;7:11253.
Lee A, Rayner SL, Gwee SSL, De Luca A, Shahheydari H, Sundaramoorthy V, et al. Pathogenic mutation in the ALS/FTD gene, CCNF, causes elevated Lys48-linked ubiquitylation and defective autophagy. Cell Mol Life Sci. 2018;75(2):335–54.
Hogan AL, Don EK, Rayner SL, Lee A, Laird AS, Watchon M, et al. Expression of ALS/FTD-linked mutant CCNF in zebrafish leads to increased cell death in the spinal cord and an aberrant motor phenotype. Hum Mol Genet. 2017;26(14):2616–26.
Ohno M, Moore R, Myers P, Negishi M. Co-chaperone-mediated suppression of LPS-induced cardiac toxicity through NFκB signaling. Shock. 2018;50(2):248–54.
Kenna KP, van Doormaal PT, Dekker AM, Ticozzi N, Kenna BJ, Diekstra FP, et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2016;48(9):1037–42.
Fang X, Lin H, Wang X, Zuo Q, Qin J, Zhang P. The NEK1 interactor, C21ORF2, is required for efficient DNA damage repair. Acta Biochim Biophys Sin Shanghai. 2015;47(10):834–41.
Brenner D, Müller K, Wieland T, Weydt P, Böhm S, Lulé D, et al. NEK1 mutations in familial amyotrophic lateral sclerosis. Brain. 2016;139(Pt 5):e28.
Gratten J, Zhao Q, Benyamin B, et al. Whole-exome sequencing in amyotrophic lateral sclerosis suggests NEK1 is a risk gene in Chinese. Genome Med. 2017;9(1):97 Published 2017 Nov 17.
Nguyen HP, Van Mossevelde S, Dillen L, De Bleecker JL, Moisse M, Van Damme P, et al. NEK1 genetic variability in a Belgian cohort of ALS and ALS-FTD patients. Neurobiol Aging. 2018;61:255.e1–7.
Higelin J, Catanese A, Semelink-Sedlacek LL, Oeztuerk S, Lutz AK, Bausinger J, et al. NEK1 loss-of-function mutation induces DNA damage accumulation in ALS patient-derived motoneurons. Stem Cell Res. 2018;30:150–62.
Forsberg K, Graffmo K, Pakkenberg B, Weber M, Nielsen M, Marklund S, et al. Misfolded SOD1 inclusions in patients with mutations in C9orf72 and other ALS/FTD-associated genes. J Neurol Neurosurg Psychiatry. 2019;90(8):861–9.
Shalom O, Shalva N, Altschuler Y, Motro B. The mammalian Nek1 kinase is involved in primary cilium formation. FEBS Lett. 2008;582(10):1465–70.
Bai SW, Herrera-Abreu MT, Rohn JL, Racine V, Tajadura V, Suryavanshi N, et al. Identification and characterization of a set of conserved and new regulators of cytoskeletal organization, cell morphology and migration. BMC Biol. 2011;9:54. https://doi.org/10.1186/1741-7007-9-54.
Iyer S, Acharya KR, Subramanian V. Prediction of structural consequences for disease causing variants in C21orf2 protein using computational approaches. J Biomol Struct Dyn. 2019;37(2):465–80. https://doi.org/10.1080/07391102.2018.1429313.
Cooper-Knock J, Moll T, Ramesh T, Castelli L, Beer A, Robins H, et al. Mutations in the glycosyltransferase domain of GLT8D1 are associated with familial amyotrophic lateral sclerosis. Cell Rep. 2019;26(9):2298–2306.e5.
Li W, Liu Z, Sun W, Yuan Y, Hu Y, Ni J, et al. Mutation analysis of GLT8D1 and ARPP21 genes in amyotrophic lateral sclerosis patients from mainland China. Neurobiol Aging. 2020;85:156.e1–4.
Rehfeld F, Maticzka D, Grosser S, Knauff P, Eravci M, Vida I, et al. The RNA-binding protein ARPP21 controls dendritic branching by functionally opposing the miRNA it hosts. Nat Commun. 2018;9(1):1235.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
Jenna M Gregory has received research grants from the Academy of Medical Sciences, the Jean Shanks Foundation, and the Pathological Society. Delphine Fagegaltier declares no conflict of interest. Hemali Phatnani has received research grants from the ALS Association, the Tow Foundation, Target ALS, and the NIH. Matthew Harms has received research grants from Biogen, the ALS Association, Target ALS Foundation, and Project ALS Foundation. He is a consultant with the Muscular Dystrophy Association.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Neurogenetics and Psychiatric Genetics
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Gregory, J.M., Fagegaltier, D., Phatnani, H. et al. Genetics of Amyotrophic Lateral Sclerosis. Curr Genet Med Rep 8, 121–131 (2020). https://doi.org/10.1007/s40142-020-00194-8
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-020-00194-8