
Abstract
Chronic rhinosinusitis with or without polyps affects more than 10 % of the population. In the last decennium, great progress was made in the elucidation of relevant pathomechanisms of innate and adaptive immunity and in the recognition of potential environmental and intramucosal triggers. Inflammatory pathways and triggers need to be integrated into a modern concept of disease differentiation beyond the nasal endoscope; in short, remodeling and inflammatory processes can be described, involving fibrosis versus edema, which are related to the clinical phenotype; the signatures of a number of T helper cells decide on the subsequent granulocytic tissue reactions beyond the level of neutrophils versus eosinophils. Based on those pathways, biomarkers allow the prediction of the likelihood of co-morbid asthma and individual selection of appropriate treatments, from glucocorticosteroids and immune-modulatory antibiotics to innovative approaches such as humanized monoclonal antibodies against IgE and interleukin-5. These approaches currently serve as proof-of-concept tools, and will expand our armamentarium tomorrow.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
There is recent interest in chronic rhinosinusitis (CRS), which is now recognized as a common disease, affecting about 11 % of the EU adult population [1], while former figures from the US point to a rate of about 14 % in the North American population [2]. CRS by its own rights is a disabling condition [3], but there is additionally a strong association between asthma and CRS at all ages; the association is stronger when CRS is associated with allergic rhinitis. Of great interest, CRS in the absence of nasal allergies is positively associated with late-onset asthma [4]. Although not determinable by epidemiologic studies, it is likely that CRS in these different conditions represents different entities.
The term CRS is now accepted by most scientists worldwide, and is used by the American and European guidelines [5, 6∙]. A recent study demonstrating that changes in the inflammatory patterns of sinus mucosa are reflected in the inferior turbinates of the same patients supports this view [7]. CRS can be understood as a dysfunctional host-environment interaction at the mucosal interfaces, resulting in inadequate mucosal responses to the environment. Environmental triggers can be cigarette smoke and indoor pollution, bacteria, viruses and fungi, and allergens or superantigens.
CRS has been typically divided into CRS without nasal polyps (CRSsNP) and CRS with nasal polyps (CRSwNP), based on nasal endoscopy. In terms of etiology and pathogenesis, CRSsNP is more tightly linked to mechanical obstruction and also recently to fibrosis of the osteo-meatal complex (OMC), while CRSwNP is generally attributed to a more diffuse mucosal edematous response [8∙]. The body of current research would suggest separate remodeling patterns, but partially overlapping inflammatory mechanisms in these diseases. For research purposes, the separation of phenotypes and eventually endotypes seems mandatory, facilitating data analysis and the determination of molecular pathways of disease. Phenotypes may be separated on clinical basis into CRSsNP and CRSwNP; the differentiation into endotypes relies on biochemical markers of inflammation such as interleukin-5 or staphylococcal enterotoxin-specific IgE antibodies, and eventually allows the prediction of co-morbidity, recurrence after surgery, or response to innovative treatment [9]. Using pheno- and endotypes, associations with environmental triggers can be studied in a meaningful way, helping us to understand the impact of the triggers and the resulting inflammatory pathways as well as their interrelation.
Inflammatory Pathways
The sinonasal epithelium provides a mechanical and innate immune barrier to the environment, and actively contributes to the regulation of the adaptive immune response. There is evidence of defects in this barrier which may foster the development of chronic mucosal inflammation in response to environmental agents and pathogenic organisms.
In CRSwNP, significantly decreased levels of the desmosomal proteins DSG2 and DSG3 [10] and tight-junction proteins claudin and occludin [11] have been reported. The expression of the epithelial protein LEKT1 is also significantly decreased in CRSwNP [12]. Lower levels of protease inhibitors like LEKT1 in the CRS epithelium may result in increased susceptibility to endogenous and exogenous protease activity [13]. This protein, encoded by the gene SPINK5, acts as a protease inhibitor involved in regulating the processing of the tight junction proteins critical to epithelial barrier function. Fungi, bacteria and many allergens all possess significant intrinsic protease activity, which, in the presence of deficient endogenous protease inhibitors such as LEKT1, may render the mechanical barrier more vulnerable to protease attack and greater mucosal penetration of foreign proteins.
Epithelial cells secrete antimicrobial molecules such as enzymes (e.g., lysozyme), opsonins (e.g., complement), anti-bacterial proteins (e.g., defensins), collectins (surfactant proteins) and other proteins (e.g., lactoferrin and mucins) [14–16]. These molecules may be increased as a sign of a response to an environmental challenge, or decreased in CRS (e.g., lactoferrin and possibly the S100 group of antimicrobials) [17–19]. S100 proteins are widely expressed in epithelial cells and have anti-microbial effects, but also effects on cell differentiation and wound healing, linking the mechanical barrier and classic anti-microbial properties [20]. Palate lung nasal epithelial clone (PLUNC), secreted by glandular epithelium with anti-biofilm properties, also has been found decreased in CRSwNP [21]. It has to be mentioned, however, that the relative contribution of those deficits to the disease is unclear, and that these observations may be a consequence of rather than a starting point for disease [19].
The epithelium also impacts on the inflammatory patterns by releasing cytokines (IL-25, IL-33 and TSLP) determining dendritic cell polarization and subsequent T cell differentiation in response to mucosal challenges [22, 23]. These cytokines may contribute to a skew in the T cell differentiation, specifically favoring Th2 cells in CRSwNP [24–27]. A whole group of proteins released upon cell death, the so-called DAMPs (danger associated molecular patterns), have been identified to mainly elicit pro-inflammatory actions in the airways [28]. The soluble form of the receptor for advanced glycation end products (RAGE) was found to be upregulated in CRSsNP and likely contributes to the induction of a Th1-biased inflammation, while in CRSwNP, high ECP protein levels would break the protein down, preventing a redirection of T helper cells into the Th1 type [29].
Dendritic cells (DCs) activate both innate and adaptive immunity via antigen capture, presentation of antigen to immature T cells, and subsequent determination of T cell responses [23]. Multiple subsets of DCs are present in the nasal mucosa [30] and seem to be increased in CRSwNP versus CRSsNP [31]. Plasmacytoid (p)DCs were found to be down-regulated in IL5+ nasal polyps, whereas high levels of mucosal IFN-γ favored those cells (L. Derycke, personal communication). This altered balance in DCs may play a crucial role in the inflammatory pathomechanism. Furthermore, there is an increase in macrophages in CRSwNP, and again a shift in certain populations can be observed: Krysko et al. [32∙] have demonstrated the presence of alternatively activated macrophages (M2) especially in IL-5 positive nasal polyps, favoring the Th2 bias by the release of mediators and chemokines such as CCL 18, a chemotaxin for DCs, naïve T cells and Th2 cells [33], but also by a deficient phagocytosis of Staphylococcus aureus, leading to an increased colonization and intramucosal survival in CRSwNP [32•]. S. aureus, by releasing superantigens, likely further skews the immune response to Th2, finally resulting in a persistent eosinophilic inflammation.
Inflammatory patterns within the CRS phenotypes can be differentiated based on the predominant T helper cell (Th1, Th2, Th17, Th21, Treg) and the consecutive granulocytic reaction (neutrophilic vs. eosinophilic) [34]. Recent data suggest that although there may be a prominent Th cell population orchestrating the inflammation, there often is a complex mixture of several CD4 and CD8 positive T cells present, which express and even co-express several key cytokines such as IFN-g, IL-4, IL-5, IL-17 and others (L Derycke, unpublished). Whereas IFN-g producing Th1 and IL-17 producing Th17 cells are associated with tissue neutrophilia, IL-5 releasing Th2 cells are orchestrating an eosinophilic reaction. It is noteworthy that even in strongly eosinophilic CRSwNP, there often are plenty of neutrophils present also due to the co-expression of IL-8, IL-17 and other cytokines. This mixture of inflammatory cells may partly explain differences in the therapeutic response to corticosteroids [35] and innovative biologicals such as anti-IL5 and anti-IgE [36, 37∙∙]. In Caucasian subjects, CRS with nasal polyps is considered to be orchestrated by Th2 cells, with IL-5 synthesis measurable in about 85 % of subjects, resulting in increased eosinophil survival and activation, which might also be associated with IgE formation [38]. In contrast, the predominant T-effector cell in the majority of Asian patients with polyps is the Th17 cell [34]. Asian and Caucasian patients also showed a marked difference in the prevalence of comorbid asthma, with this disease being rare in Chinese patients [39∙∙]. Among the CRSwNP endotypes, IL-5 expression is associated with a significant risk of asthma co-morbidity, which further increased with the mucosal presence of IgE to Staphylococcal superantigens and high total IgE concentrations [39••]. This observation was confirmed in the Caucasian and Asian patients, although the number of Th2-biased CRSwNP was significantly lower in the latter group.
Neutrophils are abundantly present in CRS tissue in cystic fibrosis patients, with the highest concentrations of IL-8, IL-17 and MPO measurable [38]. IL 8 released by epithelial cells in response to PAR-2 stimulation appears to be the main cytokine responsible for neutrophil recruitment in CRS [40]. The cellular sources of IL-17A in nasal polyps were mainly T-lymphocytes. Anti-IL-17A antibodies were able to modulate the survival of neutrophils in nasal polyps from non-CF patients; however the survival of neutrophils in CF patients was independent of IL-17A [41].
Mucosal immunoglobulin secretion by cells of the B lymphocyte lineage is an important part of the adaptive immune response. In the nasal mucosa, B cells undergo proliferation, differentiation and immunoglobulin-class switching to become mature plasma cells capable of substantial local antibody secretion. Secretion of sIgA works in concert with other innate protective factors and the mucociliary flow to limit mucosal colonization by germs without tissue-damaging inflammation [42]. In CRSwNP, homogenates demonstrate high levels of immunoglobulin production, notably IgA, IgE and IgG, in comparison with CRSsNP and control tissues [43–45]. Levels in polyp homogenates do not correlate with levels in serum, suggesting that significant immunoglobulin synthesis occurs locally in the nasal mucosa [46]; high numbers of B cells and plasma cells have been reported in nasal polyps [43, 44], and SDF-1α and BCA-1 have been identified as chemokines for the recruitment and retention of B cells in the tissue [47]. A B cell–activating factor of the TNF family (BAFF) has been found to play an important role in B cell proliferation and immunoglobulin-class switching; BAFF levels were significantly elevated in nasal polyp tissue compared to healthy tissue and CRSsNP [44]. Recent data suggest that all the necessary factors for a truly local immunoglobulin class switching and production can be found within the nasal polyp [46, 48]; specifically, high concentrations of IgE can be found in polyp homogenates independent of the presence of atopy and serum IgE antibodies. Measurements in CRSwNP demonstrated a polyclonal IgE formation, characterized by specific IgEs to hundreds of allergens; the functionality of these IgE antibodies has been shown by the degranulation of mast cells in ex vivo nasal polyp tissue models and the possibility to degranulate basophils after transfer of IgE in nasal polyp supernatants [49∙∙].
Mast cells are commonly linked to immunoglobulin E (IgE)-mediated inflammatory changes; in fact, a recent study found elevated numbers of mast cells in the sinonasal mucosa of CRSwNP patients, regardless of the atopic status, in line with observations on the role of IgE in these patients [50]. Upon activation by mucosal Ig E antibodies [49∙∙], mast cells release pre-formed granules including histamine, serotonin, proteoglycans and serine proteases; in addition, de novo synthesis and secretion of various eicosanoids, chemokines and cytokines takes place. One of the mediators found increased in CRSwNP is Prostaglandin D2 which plays a central role in the recruitment of Th2 cells into the inflamed mucosa through a CRTH2 dependent mechanism [51, 52]. In contrast, PGE2 and its primary receptor EP2, which exhibits an array of anti-inflammatory, protective effects by antagonizing leukotriene action are significantly down-regulated in nasal polyp tissue [53]. This may be linked to a deficiency in the cyclooxygenase biosynthetic pathway induced by the presence of staphylococcal superantigens which may suppress PGE2 production in structural cells [54, 55]. In line with these observations, a recent study demonstrated that in contrast to normal fibroblasts, polyp fibroblasts fail to up-regulate the cox pathway in response to inflammatory stimuli [56].
Whereas inflammatory patterns in CRSwNP showed important variations between Asia and Europe, remodeling patterns appeared to be uniform between ethnic groups [34, 57]. These observations suggest that different inflammatory patterns exist despite comparable remodeling patterns, challenging the assumption of a strong association between inflammation and remodeling. Furthermore, in CRSsNP disease limited to the OMC, remodeling markers such as TGF and collagen were increased without signs of inflammation, further separating these two pathophysiological principles [58]. Remodelling is a dynamic process that balances extracellular matrix production and degradation which is regulated by diverse mediators amongst which transforming growth factor (TGF)-β has a central role [59, 60]. Different isoforms (TGF- ß 1, 2, and 3) have been described that can bind to membrane proteins referred as receptor types Ι, ΙΙ, and ΙΙΙ; TGF-β receptor signaling is a very complex mechanism and is mediated by several signaling steps involving dimerization and phosphorylation of receptor and intermediate molecules such as Smads [61]. In CRSsNP, TGF-β1 and -ß2 concentrations are higher compared to controls, as is the expression of TGF-β Rs Ι and RΙΙΙ, and consequently, a higher number of phosphorylated Smad 2-positive cells and a higher collagen content can be observed. In contrast, a low TGF-β protein concentration, a decreased expression of TGF-β RΙΙ, and a low number of pSMad 2-positive cells in CRSwNP all indicate a low level of TGF-β signaling in nasal polyp disease reflected by a poor collagen expression and edema formation [61]. TGF-ß furthermore orchestrates the production of MMPs and of their tissue inhibitors, and is interrelated with the plasminogen activator system; those systems are further interconnected, inducing fibrinolysis in CRSwNP and favoring fibrosis in CRSsNP [62, 63].
Inflammatory Triggers
The first European multicenter CRS prevalence study by the GALEN network [1] showed a strong association between smoking and CRS. Exposure to toxins such as tobacco smoke, but also to air pollutants (e.g., diesel exhaust fumes, ozone, sulfur dioxide, nitrogen dioxide, etc.) have the potential to damage the epithelium by the production of reactive oxygen (ROS) and nitrogen species (RNS) [64]. ROS and RNS from tobacco smoke induce pro-inflammatory cytokine secretion and epithelial apoptosis and, thus, airway epithelial barrier dysfunction [65]. Cigarette smoke may also impact ciliary beat frequency [66] and favor bacterial biofilms [67]. However, the effects of tobacco smoke in the upper airway in mice appear to be well controlled [68], and the actual contribution of cigarette smoking to CRS etiology deserves further study.
Whereas the impact of viral infections on lower airway symptoms and exacerbations is well established, similar studies for the upper airways are lacking. Glorieux et al. demonstrated for the first time that herpes simplex virus type 1 (HSV1) has the capacity to breach the basement membrane of the nasal epithelium in vitro [69]. The virus was found to replicate in the nasal epithelium and lamina propria, leading to severe epithelial damage. Building up on these observations, Wang et al. [70∙] investigated the interaction between S. aureus and HSV1. Both in turbinate mucosa and nasal polyp tissue, HSV1 led to focal infection of epithelial cells and invasion into the lamina propria within 72 h. After pre-infection with HSV1, but not without, S. aureus was able to pass the basement membrane and invade the mucosa. This study demonstrated that HSV1 (and possibly other viruses) may lead to a significant damage of the nasal epithelium, and consequently may facilitate invasion of S. aureus into the nasal mucosa, and also established that nasal polyp tissue is more susceptible to the invasion compared to healthy mucosa.
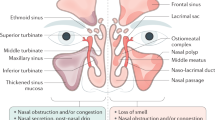
Staphylococcus aureus is the most common traditional bacterial pathogen identified in CRSwNP patients in Western countries [71, 72]. The incidence is much lower in Asia, and the germs presence is associated with a Th2 biased inflammatory reaction in CRSwNP patients [73], whereas IL-5 negative polyps rather were colonized by gram-negative bucks. We and others recently demonstrated that S. aureus may reside intra-mucosally and intra-cellularly in CRSwNP, but not in healthy controls or CRSsNP subjects [74, 75], and this may be related to the inability of macrophages to kill phagocytized germs. In line with these findings, also the presence of biofilms carrying S. aureus, but not other germs, was related to the presence of IgE to S. aureus enterotoxins and the expression of IL-5 in CRSwNP subjects [76∙], the causality of this observation is not demonstrated yet. Once introduced intramucosally, S. aureus may release enterotoxins, which have superantigenic properties and are able to polyclonally activate T- and B cells, further skewing the inflammatory response into the Th2 direction (the Staphylococcal Superantigen Hypothesis, [60, 77]. Approximately 50 % of Caucasian CRSwNP patients demonstrate B and T cell signatures consistent with prior local staphylococcal superantigen exposure [78, 79], and enterotoxins have been detected in CRSwNP patients but not in controls [80]. Enterotoxins induce the massive release of cytokines from T cells, among which typical Th2 signature cytokines [81∙∙], and stimulate the expression of IgE in B cells [60]. Those polyclonal IgE antibodies have been shown to be functional upon allergen exposure, even in the absence of serum IgE or positive skin prick testing [48]. The chronic activation of polyclonal IgE-armed mast cells may further enforce the superantigenic activation of Th2 cells and finally result in a persistent mucosal inflammation, which will spread systemically and include the lower airways [37∙∙]. The strongest predictor of comorbid asthma within polyp tissue in CRSwNP patients, SE-IgE, also plays an essential role in predicting severe asthma, low lung function, and necessary hospitalizations when measured in serum [81∙∙] Fig. 1. SE-IgE obviously is a marker for a newly discovered immune pathomechanism related to severe upper and lower airway disease [59].

Chronic rhinosinusitis can be differentiated into phenotypes (CRSsNP, CRSwNP) and endotypes based on immune markers. The endotypes are associated with different granulocytic patterns (eosinophilic vs. neutrophilic) and the presence/absence of asthma comorbidity. Novel therapeutic options can be applied for different endotypes
After initial enthusiasm for the hypothesis that all CRS is fungal, current evidence coupled with the failure of clinical trials with topical and oral antifungals indicates that a central role for fungi in CRs is unlikely [6•, 82]. Furthermore, evidence that fungal antigens are the primary initiators of mucosal T cell or B cell responses observed in CRS is lacking. There is no doubt that fungi can function as allergens and induce an allergic IgE-mediated reaction; allergic rhinitis should generally be considered a superimposed problem which contributes with a variable but relatively mild impact on the inflammation seen in most CRS patients [6•]. The term allergic fungal rhinosinusitis (AFRS) describes a specific clinical situation with fungi, mostly Aspergillus fumigatus or flavus, growing inside the sinuses and inducing immune reaction with formation of IgG and IgE to Aspergillus antigens [83]. AFRS is classically defined by nasal polyps; characteristic thick eosinophilic mucin; characteristic CT scan findings; type 1 hypersensitivity to fungal antigens by serology or skin tests; and fungal elements in the mucin detected by culture or histology [84, 85]. However, the clinical signs for diagnosis are largely unspecific, and the term AFRS implies an allergic pathomechanism, which is misleading. Aspergillus fumigatus per se induces allergic rhinitis, but not “AFS”; it is therefore likely that another stimulus such as superantigens from S. aureus are involved in this disease, explaining the high IgE and IgG production and severe eosinophilic inflammation [86]. The mechanisms seem indifferent from other severe inflammatory types of nasal polyps with high IgE concentrations, with Aspergillus preparing the ground for the impact of superantigens by creating a Th2 bias and eventually breaking the epithelial barrier. The term AFS will need to be revised accordingly once the mechanism has been clarified.
Conclusion: Pathophysiology, Phenotyping and Therapeutic Consequences
In recent years, inflammatory triggers and pathways have been identified which enable us to partially understand the pathophysiology of CRS, and at the same time guide us to differentiate pheno- and endotypes of disease, which are characterized by specific pathomechanisms (Fig. 1) [9]. Remodeling of the mucosa obviously is a key biochemical parameter differentiating CRSsNP from CRSwNP, and is widely equivalent to our endoscopic diagnosis; however, inflammatory patterns are less obvious clinically and require the use of biomarkers measured in the tissue—or, easier, in the serum of patients. Some of these pathways can even be used as targets for therapeutic interventions, such as anti-IL5 or anti-IgE. However, the understanding of phenotypes and endotypes may help us not only in innovative experimental approaches, but in everyday treatment; e.g., the response to topical glucocorticosteroids seems to be dependent on the type of inflammatory granulocytes, eosinophils or neutrophils [35]. In the same line, macrolides and doxycycline may merit a differential approach based on the predominant inflammation, the involvement of IgE immune responses, or the regulation of MMPs and their tissue inhibitors [6•].
Both, the anti-IgE and the anti-IL5 approach mark milestones in the management of CRS subtypes; both approaches not only are based on biomarkers and serve as proof-of-concept for the role of the respective pathomechanisms, but also represent innovative approaches providing new dimensions of disease control. Omalizumab, in patients with nasal polyps and comorbid asthma, demonstrated a significant therapeutic effect on upper and lower airway symptoms in allergic and non-allergic subjects [37∙∙]. The non-atopic CRSwNP patients with comorbid asthma were SE-IgE positive, as predicted from former studies [81••]. This trial serves as proof-of-concept for the SE-IgE endotype of and the role of staphylococcal superantigens in nasal polyposis. Based on protein data and in vitro experiments in nasal polyps [87, 88], identifying IL5 as key survival factor for eosinophils in polyps, a recent study confirmed the therapeutic impact of IL-5 in this disease [89∙]. The study may serve as proof-of-concept for the role of IL5 in CRSwNP; of note, it also shows the limitation of this approach in terms of the dependence of the response on a specific IL 5 positive eosinophilic polyp endotype.
Further approaches are currently in trial or may be developed based on some of the pathways mentioned here. New technical possibilities, such as proteomics, epigenetics and deep sequencing approaches to decipher the microbiome specific for CRS subtypes are already employed and will enlarge and up speed our understanding and therapeutic perspectives.
References
Papers of particular interest, published recently, have been highlighted as: ∙ Of importance •• Of major importance
Hastan D, Fokkens WJ, Bachert C, et al. Chronic rhinosinusitis in Europe—an underestimated disease. A GA2LEN in study. Allergy. 2011;66:1216–23.
Pleis JR, Lethbridge-Çejku M. Summary health statistics for U.S. adults: National Health Interview Survey, 2006. Vital Health Stat. 2007;10(235):1–155.
Ward MA. Ear, nose and throat procedures. Available at: F:\crs\Ear Nose and Throat procedures such as chronic sinusitus.htm. Accessed September 25, 2012.
Jarvis D, Newson R, Lotval J, et al. Asthma in adults and its association with chronic rhinosinusitis: the GA2LEN survey in Europe. Allergy. 2012;67:91–8.
Rosenfeld RM, Andes D, Bhattacharyya N, et al. Clinical practice guideline: adult sinusitis. Otolaryngol Head Neck Surg. 2007;137:S1–31.
∙ Fokkens WJ, Lund VJ, Mullol J, et al.: European position paper on rhinosinusitis and nasal polyps 2012. Rhinology 2012; 50(Suppl 23):1–299.
The newest updated version of the European reference in rhinosinusitis.
Van Crombruggen K, Van Bruaene N, Holtappels G, et al. Chronic sinusitis and rhinitis: clinical terminology Chronic Rhinosinusitis further supported. Rhinology. 2010;48:54–8.
∙ Van Bruaene N, Derycke L, Perez-Novo CA, et al.: TGF-b signaling and collagen deposition in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124:253–259.e2.
This paper demonstrates that remodeling may precede inflammation in the middle meatus, and underlines the fact that remodeling and inflammation are independent principles.
Van Crombruggen K, Zhang N, Gevaert P, et al. Pathogenesis of chronic rhinosinusitis: inflammation. J Allergy Clin Immunol. 2011;128:728–32.
Zuckerman JD, Lee WY, DelGaudio JM, et al. Pathophysiology of nasal polyposis: the role of desmosomal junctions. Am J Rhinol. 2008;22:589–97.
Rogers GA, Beste KD, Parkos CA, et al. Epithelial tight junction alterations in nasal polyposis. Int Forum Allergy Rhinol. 2011;1:50–4.
Richer SL, Truong-Tran AQ, Conley DB, et al. Epithelial genes in chronic rhinosinusitis with and without nasal polyps. Am J Rhinol. 2008;22:228–34.
Tieu DD, Kern RC, Schleimer RP. Alterations in epithelial barrier function and host defense responses in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124:37–42.
Avila PC, Schleimer RP. Airway epithelium. In: Kay AB, Kaplan AP, Bousquet J, et al., editors. Allergy and allergic diseases. London: Wiley-Blackwell; 2008. p. 366–97.
Ooi EH, Psaltis AJ, Witterick IJ, et al. Innate immunity. Otolaryngol Clin North Am. 2010;43:473–87.
Laudien M, Dressel S, Harder J, et al. Differential expression pattern of antimicrobial peptides in nasal mucosa and secretion. Rhinology. 2011;49:107–11.
Psaltis AJ, Bruhn MA, Ooi EH, et al. Nasal mucosa expression of lactoferrin in patients with chronic rhinosinusitis. Laryngoscope. 2007;117:2030–5.
Psaltis AJ, Weitzel EK, Ha KR, et al. The effect of bacterial biofilms on post-sinus surgical outcomes. Am J Rhinol. 2008;22:1–6.
Tieu DD, Peters AT, Carter RG, et al. Evidence for diminished levels of epithelial psoriasin and calprotectin in chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125:667–75.
Meyer JE, Harder J, Sipos B, et al. Psoriasin (S100A7) is a principal antimicrobial peptide of the human tongue. Mucosal Immunol. 2008;1:239–43.
Seshadri S, Lin D, Kato A, Carter R, et al.: Reduced expression of antimicrobial PLUNC proteins in nasal polyp tissue. J Allergy Clin Immunol. 2009;125:AB61.
Bulek K, Swaidani S, Aronica M, Li X. Epithelium: the interplay between innate and Th2 immunity. Immunol Cell Biol. 2010;88:257–68.
Hammad H, Lambrecht BN. Dendritic cells and airway epithelial cells at the interface between innate and adaptive immune responses. Allergy. 2011;66:579–87.
Nonak M, Fukumoto A, Ogihara N, et al. Synergistic induction of thymic stromal lymphopoietin by tumor necrosis factor alpha and Th2 cytokine in nasal polyp fibroblasts. Am J Rhinol. 2010;24:e14–8.
Liu T, Li TL, Zhao F, et al. Role of thymic stromal lymphopoietin in the pathogenesis of nasal polyposis. Am J Med Sci. 2011;341:40–7.
Kimura S, Pawankar R, Mori S, et al. Increased expression and role of thymic stromal lymphopoietin in nasal polyposis. Allergy Asthma Immunol Res. 2011;3:186–93.
Reh DD, Wang Y, Ramanathan M, et al. Treatment-recalcitrant chronic rhinosinusitis with polyps is associated with altered epithelial cell expression of interleukin-33. Am J Rhinol Allergy. 2010;24:105–9.
Kool M, Willart MAM, van Nimwegen M, et al. An unexpected role for uric acid as an inducer of T helper 2 cell immunity to Inhaled antigens and inflammatory mediator of allergic asthma. Immunity. 2011;34:527–40.
Van Crombruggen K, Holtappels G, De Ruyck N, et al. RAGE- processing in chronic airway conditions: involvement of Staphylococcus aureus and ECP. J Allergy Clin Immunol. 2012;129:1515–21.
Reinartz SM, van Tongeren J, van Egmond D, et al. Dendritic cells in nasal mucosa of subjects with different allergic sensitizations. J Allergy Clin Immunol. 2011;128:887–90.
Ayers CM, Schlosser RJ, O’Connell BP, et al. Increased presence of dendritic cells and dendritic cell chemokines in the sinus mucosa of chronic rhinosinusitis with nasal polyps and allergic fungal rhinosinusitis. Int Forum Allergy Rhinol. 2011;1:296–302.
∙ Krysko O, G Holtappels, N Zhang, et al.: Alternatively activated macrophages and impaired phagocytosis of S. aureus in chronic rhinosinusitis. Allergy 2011;66:396–403.
Krysko et al. show for the first time that macrophages in IL-5 positive nasal polyps are often alternatively activated and therefore not efficient in phagocytosis and killing of S. aureus. This principle may favorize the survival of this germ in those patients.
Peterson S, Poposki JA, Nagarkar DR, et al. Increased expression of CC chemokine ligand 18 in patients with chronic rhinosinusitis with nasal polyps. J Allergy Clin Immunol. 2012;29:119–127e9.
Zhang N, Van Zele T, Perez-Novo C, et al. Different types of T effector cells orchestrate mucosal inflammation in chronic sinus disease. J Allergy Clin Immunol. 2008;122:961–8.
Wen W, Liu W, Zhang L, et al.: Increased neutrophilia in nasal polyps reduces the response to oral corticosteroid therapy. J Allergy Clin Immunol. 2012;1522–1528.e5.
Gevaert P, Lang-Loidolt D, Lackner A, et al. Nasal IL-5 levels determine the response to anti-IL-5 treatment in patients with nasal polyps. J Allergy Clin Immunol. 2006;118:1133–41.
∙∙ Gevaert P, L Calus, T Van Zele, et al.: Effectiveness of Omalizumab in patients with nasal polyposis and co-morbid asthma. J Allergy Clin Immunol 2012 (in press).
This is a proof-of-concept study in allergic and non-allergic CRSwNP to demonstrate an impressive effect of anti-IgE treatment in polyp disease. The study confirms the role of IgE in the disease, and specifically confirms the functional role of polyclonal IgE in superantigen induced (SE-IgE positive, but inhalant allergen IgE negative) airway disease.
Van Zele T, Claeys S, Gevaert P, et al. Differentiation of chronic sinus diseases by measurement of inflammatory mediators. Allergy. 2006;61:1280–9.
∙∙ Bachert C, Zhang N, Holtappels G, et al.: Presence of IL-5 protein and IgE antibodies to staphylococcal enterotoxins in nasal polyps is associated with comorbid asthma. J Allergy Clin Immunol 2010;126:962–68.e6.
This paper demonstrates for the first time that asthma comorbidity in CRSwNP patients is dependent on the presence/quantity of several markers in the polyp tissue, including SE-IgE, total IgE and IL-5. The study encouraged us to test SE-IgE in severe asthma patients (see reference 81).
Rudack C, Steinhoff M, Mooren F, et al. PAR-2 activaiton regulates IL-8 and GRO-alpha synthesis by NF-kappaB, but not RANTES, IL-6, eotaxin or TARC expression in nasal epithelium. Clin Exp Allergy. 2007;37:1009–22.
Derycke L, Zhang N, Holtappels G, et al. IL-17A as a regulator of neutrophil survival in nasal polyp disease of patients with and without cystic fibrosis. J Cyst Fibros. 2012;11:193–200.
Cerutti A, Chen K, Chorny A. Immunoglobulin responses at the mucosal interface. Annu Rev Immunol. 2011;29:273–93.
Van Zele T, Gevaert P, Holtapples G, et al. Local immunoglobulin production in nasal polyposis is modulated by superantigens. Clin Exp Allergy. 2007;27:1840–7.
Kato A, Peters A, Suh L, et al. Evidence of a role for B cell-activating factor of the TNF family in the pathogenesis of chronic rhinosinusitis with nasal plyps. J Allergy Clin Immunol. 2008;121:1385–1392.e2.
Sabriov A, Hamilton RG, Jacobs JB, et al. Role of local immunoglobulin E specific for Alternaria alternata in the pathogenesis of nasal polyposis. Laryngoscope. 2008;118:4–9.
Gevaert P, Holtappels G, Johansson SGO, et al. Organization of secondary lymphoid tissue and local IgE formation to Staphylococcus aureus enterotoxins in nasal polyp tissue. Allergy. 2005;60:71–9.
Patadia M, Dixon J, Conley D, et al. Evaluation of the presence of B-cell attractant chemokines in chronic rhinosinusitis. Am J Rhinol. 2010;24:11–6.
Mechtcheriakova D, Sobanov Y, Holtappels G, et al. Activation-induced cytidine deaminase (AID)-associated multigene signature to assess impact of AID in etiology of diseases with inflammatory component. PLoS ONE. 2011;6:1–12.
∙∙ Zhang N, Holtappels G, Gevaert P, et al.: Mucosal tissue polyclonal IgE is functional in response to allergen and SEB. Allergy 2011;66:141–8.
The paper demonstrates the finding of local IgE in CRSwNP even in SPT- and serum IgE negative patients, and relates the finding to SE-IgE. Polyclonal IgE is proven to be functional upon allergen challenge, and this function is transferable to basophils. The paper forms the basis for an understanding of the therapeutic effect of anti-IgE (see reference 37).
Shaw JL, Ashoori F, Fakhri S, et al. Increased percentage of mast cells within sinonasal mucosa of chronic rhinosinusitis with nasal polyp patients independent of atopy. Int Forum Allergy Rhinol. 2012;2:233–40.
Patou J, Holtappels G, Affleck K, et al. Syk-kinase inhibition prevents mast cell activation in nasal polyps. Rhinology. 2011;49:100–6.
Pérez-Novo CA, Holtappels G, Vinall SL, et al. CRTH2 mediates the activation of human Th2 cells in response to PGD2 released from IgE/anti-IgE treated nasal polyp tissue. Allergy. 2010;65:304–10.
Pérez-Novo CA, Watelet JB, Claeys C, et al. Prostaglandin, leukotriene, and lipoxin balance in chronic rhinosinusitis with and without nasal polyposis. J Allergy Clin Immunol. 2005;115:1189–96.
Pérez-Novo CA, Waeytens A, Claeys C, et al. Staphylococcus aureus enterotoxin B regulates prostaglandin E2 synthesis growth, and migration in nasal tissue fibroblasts. J Infect Dis. 2008;197:1036–43.
Okano M, Fuijkura T, Haruna T, et al. Prostaglandin E2 suppresses staphylococcal enterotoxin-induced eosinophilia-associated cellular responses dominantly through an E-prostanoid 2-mediated pathway in nasal polyps. J Allergy Clin Immunol. 2009;123:868–71.
Roca-Ferrer J, Garcia–Garcia FJ, Pereda J, et al. Reduced expression of COXs and production of prostaglandin E2 in patients with nasal polyps with or without aspirin-intolerant asthma. J Allergy Clin Immunol. 2011;128:66–72.e1.
Li X, Meng J, Qiao X, et al. Expression of TGF, matrix metalloproteinases, and tissue inhibitors in Chinese chronic rhinosinusitis. J Allergy Clin Immunol. 2010;125:1061–8.
Van Bruaene N, Perez-Novo C, Van Crombruggen K, et al. Inflammation and remodeling patterns in early-stage chronic rhinosinusitis. Clin Exp Allergy. 2012;42:883–90.
Yang YC, Zhang N, Van Crombruggen K, et al. Transforming growth factor beta1 in inflammatory airway disease: a key for understanding inflammation and remodeling. Allergy. 2012;67:1193–202.
Bachert C, Zhang N. Chronic rhinosinusitis and asthma: novel understanding of the role of IgE “above atopy”. J Intern Med. 2012;272:133–43.
Van Bruaene N, Derycke L, Perez-Novo CA, et al. TGF-b signaling and collagen deposition in chronic rhinosinusitis. J Allergy Clin Immunol. 2009;124:253–259.e2.
Van Bruaene N, Bachert C. Tissue remodeling in chronic rhinosinusitis. Curr Opin Allergy Clin Immunol. 2011;11:8–11.
Sejima T, Holtappels G, Bachert C. The expression of fibrinolytic components in chronic paranasal sinus disease. Am J Rhinol. 2011;25:1–6.
Valko M, Leibfritz D, Moncol J, et al. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84.
Xiao C, Puddicombe SM, Field S, et al. Defective epithelial barrier function in asthma. J Allergy Clin Immunol. 2011;128:549–556.e12.
Cohen NA, Zhang S, Sharp DB, et al. Cigarette smoke condensate inhibits transepithelial chloride transport and ciliary beat frequency. Laryngoscope. 2009;119:2269–74.
Goldstein-Daruech N, Cope EK, Zhao KQ, et al. Tobacco smoke mediated induction of sinonasal microbial biofilms. PLoS ONE. 2011;6:e15700.
Huvenne W, Perez-Novo CA, Derycke L, et al. Different regulation of cigarette smoke induced inflammation in upper versus lower airways. Respir Res. 2010;11:1–9.
Glorieux S, Bachert C, Favoreel H, et al. Herpes simplex virus type 1 penetrates the basement membrane in human nasal respiratory mucosa. PLoS ONE. 2011;6:e22160.
∙ Wang XD, N Zhang, S Glorieux, et al.: Herpes simplex virus type 1 infection facilitates invasion of Staphylococcus aureus into the nasal mucosa and nasal polyp tissues. PLoS ONE 2012;7:e39875.
The paper answers the question how S. aureus can invade intact mucosa—with the help of a viral infection. The study describes the synergism between viral and bacterial infections in initiating persistent inflammation triggered by an intramucosal germ.
Van Zele T, Gevaert P, Watelet JB, et al. Staphylococcus aureus colonization and IgE antibody formation to enterotoxins is increased in nasal polyposis. J Allergy Clin Immunol. 2004;114:981–3.
Larson DA, Han JK. Microbiology of sinusitis: does allergy or endoscopic sinus surgery affect the microbiologic flora? Curr Opin Otolaryngol Head Neck Surg. 2011;19:199–203.
Ba L, Zhang N, Meng J, et al. The association between bacterial colonization and inflammatory pattern in Chinese chronic rhinosinusitis patients with nasal polyps. Allergy. 2011;66:1296–303.
Corriveau MN, Zhang N, Holtappels G, et al. Detection of Staphylococcus aureus in nasal tissue with peptide nucleic acid-fluorescence in situ hybridization. Am J Rhinol Allergy. 2009;23:461–5.
Sachse F, Becker K, von Eiff C, et al. Staphylococcus aureus invades the epithelium in nasal polyposis and induces IL-6 in nasal epithelial cells in vitro. Allergy. 2010;65:1430–7.
∙ A Foreman, G Holtappels, AJ Psaltis et al. Adaptive immune responses in Staphylococcus aureus biofilm associated chronic rhinosinusitis. Allergy 2011;66:1449–56.
Biofilms have been discussed as a basis for recurrent and difficult to treat disease; here we show that S. aureus positive biofilms do not only serve as a nidus for the germ to invade the tissue, but also cause a superantigen independent bias towards a Th2 response in nasal polyps.
Bachert C, Gevaert P, Holtappels G, et al. Total and specific IgE in nasal polyps is related to local eosinophilic inflammation. J Allergy Clin Immunol. 2001;107:607–14.
Conley DB, Tripathi A, Seiberling KA, et al. Superantigens and chronic rhinosinusitis: skewing of T-cell receptor V beta distributions in polyp-derived CD4+ and CD8+ T cells. Am J Rhinol. 2006;20:534–9.
Seiberling KA, Conley DB, Tripathi A, et al. Superantigens and chronic rhinosinusitis: detection of staphylococcal exotoxins in nasal polyps. Laryngoscope. 2005;115:1580–5.
Patou J, Van Zele T, Gevaert P, et al. Staphylococcus aureus enterotoxin B, protein A and lipoteichoic acid stimulations in nasal polyps. J Allergy Clin Immunol. 2008;121:110–5.
∙∙ Bachert C, van Steen K, Zhang N, et al.: Specific IgE against Staphylococcus aureus enterotoxins: an independent risk factor for asthma. J Allergy Clin Immunol 2012;130:376–381.e8.
This in the first paper to demonstrate that severe asthma is related to S. aureus specific IgE, which as a marker (and in contrast to grass pollen or house dust mite specific IgE) is associated with hospitalizations, oral steroid use and exacerbations within the last 12 months! The paper also delineates a group of non-atopic severe asthmatics with late onset of disease, in whom only SE-IgE can be detected.
Ebbens FA, Georgalas C, Luiten S, et al. The effect of topical amphotericin B on inflammatory markers in patients with chronic rhinosinusitis: a multicenter randomized controlled study. Laryngoscope. 2009;119:401–8.
Chakrabarti A, Denning DW, Ferguson BJ, et al. Fungal rhinosinusitis: a categorization and definitional schema addressing current controversies. Laryngoscope. 2009;119(9):1809–18.
Bent JP 3rd, Kuhn FA: Diagnosis of allergic fungal sinusitis. Otolaryngol Head Neck Surg. 1994;111:580–8.
Ryan MW. Allergic fungal rhinosinusitis. Otolaryngol Clin North Am. 2011;44:697–710.
Schubert MS. Allergic fungal sinusitis. Clin Rev Allergy Immunol. 2006;30:205–16.
Bachert C, Wagenmann M, Hauser U, et al. IL-5 synthesis is upregulated in human nasal polyp tissue. J Allergy Clin Immunol. 1997;99:837–42.
Simon HU, Yousefi S, Schranz C, et al. Direct demonstration of delayed eosinophil apoptosis as a mechanism causing tissue eosinophilia. J Immunol. 1997;99:837–42.
∙ Gevaert P, N van Bruaene, T Cattaert, et al.: Mepolizumab, a humanised anti-IL-5 monoclonal antibody, as treatment option for severe nasal polyposis. J Allergy Clin Immunol 2011;128:989–95.
Building up on a paper in 1997, in which Bachert C. et al. demonstrated the up-regulation of IL-5 in nasal polyps, this study shows that the principle of anti-IL5 is working. It also supports the need for phenotyping disease in order to predict response to innovative therapies.
Acknowledgments
This work was supported by grants to Claus Bachert from the Flemish Scientific Research Board, FWO, Nr. A12/5-HB-KH3 and G.0436.04, and the UIAP Interuniversity Attraction. Poles Program—Belgian State Belgian Science Policy, Nr. IAP P6/35. The Omalizumab/Mepolizumab studies were investigator-initiated efforts supported by grants from Novartis and GSK.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bachert, C., Al Bahrani, N., Al Dousary, S. et al. The Pathogenesis of CRS: An Update. Curr Otorhinolaryngol Rep 1, 25–32 (2013). https://doi.org/10.1007/s40136-012-0002-5
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40136-012-0002-5