
Abstract
Left ventricular hypoplasia complicates other forms of congenital heart disease in addition to the classic “hypoplastic left heart syndrome.” Within this heterogeneous group, subtle anatomic differences determine surgical management and ultimate prognosis. Thus, an individualized approach is necessary to optimize outcomes in this complex population.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
“Hypoplastic left heart syndrome” (HLHS) is historically a term reserved for a heart with normal segmental anatomy and varying degrees of hypoplasia of the left atrium, mitral valve, left ventricle (LV), aortic valve, ascending aorta and aortic arch [1]. LV hypoplasia can complicate other forms of congenital heart disease as well, broadening the spectrum of disease from the three forms of classic HLHS (involving combinations of mitral and aortic valve stenosis or atresia) to include complex double outlet right ventricles (DORV) and unbalanced complete common atrioventricular canal (CAVC) defects. As a greater understanding of the complexities of congenital heart disease associated with LV hypoplasia has been achieved, it has become clear that the preoperative evaluation must go far beyond simply assigning the label of HLHS and committing the patient to the route of single ventricle Fontan palliation. Thorough non-invasive imaging of this heterogeneous group yields critical information used to determine surgical reconstructive options and prognosis.
In the past, initial treatment for these single right ventricle (RV) lesions was limited to Norwood Stage I operation with a modified Blalock-Taussig shunt, primary cardiac transplantation, or comfort care. Today a variety of additional options exist, including the Norwood with an RV-to-pulmonary artery shunt (also known as the “Sano” modification), the “Hybrid” approach, and, if feasible, staged 2-ventricle conversions occasionally beginning with fetal intervention. The decision to pursue a particular approach rests primarily on the underlying anatomy and the surgeon’s preference, as well as important patient factors such as birth weight and extracardiac anomalies. While operative survival has improved significantly over the past two decades there is important late mortality, and about 40 % of children who undergo univentricular palliation for RV dominance will not survive to the second decade of life [2]. Some studies have shown that patients with atypical variants may fare the worst [3, 4, 5•], and thus an individualized approach to the management of LV hypoplasia is emerging.
Classic Hypoplastic Left Heart Syndrome
The classic form of HLHS is divided into three subtypes, including aortic and mitral stenosis (AS/MS), aortic atresia with mitral stenosis (AA/MS), and aortic and mitral atresia (AA/MA). Most often HLHS is characterized by severe hypoplasia of all left-sided structures, including a rudimentary LV and a diminutive ascending aorta. In the fetus with HLHS, very little (if any) pulmonary venous return enters the LV, and most crosses the atrial septal communication in an abnormally left-to-right direction. Fetal survival is permitted by patency of this communication as well as the ductus arteriosus, which supplies systemic blood flow from the RV to the descending aorta and often retrograde flow to the head and neck vessels and coronary arteries via the aortic arch. After birth, the neonate relies on continued patency of both the atrial communication and the ductus arteriosus, or inevitable demise will occur.
Many theories exist about the development of HLHS, one of which is that of flow dynamics. Normal embryologic development of the cardiovascular system requires the shear stress of blood flow to trigger growth and differentiation of the vasculature and intracardiac valves and chambers [6]. Thus, altered blood flow in utero due to downstream obstruction may result in hypoplasia of more upstream structures, as seen in fetuses with aortic valve stenosis and an initially dilated LV who suffer arrested left heart growth and progress to HLHS later in gestation [7]. Perturbed flow across an intrinsically abnormal atrial septum or mitral valve may also be an inciting factor [8]. A genetic component undoubtedly provides an important substrate for the development of HLHS, as demonstrated by its recognized association with genetic abnormalities such as Turner, Down, and Holt-Oram syndromes [9], and in reports of increased prevalence of anomalies such as bicuspid aortic valve in first-degree relatives of those with HLHS [10]. However, in contrast to the higher prevalence of extracardiac anomalies in some other single ventricle lesions, only about 5–10 % of patients with classic HLHS have an identifiable syndrome or chromosomal aberration [4, 11•].
Preoperative evaluation includes careful assessment of multiple anatomic details, including precise measurements of all left-sided structures, mitral and aortic valve structure and function, and mitral valve attachments. The adequacy of the right heart to support the systemic circulation must be assessed by detailed imaging of the RV and tricuspid valve. Associated anomalies such as pulmonary venous obstruction or anomalous drainage, abnormal coronary configuration, and “sinusoids” connecting the coronary circulation with the hypertensive LV cavity must be identified. Endocardial fibroelastosis, a stiff, fibrotic, echogenic layer along the endocardium of the LV cavity, causes both systolic and diastolic dysfunction in HLHS. This type of dysfunction typically does not complicate other single ventricle lesions in which LV afterload is reduced by a ventricular septal defect (VSD), but plays a major role in classic HLHS [12].
A restrictive or intact atrial septum is found in a small percentage of newborns with HLHS and related lesions, resulting in hypertension of the left atrium and pulmonary vasculature. Rightward bowing of the atrial septum, turbulent flow and elevated mean gradients across the atrial communication, abnormally high estimates of left atrial pressures, and clinical findings such as profound cyanosis and poor perfusion suggest the need for emergent intervention. Even after catheter septoplasty of a restrictive atrial septum, however, neonatal mortality remains high [6]. Pulmonary venous hypertension in fetal life promotes “arterialization” and parenchymal abnormalities of the pulmonary vasculature, blunted pulmonary vasoreactivity, and dilated lymphatics, which may greatly influence outcomes [6, 13]. Many centers have incorporated routine maternal hyperoxygenation during echocardiography of the late gestation fetus with HLHS, for evaluating pulmonary vascular health and predicting which neonates will require emergent intervention. Fetuses with HLHS and a restrictive atrial septum have demonstrated Doppler evidence of significantly less pulmonary vasoreactivity than those with an open atrial communication [13].
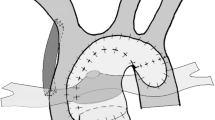
In the most common management strategy of classic HLHS, a series of complex surgeries starting in the neonatal period culminates in the univentricular Fontan circulation. The newborn is maintained on a continuous infusion of intravenous prostaglandin to maintain ductal patency until the Norwood stage 1 operation (Fig. 1), which is typically performed within the first week of life for those without delayed diagnosis. The goals of the Norwood procedure are to restore reliable systemic blood flow while protecting the pulmonary vascular bed. This is accomplished by division of the main pulmonary artery, anastomosis of the ascending aorta to this segment, and reconstruction of a “neo-aorta” to receive the RV output. Controlled blood flow is provided to the now isolated pulmonary arteries through either a modified Blalock-Taussig shunt or RV-to-pulmonary artery shunt [14, 15]. An atrial septectomy is performed to allow unobstructed left-to-right flow.

In most centers the Norwood stage 1 operation is the first intervention for classic HLHS, with a modified Blalock-Taussig shunt (a) or an RV-to-pulmonary artery shunt (b) supplying pulmonary blood flow. Some centers utilize the “Hybrid” approach (c), which includes stenting of the ductus arteriosus and bilateral pulmonary artery banding
The bidirectional Glenn procedure (superior cavopulmonary connection), which is typically performed at 4–6 months of age, is an extracardiac surgery in which the superior vena cava is anastomosed to the pulmonary artery. This provides more reliable pulmonary blood flow, decreases the pressure and volume load of the single RV, and relieves systemic output (importantly, the coronary arteries) of the diastolic flow reversal that occurs in cases of Blalock-Taussig shunts. Finally, the Fontan procedure (total cavopulmonary connection) is employed at 2–4 years of age. Here, the inferior vena cava is connected to the pulmonary artery by means of an intracardiac baffle or an extracardiac conduit, which may be fenestrated to allow preserved (although hypoxemic) cardiac output in cases where increased pulmonary vascular resistance is of concern.
The “Hybrid” approach (Fig. 1) has been employed as the first stage of HLHS palliation in some centers, while most have reserved it for neonates with major extracardiac anomalies or complicating factors such as low birth weight or prematurity [16, 17]. The Hybrid procedure currently involves stenting of the ductus arteriosus (via sternotomy) and bilateral pulmonary artery banding to protect the pulmonary vasculature. Retrograde arch obstruction is a complication that occurs in 24–29 % of patients (especially those with aortic atresia) after this approach, and has been identified as a risk factor for morbidity and mortality [18, 19]. Initial enthusiasm for the Hybrid technique as a means of avoiding the sequelae of neonatal cardiopulmonary bypass was tempered by equally high mortality rates [6, 16, 17].
There is a subset of patients with AS/MS in whom borderline LV hypoplasia exists, due to less severe valvar stenosis or occasionally an alternate exit of blood flow in the form of a VSD [6]. Patients with Shone complex, a combination of multiple left-sided obstructive lesions, may also fit into this “borderline” group. These anatomic variants may be amenable to biventricular repair, either as initial surgery if mitral valve and LV dimensions are deemed adequate, or as part of “staged LV recruitment” to promote growth of the left heart [20, 21]. This entails, during stage 1–3 of palliation, concomitant performance of mitral and aortic valvuloplasty, atrial septal defect restriction, and resection of LV endocardial fibroelastosis, breaking away to biventricular conversion at any interstage point [20]. In select patients, this strategy may allow a patient with a borderline LV to achieve biventricular circulation.
Since it was first reported in 1991, fetal aortic balloon valvuloplasty has been utilized in select cases with the goal of relieving aortic outflow obstruction, promoting growth of the left heart, and achieving a postnatal biventricular circulation [7]. Candidates are chosen quite carefully, as the procedure carries important risks of maternal morbidity and fetal demise. Recent studies suggest that while fetal intervention may result in enhanced prenatal growth of the aortic and mitral valves, growth of the LV does not improve to the same degree [7]. Some authors postulate that reduced hyperplastic potential of the LV occurs at a certain point in fetal development, rendering intervention after that time less successful [6].
Falling out of favor as palliative options for HLHS improve is the tactic of primary orthotopic heart transplantation, first developed in the mid-1980s [22]. Today, most centers reserve transplantation for those with significant RV dysfunction or severe tricuspid regurgitation that is not amenable to further intervention [14], or as a “rescue” treatment for those who have failed palliation [23]. Hesitation to use primary transplant is influenced by many factors, including the improved outcomes for palliated single ventricle patients and the shortage of donors, as well as wait-list and post-transplant morbidity and mortality [14].
Early outcomes of patients with HLHS after the first stage of palliation have been described by reports from the prospective single ventricle reconstruction (SVR) trial [5•, 11•, 24]. Mortality during the Norwood hospitalization was 16 % in this trial, and the strongest predictors of mortality (which have been corroborated by other studies) were need for extracorporeal membrane oxygenation support [14, 15] and open sternum on the day of Norwood operation. Additional risk factors for death during the initial hospitalization included lower birth weight [3, 14], genetic abnormalities [14, 15], and lower surgical center volume. Risk factors identified in this trial for intermediate mortality (at 2.7 years) have also been previously described [11•], including prematurity [1, 15], a smaller ascending aorta, a restrictive atrial septum [1], and lower socioeconomic status. The Sano modification, while providing improved transplant-free survival at 1 year when compared to the modified Blalock-Taussig shunt (74 vs. 64 %), was not associated with improved survival at intermediate follow-up [5•, 24]. Although previous smaller single center studies had found no difference in mortality between the anatomic subtypes of HLHS [1, 3, 11•], the SVR trial showed a clear survival benefit in patients with AS/MS compared to other HLHS subtypes and other single RV variants at intermediate follow-up [5•]. Long-term outcomes of classic HLHS vary in the literature, but it is anticipated that about 70 % of patients born today will survive to adulthood [8].
Complex Double Outlet Right Ventricle
By consensus of the Congenital Heart Surgery Nomenclature and Database Project, DORV is defined as “a type of ventriculoarterial connection in which both great vessels arise entirely or predominantly from the RV” [25]. DORV encompasses a wide range of pathology, with the orientation of the great arteries, the relationship of the VSD to the great arteries, the degree of aortic or pulmonic outflow obstruction, and associated defects influencing surgical options [26]. Complex DORV, especially in cases of heterotaxy syndrome, may also be associated with unbalanced CAVC defects [27, 28].
The mitral valve in complex DORV may be normal, mildly hypoplastic and stenotic, or fully atretic. Similar to the newborn with the AA/MA subtype of HLHS, survival with DORV and mitral atresia depends on an unrestrictive atrial communication for left atrial egress. In those with patency of the mitral valve and LV output directed through a subpulmonary VSD, aortic outflow tract obstruction and varying degrees of aortic valve and arch pathology may occur. In DORV with a subaortic VSD, pulmonary outflow tract obstruction may ensue.
The causes of congenital heart disease are often multi-factorial, and in DORV there are distinct chromosomal abnormalities in about 10 % and extracardiac anomalies in 34 % [26]. This lesion’s association with heterotaxy syndrome and right atrial isomerism, in some instances, has linked it to specific genetic mutations and autosomal recessive inheritance [29]. In these patients, extracardiac anomalies such as asplenia and intestinal malrotation are common.
While most forms of DORV are amenable to biventricular repair, consideration must be paid to the initial risk compared to the conceivable late benefit of attempting biventricular circulation in the complex DORV [26, 27]. An adequately sized mitral valve with normal attachments, an adequately sized LV, and a VSD type other than noncommitted have been recognized as the main determinants of a successful biventricular repair in DORV [26], but those with significant mitral valve stenosis or atresia and LV hypoplasia require univentricular palliation. In cases of complex DORV and aortic outflow tract obstruction, neonatal surgery would include a modified Norwood Stage I palliation similar to those with classic HLHS. Mostly reported in classic HLHS, the Hybrid procedure may be practical in some complex DORVs as well. At this time, primary cardiac transplantation and fetal intervention are not typically undertaken for this lesion.
Patients with complex DORV and LV hypoplasia are often grouped with classic HLHS in outcomes studies, and small numbers of patients preclude accurate survival estimates of this entity. The sheer heterogeneity of the complex DORV highlights the utility of an individualized surgical approach. The association of DORV with highly complex laterality defects may influence outcomes, and studies of patients with right atrial isomerism (associated with DORV in almost half, most of which require single ventricle palliation) report dismal survival rates of as low as 22 % at a median of about 14 years [29]. This may be largely due to the frequent finding of total anomalous pulmonary venous return in these patients, which has been shown to be an independent risk factor for mortality in cohorts of complex DORV with or without LV hypoplasia [28]. The more “straightforward” DORV undergoing univentricular Fontan palliation, however, will likely experience outcomes more comparable to those with HLHS. Interestingly, some studies of DORV have demonstrated higher rates of mortality and reintervention after biventricular repair compared to single ventricle palliation, perhaps due to misjudgements about the adequacy of the left heart to support the systemic circulation [26, 27]. This emphasizes the importance of a meticulous preoperative evaluation.
Right Ventricle Dominant Unbalanced Common Atrioventricular Canal Defect
CAVC defects result from failure of the embryologic endocardial cushions to develop, leaving a common atrioventricular (AV) valve, a primum atrial septal defect, and often an inlet VSD. The defect is “unbalanced” if the common AV valve is positioned predominantly over one ventricle, with varying degrees of hypoplasia of the contralateral ventricle [12, 30, 31]. Unbalanced CAVC defects are complex lesions with highly variable morphology. About 10 % of CAVC defects are considered unbalanced, almost 90 % of which are RV dominant with LV hypoplasia [4, 30, 31]. Of these patients, the majority have some degree of AV valve regurgitation. The LV outflow tract, which is elongated with an unusual course in the CAVC, is frequently obstructed in the unbalanced forms [4, 12]. The aortic valve may be severely affected and even atretic, and the aortic arch may be hypoplastic with discrete coarctation.
Of patients with CAVC, 76 % (significantly more than those with HLHS or complex DORV) have an associated chromosomal abnormality, identifiable syndrome, or heterotaxy [4]. Up to 35 % of unbalanced CAVC defects are associated with Trisomy 21, and about 40 % are associated with heterotaxy syndromes and atrial isomerism [4]. In addition, extracardiac anomalies in the gastrointestinal and urologic systems are common.
Because of the wide range of LV size in CAVC defects, and the implications of committing patients (especially those considered high-risk) to univentricular palliation, the primary focus of the preoperative evaluation is the adequacy of left AV valve and LV size. Complicating the decision of which patients will tolerate a biventricular repair, the degree of ventricular hypoplasia quite often does not correlate with the degree of AV valve unbalance [30, 32]. Cohen et al. [30] thus sought to refine the definition by measuring the ratio of the smaller AV valve area to the larger AV valve area, and identified a ratio of less than or equal to 0.67 as diagnostic of unbalance (with 1.0 representing equal balance). Alternatively, Jegatheeswaran et al. [32] modified this “AV valve index,” dividing the left AV valve area by the total AV valve area such that a value of 0.5 represented equal balance. At a ratio of less than about 0.2 most clinicians in this study opted for staged univentricular palliation, while more unpredictable clinical decision-making occurred at ratios of 0.2–0.4. At ratios greater than 0.4, a clearer transition in surgical strategy from univentricular to biventricular repair occurred [32]. It is important to note that significant overlap in the ventricular cavity size ratio occurs between unbalanced and balanced defects, suggesting that assessment of ventricular size alone is not adequate in the preoperative evaluation of the CAVC [12, 30].
In addition to AV valve size, assessment of valve structure and function is critical in the preoperative evaluation. The degree of valvar insufficiency (which is more common in unbalanced than in balanced defects [30]), leaflet anomalies and clefts, AV valve inflow (which may be directed through the cleft), chordal attachments, and papillary muscle architecture should be systematically assessed, keeping in mind that Doppler gradients across the left AV valve are often unreliable in the setting of the large primum atrial septal defect [12]. Noninvasive imaging of the beating heart can provide a precise understanding of the dynamic valve structure and function, which helps prepare for technical modifications during repair of the intraoperative arrested, empty heart.
The optimal surgical strategy for the unbalanced CAVC defect is largely unknown, and there are no concrete guidelines for clinical decision-making [32]. Options include biventricular repair if the left heart is believed to be adequate, single ventricle palliation beginning with a modified Norwood procedure for those with severe LV outflow tract or aortic valve obstruction, or a temporizing pulmonary artery band in cases with an acceptable systemic outflow [4]. Because of the relatively high incidence of AV valve regurgitation, concomitant repair of the valve may be necessary. Staged biventricular repairs have also been attempted in unbalanced CAVC defects [33], although long-term outcomes have not yet been reported. As in HLHS, palliation continues with a bidirectional Glenn and concludes with a Fontan procedure. Of note, in cases of significant AV valve regurgitation, it has been postulated that the insufficiency may be improved by earlier progression to the second stage, reducing the volume load of the RV [31].
Compared to balanced CAVC defects, unbalanced lesions are associated with higher morbidity and mortality [4, 12, 31, 32]. Mortality in patients with unbalanced CAVC defects (after biventricular repair, univentricular repair, or pulmonary artery banding) is reported at 22.4 % compared to only 6.9 % of balanced CAVC defects (after biventricular repair or banding) [32]. Although not all studies concur [2], there is also evidence to suggest that patients with palliated RV-dominant CAVC defects may experience worse outcomes than those with classic HLHS, with significantly lower mid-term survival reported (50 vs. 74 % survival to a median of about 2 years) [4]. Disparities in survival may be due to unique features of this lesion when compared to other forms of LV hypoplasia, including a malaligned AV junction, abnormal AV valve leaflets, and the higher incidence of subsequent AV valve repair or replacement in unbalanced CAVC patients (as high as 30 %) with attrition after reoperation [4]. The higher incidence of total anomalous pulmonary venous return in the unbalanced CAVC compared to HLHS may also influence outcomes [4]. Trisomy 21 complicates this lesion further, as these patients have a predilection for pulmonary vascular disease due to hypotonia with hypoventilation, upper airway obstruction, and intrinsic lung disease. Since the Fontan circulation relies on low pulmonary vascular resistance, patients with Trisomy 21 are considered high-risk candidates for univentricular palliation, and if feasible, a biventricular repair should be the goal [32].
Discussion
A number of congenital heart diseases associated with LV hypoplasia, most requiring a series of surgeries culminating in the Fontan palliation, are often classified together as “single RV” lesions. It is apparent, however, that these patients must be approached on an individual basis, recognizing the crucial disparities within the lesion types (and subtypes) that alter management and prognosis.
The single RV has been identified as an independent risk factor for mortality when compared to single LV lesions [1], with 10-year survival rates reported at 60 % (vs. 85 %) [2]. This may be due to differences in the potential for cellular hyperplasia between the RV and LV, as well as inherent properties of the tricuspid valve that make functional valve abnormalities more common [1].
Although palliation of the diverse group of congenital heart diseases associated with LV hypoplasia may conclude with a similar third stage operation, the years leading up to Fontan completion are highly variable. The criteria used to determine optimal surgical strategy in one variant may not be applicable to another. Wide variations in anatomy make surgical outcomes of large groups of single RVs difficult to extrapolate to a single, unique patient. Indeed, within studied cohorts of palliated patients, conclusions about survival are often inconsistent. Some suggest no significant difference in survival between classic HLHS and the more atypical forms of single RV in the recent era [2]. Others point out significantly worse outcomes for atypical lesions [3, 4], with a death hazard ratio of 4 for SVR trial patients with non-HLHS forms of single RV compared to those with the AS/MS subtype of classic HLHS [5•].
Setting these lesions apart from one another are complexities such as AV valve anatomy, the incidence of compromised LV function, associated cardiac abnormalities such as anomalous pulmonary venous return, and important differences in the prevalence of genetic syndromes and heterotaxy. Management strategies and univentricular or biventricular surgical options depend largely on these individual patient and anatomic characteristics.
Finally, as survival of patients with LV hypoplasia has increased, so has the recognition that neurodevelopmental impairments are exceedingly common in this group. The SVR trial revealed that 14-month developmental indices of the Bayley Scales of Infant Development-II in a large cohort of single RV patients are inferior to the general population (Psychomotor Development Index >1 and >2 standard deviations below the predicted mean in 65 and 44 % of patients, respectively) [34•]. Factors such as the presence of a genetic syndrome, lower birth weight, the experience of the surgical center, longer hospitalization and period of mechanical ventilation after Norwood procedure, and post-discharge complications were identified as predictors of lower neurodevelopmental scores [34•]. While inherent patient factors are responsible for a significant proportion of such impairment, optimizing neurodevelopmental outcomes must nonetheless become the new focus of management strategies.
Conclusion
LV hypoplasia complicates other forms of congenital heart disease in addition to the classic “HLHS.” Lesions such as complex DORV and RV-dominant CAVC defects are also associated with hypoplastic left-sided structures, and often join HLHS in the surgical route that ultimately ends with Fontan palliation. However, it is clear that within this heterogeneous group, and indeed within the smaller subgroups, subtle anatomic differences exist that complicate preoperative management, surgical decision-making, and ultimate prognosis. A highly individualized approach to each of these cardiac lesions, from the echocardiographic evaluation to the operative strategy, is crucial to maximize outcomes in this complex population.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Daebritz SH, Nollert GD, Zurakowski D, et al. Results of Norwood stage I operation: comparison of hypoplastic left heart syndrome with other malformations. J Thorac Cardiovasc Surg. 2000;119(2):358–67.
d’Udekem Y, Xu MY, Galati JC, et al. Predictors of survival after single-ventricle palliation: the impact of right ventricular dominance. J Am Coll Cardiol. 2012;59(13):1178–85.
Murtuza B, Stumper O, Wall D, et al. The effect of morphologic subtype on outcomes following the Sano-Norwood procedure. Eur J Cardiothorac Surg. 2012;42(5):787–93.
Owens GE, Gomez-Fifer C, Gelehrter S, et al. Outcomes for patients with unbalanced atrioventricular septal defects. Pediatr Cardiol. 2009;30(4):431–5.
• Tweddell JS, Sleeper LA, Ohye RG, et al.: Intermediate-term mortality and cardiac transplantation in infants with single-ventricle lesions: risk factors and their interaction with shunt type. J Thorac Cardiovasc Surg 2012;144(1):152–9. This is an important analysis of data from the Single Ventricle Reconstruction Trial, evaluating risk factors associated with death and cardiac transplantation in single ventricle patients at intermediate follow-up.
Hickey EJ, Caldarone CA, McCrindle BW. Left ventricular hypoplasia: a spectrum of disease involving the left ventricular outflow tract, aortic valve, and aorta. J Am Coll Cardiol. 2012;59(1 Suppl):S43–54.
McElhinney DB, Marshall AC, Wilkins-Haug LE, et al. Predictors of technical success and postnatal biventricular outcome after in utero aortic valvuloplasty for aortic stenosis with evolving hypoplastic left heart syndrome. Circulation. 2009;120(15):1482–90.
Feinstein JA, Benson DW, Dubin AM, et al. Hypoplastic left heart syndrome: current considerations and expectations. J Am Coll Cardiol. 2012;59(1 Suppl):S1–42.
Natowicz M, Chatten J, Clancy R, et al. Genetic disorders and major extracardiac anomalies associated with the hypoplastic left heart syndrome. Pediatrics. 1988;82(5):698–706.
Brenner JI, Berg KA, Schneider DS, et al. Cardiac malformations in relatives of infants with hypoplastic left-heart syndrome. Am J Dis Child. 1989;143(12):1492–4.
• Tabbutt S, Ghanayem N, Ravishankar C, et al. Risk factors for hospital morbidity and mortality after the Norwood procedure: A report from the Pediatric Heart Network Single Ventricle Reconstruction trial. J Thorac Cardiovasc Surg 2012;144(4):882–95. This is an analysis of data from the Single Ventricle Reconstruction Trial, evaluating risk factors associated with morbidity and mortality during the Norwood hospitalization.
Cohen MS, Spray TL: Surgical management of unbalanced atrioventricular canal defect. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2005;8(1):135–44.
Szwast A, Tian Z, McCann M, et al. Vasoreactive response to maternal hyperoxygenation in the fetus with hypoplastic left heart syndrome. Circ Cardiovasc Imaging. 2010;3(2):172–8.
Gaynor JW, Mahle WT, Cohen MI, et al. Risk factors for mortality after the Norwood procedure. Eur J Cardiothorac Surg. 2002;22(1):82–9.
Jacobs JP, O’Brien SM, Chai PJ, et al. Management of 239 patients with hypoplastic left heart syndrome and related malformations from 1993 to 2007. Ann Thorac Surg 2008;85(5):1691–6; discussion 1697.
Bacha EA, Daves S, Hardin J, et al. Single-ventricle palliation for high-risk neonates: the emergence of an alternative hybrid stage I strategy. J Thorac Cardiovasc Surg. 2006;131(1):163e2–171e2.
Galantowicz M, Cheatham JP, Phillips A, et al. Hybrid approach for hypoplastic left heart syndrome: intermediate results after the learning curve. Ann Thorac Surg 2008;85(6):2063–70; discussion 2070–1.
Egan MJ, Hill SL, Boettner BL, et al. Predictors of retrograde aortic arch obstruction after hybrid palliation of hypoplastic left heart syndrome. Pediatr Cardiol. 2011;32(1):67–75.
Stoica SC, Philips AB, Egan M, et al. The retrograde aortic arch in the hybrid approach to hypoplastic left heart syndrome. Ann Thorac Surg 2009;88(6):1939–46; discussion 1946–7.
Emani SM, McElhinney DB, Tworetsky W, et al. Staged left ventricular recruitment after single-ventricle palliation in patients with borderline left heart hypoplasia. J Am Coll Cardiol. 2012;60(19):1966–74.
Hansen JH, Petko C, Bauer G, et al. Fifteen-year single-center experience with the Norwood operation for complex lesions with single-ventricle physiology compared with hypoplastic left heart syndrome. J Thorac Cardiovasc Surg. 2012;144(1):166–72.
Bailey LL. Origins of neonatal heart transplantation: an historical perspective. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu. 2011;14(1):98–100.
Murtuza B, Dedieu N, Vazquez A, et al. Results of orthotopic heart transplantation for failed palliation of hypoplastic left heart. Eur J Cardiothorac Surg 2013;43(3):597–603.
Ohye RG, Sleeper LA, Mahony L, et al. Comparison of shunt types in the Norwood procedure for single-ventricle lesions. N Engl J Med. 2010;362(21):1980–92.
Walters HL 3rd, Mavroudis C, Tchervenkov CI, et al. Congenital Heart Surgery Nomenclature and Database Project: double outlet right ventricle. Ann Thorac Surg. 2000;69(4 Suppl):S249–63.
Bradley TJ, Karamlou T, Kulik A, et al. Determinants of repair type, reintervention, and mortality in 393 children with double-outlet right ventricle. J Thorac Cardiovasc Surg. 2007;134(4):967e6–973e6.
Kleinert S, Sano T, Weintraub RG, et al. Anatomic features and surgical strategies in double-outlet right ventricle. Circulation. 1997;96(4):1233–9.
Takeuchi K, McGowen FX Jr, Bacha EA, et al. Analysis of surgical outcome in complex double-outlet right ventricle with heterotaxy syndrome or complete atrioventricular canal defect. Ann Thorac Surg. 2006;82(1):146–52.
Eronen MP, Aittomaki KA, Kajantie EO, et al. The Outcome of Patients With Right Atrial Isomerism is Poor. Pediatr Cardiol. doi:10.1007/s00246-012-0445-y.
Cohen MS, Jacobs ML, Weinberg PM, Rychik J. Morphometric analysis of unbalanced common atrioventricular canal using two-dimensional echocardiography. J Am Coll Cardiol. 1996;28(4):1017–23.
Mitchell ME, Litwin SB, Tweddell JS. Complex atrioventricular canal. Semin Thorac Cardiovasc Surg Pediatr Card Surg Annu 2007;10(1):32–41.
Jegatheeswaran A, Pizarro C, Caldarone CA, et al. Echocardiographic definition and surgical decision-making in unbalanced atrioventricular septal defect: a Congenital Heart Surgeons’ Society multiinstitutional study. Circulation. 2010;122(11 Suppl):S209–15.
Foker JE, Berry JM, Harvey BA, Pyles LA. Mitral and tricuspid valve repair and growth in unbalanced atrial ventricular canal defects. J Thorac Cardiovasc Surg. 2012;143(4 Suppl):S29–32.
• Newburger JW, Sleeper LA, Bellinger DC, et al. Early developmental outcome in children with HLHS and related anomalies: the single ventricle reconstruction trial. Circulation 2012;125(17):2081–91. This is a study of the 14-month neurodevelopmental outcomes of transplant-free survivors of the Norwood operation, evaluating risk factors associated with lower development indices.
Disclosure
Denise A. Hayes, Wyman W. Lai, Peter Frommelt, and Emile Bacha declare no conflicts of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Hayes, D.A., Lai, W.W., Frommelt, P. et al. Beyond Hypoplastic Left Heart Syndrome: The Spectrum of Congenital Heart Disease Associated with Left Ventricular Hypoplasia. Curr Pediatr Rep 1, 102–108 (2013). https://doi.org/10.1007/s40124-013-0016-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-013-0016-6