
Abstract
The use of cerebrospinal fluid (CSF) levels of Aβ42 and Tau phosphorylated at threonine 181 (pTau181) as endophenotypes for genetic studies of Alzheimer’s disease (AD) has led to successful identification of both rare and common AD risk variants. In addition, this approach has provided meaningful hypotheses for the biological mechanisms by which known AD risk variants modulate the disease process. In this article, we discuss these successes and outline challenges to effective and continued applications of this approach. We contrast the statistical power of this approach with traditional case-control designs and discuss solutions to address challenges in quality control and data analysis for these phenotypes. Finally, we discuss the potential for the use of this approach with larger samples as well as the incorporation of next generation sequencing and for future work with other endophenotypes for AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
A variety of innovative approaches will be necessary to solve the complex etiology of Alzheimer’s disease (AD). One recent approach is using cerebrospinal fluid levels of Aβ42 and pTau181 as endophenotypes to study the genetics of AD. Using endophenotypes to study psychiatric disease was introduced by Gottesman and Shields [1], who defined endophenotypes as “internal phenotypes discoverable by a biochemical test or microscopic examination.” Gottesman and Gould [2] later provided five criteria specific to endophenotypes that were adapted from primary phenotypes in psychiatric genetics outlined by Gershon and Gouldin [3]: (1) an endophenotype is associated with illness in the population; (2) an endophenotype is heritable; (3) an endophenotype is state-independent (present whether or not illness is active in the individual); (4) within families, an endophenotype and illness co-segregate; (5) for diseases with complex inheritance, an endophenotype found in affected family members is found in non-affected family members at a higher rate than in the general population. Endophenotype-driven studies have several advantages over qualitative disease status traits, including increased statistical power, reduced heterogeneity, and more specific biological hypotheses for associated variants [4••, 5, 6].
Aβ42 and pTau181 levels in CSF fit these criteria and are among the best studied and most reliable biomarkers for AD [7–9]. These powerful endophenotypes have been used in numerous recent studies to identify novel loci associated with AD and related phenotypes. Here we review the recent discoveries in AD genetics that have been facilitated by this endophenotype-based approach.
Novel Discoveries
DNA Sequencing
While technology has made whole-exome and -genome sequencing a tractable experimental design, published work to date has been mainly limited to sequencing candidate genes. Despite this limitation, two studies have successfully identified AD risk variants by sequencing individuals with extreme levels of CSF Aβ42 and pTau181, then examining variants with likely functional significance for effects on risk and other aspects of AD. The earliest study of this kind was conducted by Kauwe et al. [10]. In this experiment, the promoter and coding regions of PSEN1 were genotyped in individuals with CSF Aβ levels in the top and bottom 5 % of the population distribution. Among the variants identified was an alanine to valine change at the 79th amino acid (A79V). This variant was shown to segregate in the family of the proband and results in increased secretion of Aβ42 in cell lines. Subsequent work has identified additional families in which A79V segregates with disease in an autosomal dominant manner [11].
More recently, Benitez et al. [4••] expanded this approach and used next-generation sequencing technologies to evaluate coding and promoter regions in APP, PSEN1, PSEN2, GRN, APOE and MAPT in individuals exhibiting extreme Aβ42 and pTau181 CSF levels. They identified several known pathogenic variants, several known high-risk variants and nine novel variants. One known variant, PSEN1 p.E318G (rs17125721), was previously classified as non-pathogenic because it did not segregate with disease status in some families. This coding variant is strongly associated with CSF tau and pTau181 levels. Benitez et al. also found that p.E318G is associated with Aβ accumulation in APOE-ε4 carriers and that APOE-ε4 carriers have increased risk of developing AD (OR 10.7, 95 % CI 4.7–24.6). APOE-ε4 non-carriers have half the risk of developing AD (OR 3.9, 95 % CI 3.4–4.4).
These studies demonstrate that combining CSF endophenotypes with next-generation sequencing empowers researchers to identify known pathogenic mutations in “sporadic” AD cases. Additionally, using CSF endophenotypes allowed researchers to identify a low-frequency variant that has a large effect on AD risk and interacts with APOE. Analysis of variants discovered in recent genome-wide association studies (GWAS) for AD suggests that a large proportion of the genetic component of AD may be explained by rare and low-frequency variants [12]. Rare variants have a minor allele frequency (MAF) between <1 %, while low-frequency variants have an MAF between 1 and 5 %. Identifying these variants is challenging and requires sequencing data in large populations. Using quantitative traits, including CSF biomarker levels, is a powerful approach to identify rare and low-frequency variants.
SNPs for Candidate Gene and Genome-Wide Association Studies
Experiments based on SNP arrays have also led to novel AD gene discoveries. In 2010, Cruchaga et al. [13] analyzed 384 SNPs across 34 genes associated with tau metabolism, testing for association with pTau181 CSF levels. The authors identified an SNP (rs1868402) in intron 5 of PPP3R1 and six surrounding SNPs associated with pTau181 CSF levels. Subsequent analyses found that this SNP was also associated with the rate of progression in AD cases. In the same data set, however, no association for the rate of progression was found for APOE. Based on these results and what is known about the AD pathogenic pathway and amyloid cascade hypothesis, the authors postulated that genetic variants associated with tau levels are more likely to affect the rate of progression and variants associated with Aβ42 levels should be associated with risk for disease. This association was replicated in multiple data sets. Peterson et al. [14] found that rs1868402 carriers have an elevated AD progression rate compared to non-carriers. Elias-Sonnenschein et al. [15•] later replicated the association between rs1868402 and increased tau levels in a Finnish data set. These studies implicate PPP3R1, or another proximal gene, in AD's etiology.
In 2012, Bekris et al. [16] conducted a candidate gene study in 270 samples. They reported that both rs913275 in the PPP2R4 and rs7768046 in FYN are associated with both tau and pTau181 CSF levels [16]. They studied 18 SNPs within the 5′ and 3′ regions of 5 kinase and 4 phosphatase genes using 101 AD patients and 169 cognitively normal controls. This study was conducted using a small subset of the sample used in the candidate gene screen conducted by Cruchaga et al. [13]. Rs7768046 did not pass imputation quality control filtering, and rs913275 is marginally associated with tau levels in the larger sample that was analyzed by Cruchaga et al. [17••] (tau p < 0.05; pTau p < 0.072).
Several genome-wide association studies of CSF Aβ42 and pTau181 levels have been published [17••, 18, 19]; unfortunately, the earliest studies suffered from lack of power and succeeded only in validating the known association between these phenotypes and the APOE-ε4 allele. The first GWAS of CSF Aβ42 and tau levels was conducted by Han et al. [19], where they conducted GWAS of each phenotype in the normal (n = 109), MCI (n = 109) and AD (n = 172) subsets in the ADNI samples. They confirmed the association of the APOE-ε4 allele with both CSF Aβ42 and tau levels and reported several putative loci for association with Aβ42 and tau levels including association between CSF Aβ42 levels and rs1022442 in NCAM2, which has been previously implicated in AD and other diseases [20–22]. Han et al. acknowledge the limited size and lack of replication in their initial report. Cruchaga et al. further evaluated these loci in nearly 1,300 samples and were unable to replicate this result (p < 0.57).
In 2011, Kim et al. [18] published another analysis using the ADNI cohort combining samples of all clinical diagnoses for a total sample size of 374. This analysis again confirmed the association with the APOE-ε4 allele. In addition, Kim et al. reported a genome-wide significant association between rs4499362 (EPC2) and tau levels. Unfortunately, this association did not replicate in the Cruchaga et al. [17••] study (p < 0.09).
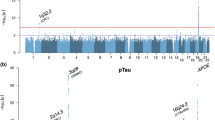
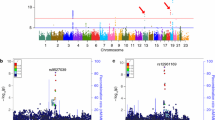
In the largest genome-wide association study of these phenotypes conducted to date, Cruchaga et al. [17••] used nearly 1,300 samples to identify three novel loci that showed genome-wide significance with tau and pTau181 endophenotypes. Cruchaga et al. identified rs9877502 located between GEMC1 and OSTN (3q28; p < 4.89 × 10−9) associated with tau levels, rs514716 located within GLIS3 (9p24.2; p < 1.07 × 10−8 and p < 3.22 × 10−9) associated with tau and pTau181 and rs6922617 located within the TREM gene cluster (6p21.1; p < 3.58 × 10−8). Of these four genome-wide significant loci (APOE, 3q28, 9p24.2, and 6p21.1), three of them also show an association with AD risk and/or progression. APOE is a well-known risk factor for AD that current hypotheses suggest affects AD risk through an Aβ-dependent mechanism. Cruchaga et al. used several statistical analyses to correct for the effect of the APOE levels on Aβ42. Even after stringent correction, APOE was still highly correlated with CSF tau and pTau181 levels, indicating that APOE also exerts it pathogenic mechanism by increasing tau pathology. Several studies also support this hypothesis: (1) Gibb et al. [23] and Zhou et al. [24] demonstrated that APOE shows isoform-specific differences in its interaction with tau in vitro; (2) Brecht et al. [25] and Andrews-Zwilling et al. [26] demonstrated neuron-specific differences in APOE isoform proteolysis is associated with increased tau phosphorylation and pathology in transgenic mice.
The SNP in 3q28 (rs9877502) also showed a consistent association with AD risk, tangle pathology and global cognitive decline in separate data sets. The association of this SNP was stronger with global cognitive decline than with any other AD phenotype, as predicted previously [13]. The variant located on 6p24.2 is located in the TREM gene cluster, where a low frequency variant (R47H) in TREM2 with a large effect on AD was found by two groups in late 2012 [27, 28]. More recently, a large GWAS published by the International Genomics of Alzheimer’s Project (IGAP) also identified several SNPs in the same region with a strong association to AD risk (p < 1 × 10−7) [29]. By using CSF levels and conditional statistical analyses, Benitez et al. [30] were able to demonstrate that the IGAP signal and R47H (TREM2) variant represent independent signals. In addition, the IGAP signal is likely driven by the TREML2 coding missense variant S144G (rs3747742), and this variant is protective, in contrast to the R47H (TREM2), which is a risk factor with an OR similar to that of APOE.
In summary, for three of the four genome-wide significant loci for CSF tau levels in Cruchaga et al. [17••], there is strong evidence that they are involved in the AD etiology. Therefore, it is possible that the fourth genome-wide significant locus (GLIS3; 9p24.2) for CSF tau also plays a role in AD, but the mechanism is still unknown. Several common and rare variants in GLIS3 are associated with diabetes, which itself is an AD risk factor (for a summary of the literature on the AD-diabetes relationship, see AlzRisk.org). Therefore, it is possible that variants in GLIS3 affect disease duration or age at onset. Cruchaga et al. also estimated these genome-wide significant loci (including APOE) explain approximately 21 % of genetic variability for CSF tau and pTau181 levels, suggesting that other common and rare variants may reach the genome-wide significance threshold in larger studies.
Functional Hypotheses for Known AD Variants
One of the main advantages of using CSF levels in endophenotype-based genetic studies is that they provide evidence for biological mechanisms for variants involved in the AD etiology. Therefore, the CSF levels have been used to analyze the most likely functional mechanism for variants identified by GWAS or other studies. Here, we discuss specific examples.
APOE
As discussed before, APOE is the strongest risk factor for AD, and several studies performed in cellular and animal models clearly indicate that APOE affects Aβ clearance, and the CSF studies also found a strong association between APOE and CSF Aβ levels (Table 1). This association is much stronger with Aβ42 than with case-control status or CSF tau levels. It is interesting, however, that APOE is also associated with tau. As explained above, further statistical and functional studies support the hypothesis that APOE affects tau pathology. This could also explain why APOE is a strong risk factor for AD since APOE increases the risk of AD through an Aβ- and tau-dependent mechanism.
MAPT
In 2007, Laws et al. [31] sought to provide further evidence that the microtubule-associated protein tau (MAPT) was in fact playing a role in AD. The authors used a fine mapping approach consisting of traditional phenotypic association and quantitative trait (QT) analysis with CSF tau levels. The authors suggest the causal variant is proximal to rs242557. Kauwe et al. [32] later tested whether Aβ42 and pTau181 CSF levels could successfully be used to identify loci involved in AD, using SNPs located in a known AD risk gene MAPT as a proof of concept. Several SNPs were associated with increased CSF tau and pTau181 levels. Association signals for these SNPs were strongest in individuals with evidence of Aβ deposition in the brain. Alleles associated with increased CSF tau and pTau181 levels were also associated with earlier age of onset. These data demonstrated that Aβ42 and pTau181 CSF levels could successfully identify AD risk loci and suggested that they could be used to identify novel loci. Elias-Sonnenschein et al. [15•] found in 2013 that rs2435211 was associated with both increased tau and pTau181 CSF levels.
GWAS Loci/Genes
Several efforts have focused on identifying the functional mechanism of GWAS loci, but not with strong results. Elias-Sonnenschein et al. [15•] analyzed 25 SNPs in 222 AD patients with CSF levels and found that CLU (rs11136000) and MS4A4A (rs2304933) correlated with significantly decreased Aβ42, while SORL1 (rs73595277) and MAPT (rs16940758) correlated with increased pTau181. On the other hand, Guo et al. analyzed the association of SORL1 SNPs with CSF levels in 1,005 MCI and AD patients and found several SNPs and haplotypes in SORL1 associated with Aβ42, but not tau [33]. Brouwers et al. [34] analyzed the association of several BIN1 SNPs with CSF levels in 339 AD cases and found four SNPs correlated with increased CSF amyloid Aβ42 levels, suggesting a role for the CR1 protein in Aβ metabolism. However, in a larger study, Kauwe et al. [35•] did not identify any variant in CR1, BIN1, CLU or PICALM passing stringent multiple test correction in a study including CSF levels from more than 600 individuals. Several variants in CLU and PICALM did show a nominal association with CSF Aβ42 and tau. These discrepant results indicate that additional studies in larger data sets are needed to identify real associations with GWAS loci.
Considerations for Analysis and Study Design
The main advantages of CSF levels as an endophenotype for AD are the increased statistical power and development of biological hypotheses for genetic effects. In previous sections, we already showed examples of how the CSF levels have been used to identify potential functional mechanisms, as in the case of APOE, MAPT and TREM2. The CSF levels have been used successfully to identify functional and independent signals in complex GWAS loci, as in the case of TREM2.
CSF endophenotypes also provide approximately five-fold more power than the regular case-control studies. When comparing the p value for APOE associated with case-control versus APOE associated with CSF Aβ42 and tau, the p-value for CSF Aβ42 is five times more significant than the p-value for case-control analysis. Likewise, even CSF tau provides twice the power as the case-control analysis, even though tau is not the main pathogenic mechanism for APOE. As a practical example to compare the power of an endophenotype-based study, approximately 15,000 individuals are needed to achieve similar power as in the currently largest GWAS (74,000) [29]. Unfortunately, the largest GWAS published for CSF endophenotypes to date includes only 1,269 individuals [17••]. Despite this problem, using CSF Aβ and tau as endophenotypes for AD has proven successful given the previously discussed studies and the advantages in terms of both statistical power and development of biological hypotheses for genetic effects.
Notwithstanding the advantages of endophenotype-based approaches and the success that has been realized to date, there are serious challenges in gathering and analyzing CSF Aβ and tau phenotypes. These challenges should be considered in future study designs. First, obtaining samples and CSF measurements is expensive and difficult, limiting sample collection. This has left CSF GWAS studies underpowered relative to the massive data sets that have been assembled for studies using case-control status as the phenotype [29].
Measuring CSF Aβ and tau levels at different sites can be inconsistent because of sample handling, preparation, antibody selection and other methodological practices [36]. While there have been concerted efforts to standardize these practices, combining data from multiple sites remains challenging and must be done with great care. Cruchaga et al. [17••] dealt with these challenges by standardizing phenotype data from each series to a mean of zero, then including adjustments for site in the analysis. Significant markers are then carefully checked to confirm consistent signals for association from each individual data set. This approach has been used successfully in several studies [13, 17••, 32, 35•, 37, 38]. A more conservative approach would be to analyze each data set separately, then use meta-analysis methods to look for consistent signals. In practice, these methods produced similar results when applied to the recent GWAS by Cruchaga et al. [17••] (Table 2).
In addition, these phenotypes are rarely normally distributed. As a normal distribution is a fundamental assumption of linear regression, this is an important consideration when analyzing CSF Aβ and tau levels. In some cases, simple transformations are sufficient to approximate normality. When they do not, researchers must interpret the results cautiously. Replication can be a simple solution; if the result is consistent in independent data sets that do not violate the assumptions of the model, then the result is likely to be valid. In cases where replication cannot be conducted, non-parametric approaches may be used to avoid violating the normality assumption.
Including appropriate covariates is also an important consideration. While including variables with known effects on the phenotype and adjusting for population stratification are common practice, there are differing opinions on how clinical status should be incorporated into the genetic analysis of CSF Aβ42 and tau levels. On one hand, it can be argued that including clinical status as a covariate is necessary since it clearly explains variance in CSF Aβ and tau levels because it would help identify variants that are associated with CSF Aβ42 and tau levels independently of clinical status. Similarly, some researchers opt for subset analyses of each clinical phenotype class. This allows researchers to analyze and presumably identify variants that are associated with CSF Aβ42 and tau levels either independently of disease or specifically in the context of disease. Unfortunately, this also results in drastically reduced power as sample size is reduced in each subset, and the variance in both Aβ42 and tau is drastically reduced in AD cases.
On the other hand, the purpose of an endophenotype-based approach is to find markers that influence the risk for disease by leveraging the statistical advantages of the endophenotype. As that is the case, there is no reason to adjust for the variance explained by clinical status. That variance is, in fact, the variance that is most likely to be explained by genes that alter the risk for disease via effects on CSF Aβ42 and tau levels. As such, we recommend that studies using these endophenotypes to find novel AD risk genes do not adjust for clinical status or APOE ε4 genotype.
Finally, identifying variants that are associated with CSF Aβ42 and tau levels does not constitute a discovery in Alzheimer’s disease. Findings must be brought back to a disease context. Associated variants should be tested for association with AD risk, rate of decline in AD patients, measures of AD pathology and other aspects of disease that may be available. This key aspect of an endophenotype-based approach to studying AD genetics provides the confirmation that the variant indeed alters disease. This confirmation allows the realization of the full benefits of this approach—the identification of novel variants that alter aspects of AD through a clearly predicted biological pathway.
Future Directions
Current published studies have already demonstrated the utility of CSF biomarker levels as endophenotypes for AD. Several common variants [13, 17••, 32] and rare variants [10] have been identified using this approach. We expect that larger GWASs for CSF tau and Aβ will identify additional significant genome-wide signals, although this may require meta-analyses from different studies.
Low-frequency and rare variant studies have been challenging because such studies require re-sequencing large populations. New “exome-genotyping arrays,” have enabled researchers to analyze genome-wide, low-frequency and rare coding variants at an affordable cost. These genotyping arrays contain more than 250,000 coding variants that were identified by exome sequencing in more than 6,000 individuals. We expect that this new genotyping technology will help researchers identify additional variants and genes involved in AD's etiology, although at this moment there are not any reports using the exome chip for AD for CSF biomarker studies. Similarly, whole-exome or whole-genome sequencing experiments are expected to unravel novel variants and genes associated with complex diseases, including CSF tau and Aβ levels. This approach has already identified novel variants and genes associated with changes in the hippocampal volume and MCI status. Normally these studies require extremely large data sets in order to identify significant associations, but there are several approaches to avoid sequencing large populations. One approach is to sequence selected individuals with extreme CSF tau and Aβ levels in the population. Examples of this approach include studies by Kauwe et al., Benitez et al. [4••] and Nho et al. [39], who selected individuals in the extreme of the distribution for hippocampal volume change. Similar approaches may lead to the identification of novel genes associated with CSF levels and AD.
Many AD researchers have also turned to interaction studies to better understand AD's etiology. While some highly penetrant variants have been identified, such as the TREM2 variant and APOE, Ebbert et al. [40] demonstrated that most genes implicated in AD's etiology do not predict disease status at a level that is diagnostically useful and that they may interact in a non-additive manner. This highlights the importance of approaches that seek to address the role of complex gene-gene interactions in the risk for AD. As endophenotypic-based studies identify novel loci involved in AD, researchers need to also explore how the loci interact in order to truly understand AD's etiology.
Conclusions
In summary, using CSF Aβ and tau levels as endophenotypes for genetic studies of AD has resulted in the discovery of novel AD risk variants and provided evidence for the biological mechanism of risk effects of variants that have been discovered using different approaches. This work has been of great value to the AD genetics community. Success with these phenotypes also highlights the importance of applying other endophenotypes to the study of AD. As data sets grow in size and efforts are made to incorporate whole-genome and -exome sequencing approaches, we expect that important and novel insights into the genetic etiology of AD will be made by leveraging imaging, fluid and psychometric, and other types of endophenotypes for AD.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gottesman II, Shields J. Genetic theorizing and schizophrenia. Br J Psychiatry. 1973;122:15–30.
Gottesman II, Gould TD. The endophenotype concept in psychiatry: etymology and strategic intentions. Am J Psychiatry. 2003;160:636–45.
Gershon ES, Goldin LR. Clinical methods in psychiatric genetics. I. Robustness of genetic marker investigative strategies. Acta Psychiatr Scand. 1986;74:113–8.
•• Benitez BA, Karch CM, Cai Y, et al. The PSEN1, p.E318G variant increases the risk of Alzheimer’s disease in APOE-ε4 carriers. PLoS Genet. 2013;9:e1003685. doi: 10.1371/journal.pgen.1003685. This article illustrates the successful use of next generation sequencing technologies with CSF biomarker data to discover novel AD risk variants. Here APP, PSEN1, PSEN3, GRN, MAPT and APOE were sequenced in 212 individuals with extreme CSF Aβ42 or tau levels. Further analyses demonstrate that the effect of PSEN1, p.E318G on AD risk depends upon interaction with APOE ε4 and is mediated by Aβ deposition.
Nolan DK, Sutton B, Haynes C, et al. Fine mapping of a linkage peak with integration of lipid traits identifies novel coronary artery disease genes on chromosome 5. BMC Genet. 2012;13:12. doi:10.1186/1471-2156-13-12.
Shulman JM, Chibnik LB, Aubin C, et al. Intermediate phenotypes identify divergent pathways to Alzheimer’s disease. PLoS ONE. 2010;5:e11244. doi:10.1371/journal.pone.0011244.
Fagan AM, Roe CM, Xiong C, et al. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007;64:343–9. doi:10.1001/archneur.64.3.noc60123.
Hansson O, Zetterberg H, Buchhave P, et al. Prediction of Alzheimer’s disease using the CSF Abeta42/Abeta40 ratio in patients with mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;23:316–20. doi:10.1159/000100926.
Kapaki EN, Paraskevas GP, Tzerakis NG, et al. Cerebrospinal fluid tau, phospho-tau181 and beta-amyloid1-42 in idiopathic normal pressure hydrocephalus: a discrimination from Alzheimer’s disease. Eur J Neurol. 2007;14:168–73. doi:10.1111/j.1468-1331.2006.01593.x.
Kauwe JSK, Jacquart S, Chakraverty S, et al. Extreme cerebrospinal fluid amyloid beta levels identify family with late-onset Alzheimer’s disease presenilin 1 mutation. Ann Neurol. 2007;61:446–53. doi:10.1002/ana.21099.
Brickell KL, Leverenz JB, Steinbart EJ, et al. Clinicopathological concordance and discordance in three monozygotic twin pairs with familial Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2007;78:1050–5. doi:10.1136/jnnp.2006.113803.
Ridge PG, Mukherjee S, Crane PK, et al. Alzheimer’s disease: analyzing the missing heritability. PLoS ONE. 2013;8:e79771. doi:10.1371/journal.pone.0079771.
Cruchaga C, Kauwe JSK, Mayo K, et al. SNPs associated with cerebrospinal fluid phospho-tau levels influence rate of decline in Alzheimer’s disease. PLoS Genet. 2010;6:e1001101. doi:10.1371/journal.pgen.1001101.
Peterson D, Munger C, Crowley J, et al. Variants in PPP3R1 and MAPT are associated with more rapid functional decline in Alzheimer’s disease: the Cache County Dementia Progression Study. Alzheimers Dement. 2013;. doi:10.1016/j.jalz.2013.02.010.
• Elias-Sonnenschein LS, Helisalmi S, Natunen T, et al. Genetic loci associated with Alzheimer’s disease and cerebrospinal fluid biomarkers in a finnish case–control cohort. PLoS ONE. 2013;8:e59676. doi: 10.1371/journal.pone.0059676. This study is one of just two that have systematically evaluated known AD risk variants for association with CSF biomarker levels. Evidence for association with CSF Aβ42 was reported for variants in CLU and MS4A4A in 222 AD patients. These results suggest that these genes act to alter AD risk through an amyloid-related mechanism.
Bekris LM, Millard S, Lutz F, et al. Tau phosphorylation pathway genes and cerebrospinal fluid tau levels in Alzheimer’s disease. Am J Med Genet B Neuropsychiatr Genet. 2012;159B:874–83. doi:10.1002/ajmg.b.32094.
•• Cruchaga C, Kauwe JSK, Harari O, et al. GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer’s disease. Neuron. 2013;78:256–268. doi: 10.1016/j.neuron.2013.02.026. This article describes the largest genome-wide association study of CSF Aβ42 and tau levels to date. The study included 1,269 samples with both CSF biomarker data and genetic data, and reported three genome-wide significant variants in addition to the APOE e4 allele. One variant, rs9877502, also showed association with risk for AD, tangle pathology and global cognitive decline in independent data sets.
Kim S, Swaminathan S, Shen L, et al. Genome-wide association study of CSF biomarkers Abeta1-42, t-tau, and p-tau181p in the ADNI cohort. Neurology. 2011;76:69–79. doi:10.1212/WNL.0b013e318204a397.
Han M-R, Schellenberg GD, Wang L-S, et al. Genome-wide association reveals genetic effects on human Aβ42 and τ protein levels in cerebrospinal fluids: a case control study. BMC Neurol. 2010;10:90. doi:10.1186/1471-2377-10-90.
Paoloni-Giacobino A, Chen H, Antonarakis SE. Cloning of a novel human neural cell adhesion molecule gene (NCAM2) that maps to chromosome region 21q21 and is potentially involved in Down syndrome. Genomics. 1997;43:43–51. doi:10.1006/geno.1997.4782.
Wang K, Li W-D, Zhang CK, et al. A genome-wide association study on obesity and obesity-related traits. PLoS ONE. 2011;6:e18939. doi:10.1371/journal.pone.0018939.
Fox CS, Liu Y, White CC, et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012;8:e1002695. doi:10.1371/journal.pgen.1002695.
Gibb GM, Pearce J, Betts JC, et al. Differential effects of apolipoprotein E isoforms on phosphorylation at specific sites on tau by glycogen synthase kinase-3 beta identified by nano-electrospray mass spectrometry. FEBS Lett. 2000;485:99–103.
Zhou J, Chen J, Feng Y. Effect of truncated-ApoE4 overexpression on tau phosphorylation in cultured N2a cells. J Huazhong Univ Sci Technol Med Sci. 2006;26:272–4.
Brecht WJ, Harris FM, Chang S, et al. Neuron-specific apolipoprotein e4 proteolysis is associated with increased tau phosphorylation in brains of transgenic mice. J Neurosci. 2004;24:2527–34. doi:10.1523/JNEUROSCI.4315-03.2004.
Andrews-Zwilling Y, Bien-Ly N, Xu Q, et al. Apolipoprotein E4 causes age- and tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci. 2010;30:13707–17. doi:10.1523/JNEUROSCI.4040-10.2010.
Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer’s disease. N Engl J Med. 2013;368:117–27. doi:10.1056/NEJMoa1211851.
Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med. 2013;368:107–16. doi:10.1056/NEJMoa1211103.
Lambert J-C, Ibrahim-Verbaas CA, Harold D, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;. doi:10.1038/ng.2802.
Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer’s disease in Spanish population. Neurobiol Aging. 2013;34(1711):e15–7. doi:10.1016/j.neurobiolaging.2012.12.018.
Laws SM, Friedrich P, Diehl-Schmid J, et al. Fine mapping of the MAPT locus using quantitative trait analysis identifies possible causal variants in Alzheimer’s disease. Mol Psychiatry. 2007;12:510–7. doi:10.1038/sj.mp.4001935.
Kauwe JSK, Cruchaga C, Mayo K, et al. Variation in MAPT is associated with cerebrospinal fluid tau levels in the presence of amyloid-beta deposition. Proc Natl Acad Sci USA. 2008;105:8050–4. doi:10.1073/pnas.0801227105.
Guo L-H, Westerteicher C, Wang X-H, et al. SORL1 genetic variants and cerebrospinal fluid biomarkers of Alzheimer’s disease. Eur Arch Psychiatry Clin Neurosci. 2012;262:529–34. doi:10.1007/s00406-012-0295-x.
Brouwers N, Van Cauwenberghe C, Engelborghs S, et al. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol Psychiatry. 2012;17:223–33. doi:10.1038/mp.2011.24.
• Kauwe JSK, Cruchaga C, Karch CM, et al. Fine mapping of genetic variants in BIN1, CLU, CR1 and PICALM for association with cerebrospinal fluid biomarkers for Alzheimer’s disease. PLoS ONE. 2011 6:e15918. doi:10.1371/journal.pone.0015918. This study is the largest evaluation of known AD risk variants for association with CSF Aβ42 and tau levels to date; 664 samples with biomarker and genetic data were used to test for association with variants in BIN1, CLU, CR1 and PICALM. There was marginal association between variants in CLU and PICALM and CSF Aβ42, but the strongest findings suggest an assocoation between variants in PICALM and CSF pTau181 levels.
Mattsson N, Andreasson U, Persson S, et al. CSF biomarker variability in the Alzheimer’s association quality control program. Alzheimers Dement. 2013;9:251–61. doi:10.1016/j.jalz.2013.01.010.
Kauwe JSK, Cruchaga C, Bertelsen S, et al. Validating predicted biological effects of Alzheimer’s disease associated SNPs using CSF biomarker levels. J Alzheimers Dis. 2010;21:833–42. doi:10.3233/JAD-2010-091711.
Cruchaga C, Kauwe JSK, Nowotny P, et al. Cerebrospinal fluid APOE levels: an endophenotype for genetic studies for Alzheimer’s disease. Hum Mol Genet. 2012;21:4558–71. doi:10.1093/hmg/dds296.
Nho K, Corneveaux JJ, Kim S, et al. Whole-exome sequencing and imaging genetics identify functional variants for rate of change in hippocampal volume in mild cognitive impairment. Mol Psychiatry. 2013;18:781–7. doi:10.1038/mp.2013.24.
Ebbert MTW, Ridge PG, Wilson AR, et al. Population-based analysis of Alzheimer’s disease risk alleles implicates genetic interactions. Biol Psychiatry. 2013;. doi:10.1016/j.biopsych.2013.07.008.
Disclosures
C. Cruchaga has received research support from grant NIA-R01-AG044546. M.T.W. Ebbert declares no conflicts of interest. J.S.K. Kauwe has received research support from grant NIA-R01-AG042611.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cruchaga, C., Ebbert, M.T.W. & Kauwe, J.S.K. Genetic Discoveries in AD Using CSF Amyloid and Tau. Curr Genet Med Rep 2, 23–29 (2014). https://doi.org/10.1007/s40142-014-0031-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40142-014-0031-0