
Abstract
Childhood obesity and its related comorbidities, such as dyslipidemia, arterial hypertension and impaired glucose metabolism, which constitute the Metabolic Syndrome, are associated with an increased risk for early onset endothelial dysfunction, atherosclerosis, and cardiovascular disease later in life. Genetic, epigenetic and developmental influences, such as maternal weight status during pregnancy, birth weight, nutrition and early stress, interact with modern lifestyle factors producing the obese phenotype. Increased insulin secretion, together with disturbed concentrations of adipokines and inflammation-related molecules secreted by adipose tissue contribute, through complex pathophysiologic pathways, to the development of endocrine and metabolic complications of childhood obesity.
Similar content being viewed by others
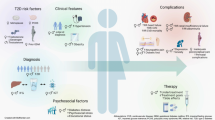


Avoid common mistakes on your manuscript.
Introduction
During the last three to four decades, the prevalence of childhood obesity has been rising dramatically worldwide, leading to an increase in the prevalence of obesity in adulthood, and, consequently, increased cardiovascular morbidity and mortality. Childhood obesity has emerged as a complex phenotype that results from gene–environment interactions during developmentally vulnerable periods of life. Although endocrine causes of pediatric obesity, such as Cushing syndrome, growth hormone deficiency or hypothyroidism, are rare, obesity-related endocrine and metabolic manifestations represent the most common concurrent and/or long-term consequences of childhood obesity, and consist a major risk for atherosclerotic cardiovascular disease later in life. The present overview focuses on the endocrine and metabolic aspects of simple pediatric obesity.
The Pediatric Metabolic Syndrome (MetS)
The increasing prevalence of childhood obesity over the last few decades and the parallel increase in the prevalence of diabetes type 2 in the young underscore the importance of recognizing that pediatric MetS is associated with an increased risk for the development of diabetes type 2 and cardiovascular disease (CVD) later in life [1, 2]. MetS was originally defined in adults as the clustering of obesity and related cardiometabolic risk factors that directly increased the risk for diabetes type 2 and CVD [3]. Its main components include central obesity, glucose intolerance, dyslipidemia [increased triglycerides and decreased high density lipoprotein (HDL)] and increased arterial blood pressure (BP), whereas insulin resistance (IR) is considered as a core pathophysiologic mechanism linking most of the above described abnormalities. Reaven and colleagues [4] first described the concept of MetS, hypothesizing that it was the main feature in the development of CVD, mainly through IR of target tissues. The Third National Health and Nutrition Examination Survey (NHANES 1988–1994) [5] was the first to reveal that 4.2 % of adolescents in general, and almost 30 % of overweight and obese adolescents in the US fulfilled the criteria for the diagnosis of MetS. The NHANES in 1999–2000 identified a further increase in the prevalence of MetS among US adolescents, from 4.2 % in NHANES III (1988–1994) to 6.4 % in NHANES (1999–2000) [6]. The prevalence of MetS was almost exclusively high among obese adolescents.
In children and adolescents, similarly to adults, there is no general consensus regarding the definition of MetS [7, 8]. A variety of published pediatric studies have used diverse sets of variables, number of criteria (three or four) and cut-off points to define risk factors associated with MetS. However, most definitions of pediatric MetS do not take into account the alterations in the hormonal milieu during childhood and adolescence, for example the developmentally-related increase of insulin secretion [9], the pubertal changes in fat and fat-free mass or the alterations in growth hormone and sex steroid secretion during puberty. One of the most prevailing definitions is in the consensus report, which was published by the International Diabetes Federation (IDF) group in 2007 [10]. This definition includes three age groups: 6 to <10, 10 to <16 and >16 years (adult criteria). In the IDF consensus report, obesity is defined as waist circumference (WC) ≥90th percentile, or adult cut-off if lower, while absolute numbers, rather than percentiles, are used for all other parameters. The rationale for using absolute numbers for cut-offs instead of percentiles is based on the fact that clinical, biochemical and hormonal values are highly inconsistent and depend upon maturation stages during childhood and adolescence. The absolute cut-offs are as follows: ≥150 mg/dL for triglycerides (or specific treatment for triglycerides), <40 mg/dL for HDL and <50 mg/dL in females older than 16 (or specific treatment for HDL), ≥130 mmHg for systolic and ≥85 mmHg for diastolic BP (or treatment of previously diagnosed hypertension) and fasting plasma glucose ≥100 mg/dL or known diabetes type 2.
More recent studies have examined the stability of MetS diagnosis during adolescence. Epidemiological studies showed that although the metabolic risk factor clustering was consistent, the categorical diagnosis of MetS was not stable during adolescence [11], including both gain and loss of this diagnosis. Studies that examined the consistency of three alternative models of MetS factor structure across three developmental changes suggested that the concepts used to support the utility of MetS diagnosis in the young may vary by maturation, thus, they do not fit the pediatric population [12•]. These studies reveal a high level of diagnostic inconsistency of MetS through adolescence, and suggest that defining the MetS may not be an accurate method for risk identification in the young. Therefore, clinicians should focus their efforts on healthy lifestyle promotion and obesity prevention rather than targeting specific hormonal and biochemical alterations.
Adipokines in Childhood Obesity
The adipose tissue, which consists of the white adipose tissue (WAT), the most prevalent type, and the brown adipose tissue that participates mainly in thermogenesis, has now emerged as an active endocrine organ regulating a variety of physiological processes [13, 14]. The adipokines, such as leptin, adiponectin, resistin, adipsin and visfatin, are proteins secreted by the adipocytes and released into the systemic circulation, regulating tissue insulin sensitivity, food intake, inflammation, immunity and vascular tone [14] (Fig. 1). The WAT consists mainly of adipocytes and a small number of macrophages. The number of macrophages infiltrated in WAT is directly associated with the level of adiposity and the size of the adipocytes in humans and rats [15]. Macrophages are the main source of WAT-derived TNF-alpha and interleukin-6, and circulating concentrations of these cytokines are directly associated with adiposity, IR, and the MetS [15, 16].

Obesity is associated with increased secretion of adipokines, such as leptin, resistin, RBP-4, TNF-alpha, IL-6 and visfatin, and decreased secretion of adiponectin. These proteins are released into the systemic circulation, and they are involved in the pathophysiology of changes in insulin sensitivity and the Metabolic Syndrome, as well as in food intake, energy consumption, neuroendocrine function, inflammation, immunity and altered vascular tone
Leptin, the protein encoded by the ob gene, belongs to the class 1 cytokine superfamily, and is mainly produced by adipocytes [17]. Circulating leptin concentrations are directly associated with the adipose tissue mass and this hormone is mainly involved in the control of appetite and energy consumption. Leptin acts on the hypothalamus and causes a decrease in food intake and increase in energy consumption by inducing anorexigenic and inhibiting orexigenic neuropeptides [18]. Typically, serum leptin concentrations are elevated in obese children and decrease following weight reduction [19, 20]. Recent studies that examined the relation between plasma leptin concentrations and various adiposity measurements, have demonstrated similar associations between leptin concentrations and BMI, WC, fat mass index (FMI), and percentage body fat (%BF) in females [21•]. In males, there was a stronger association of leptin concentrations with the percentage of BF and FMI than with BMI and WC. Furthermore, leptin was the most sensitive adipokine for predicting the accumulation of cardiovascular risk factors and the presence of MetS [22].
Adiponectin is a 244-residue protein, mainly produced in WAT, which increases the oxidation of fatty acids and reduces the synthesis of glucose in the liver [23]. Adiponectin circulates in human blood in multimers, a trimeric low-molecular-weight, a hexameric medium-molecular-weight, and a high-molecular-weight (HMW) adiponectin [24]. It has been suggested that these adiponectin multimers have different biological actions and that the HMW-form is the one with the highest activity. Apiponectin has profound insulin-sensitizing effects and plays an important role in regulating energy metabolism. Circulating adiponectin concentrations are decreased in obese patients, particularly in patients with abdominal obesity, while they increase following weight loss [25, 26].
Adiponectin has been associated with inflammatory markers and the MetS in childhood obesity [27]. In a multiethnic cohort of obese children, an association between adiponectin and C-reactive protein (CRP) concentrations was noted, which was independent of IR and adiposity [28]. Low adiponectin concentrations were also associated with components of the MetS, such as low HDL and high triglyceride-to-HDL ratio. Hypoadiponectinemia appears to be related to a low-grade systemic chronic inflammatory state and it is thought to represent a link between inflammation and obesity.
In addition to the above, hypoadiponectinemia is an independent risk factor for MetS in children [29]. Ethnic differences have been reported in adiponectin concentrations in children: obese African–American and Hispanic children have lower adiponectin concentrations, owing to their decreased insulin sensitivity, which may account for the increased risk of diabetes type 2 in these ethnic groups [27, 29]. Age and puberty also have an impact on adiponectin concentrations: adiponectin is lower in older children [27], and its decline is associated with alterations in the concentrations of sex steroids and growth factors at puberty [30].
Retinol Binding Protein-4 (RBP-4) is a 21 kDa protein that circulates bound to transthyretin (TTR) in a form of a 80 kDa protein complex. The soluble form of RBP-4 is an adipokine involved in systemic IR: high concentrations of RBP-4 were noted in obese, insulin resistant, type 2 diabetic adults, while reduced concentrations were associated with improved insulin action. In children, a study in 80 girls showed that, in contrast to adult studies, RBP-4 concentrations correlated negatively with BMI-SDS [31]. This discrepancy in the RBP-4 concentrations between adult and pediatric obese subjects might be due to the development of prevailing protective mechanisms of the organism during childhood.
Finally, chemerin is a recently identified chemo-attractant adipokine, which acts via the G-coupled chemokine-like receptor 1 (CMKLR1 or ChemR23). Chemerin and its receptor are mainly located in adipose tissue [20]. Chemerin concentrations are significantly higher in obese than in lean children, and correlate with leptin concentrations, skinfold thickness, high-sensitivity CRP, as well as with the markers of endothelial activation, intercellular adhesion molecule-1 (ICAM-1) and E-selectin [32, 33•].
Biomarkers of Atherosclerosis and CVD
CVD is the leading cause of death in modern societies, whereas well recognized risk factors, such as obesity and related comorbidities, are associated to accelerated atherosclerosis [34]. The atherosclerotic process is initiated during childhood [35], while in children with obesity and related risk factors this process develops more rapidly [36, 37]. In pathology studies of children who died from other causes, fatty streaks and fibrous plaques were associated with previously measured BMI [36, 37]. Recently, newer noninvasive techniques have revealed the precursors of CVD in children and youth, whereas one of the first abnormalities seen was endothelial dysfunction, which is a biomarker of arterial damage [38, 39]. The intima-media thickness (IMT) of the common carotid artery is a reliable method for the prediction of CVD [40]. Increased IMT has been reported in obese children with features of the MetS, such as hypertension, dyslipidemia, or impaired glucose tolerance [40–42]. Given that early vascular changes occur in obese children with the MetS, it seems that obesity-related cardiovascular risk factors, such as hypertension, dyslipidemia, IR or disturbed glucose metabolism are associated with increased IMT, rather than obesity per se. Indeed, although obesity itself is an independent predictor of CVD in adults, there is no sufficient evidence to link directly obesity to CVD in children [43].
In addition to the traditional risk biomarkers for CVD in children (including glucose, insulin, and lipid profile concentrations), other risk factors, such as left ventricular hypertrophy [44], increased concentrations of homocysteine, CRP and lipoprotein [45, 46] have been associated with CVD. Circulating high sensitivity (hs) CRP concentrations are increased in central adiposity, consistent with the proinflammatory state associated with visceral adiposity [47]. In addition, the increased hsCRP concentrations may play an important role in the progression of arterial thrombosis and endothelial injury repair [48].
Newer biomarkers related to the atherosclerotic process or subsequent deteriorations in cardiac function have been investigated in young individuals with obesity and MetS. These include troponin and B-type natriuretic peptide, which were traditionally used in the evaluation of cardiac processes, such as acute coronary syndrome, myocardial infarction, and heart failure [49, 50•]. We have shown that circulating high-sensitivity troponin T (hs-TnT), a sensitive biomarker of cardiac dysfunction, is increased in children and adolescents with MetS compared with obese children without the MetS and children with normal BMI [51]. This finding implies that it is the unfavorable metabolic profile, rather than childhood obesity per se, that is associated with increased hs-TnT concentrations. In adults, minimally increased troponin concentrations have been determined in individuals without typical ischemic chest pain, whereas individuals with TnT elevation had underlying CVD or a high-risk phenotype for CVD [52].
The Hs-TnT and N-terminal pro-brain natriuretic peptide (NT-proBNP) represent direct indicators of functional and structural damage in the cardiovascular system and they are predictors of myocardial damage [53]. We have previously reported, in a different cohort of obese children, that NT-proBNP concentrations were lower in the obese males than in the normal BMI group and higher in the obese hypertensive than the obese normotensive group, pointing to a potential role of NT-proBNP in obesity-related hypertension [54]. Furthermore, we demonstrated a positive association between NT-proBNP and adiponectin concentrations only in female adolescents, and this association was stronger in girls with a BMI-z-score >2.5, suggesting that although obesity does not influence directly NT-proBNP concentrations in female adolescents, the severity of obesity affects the association between NT-proBNP and adiponectin [55].
Fetal Programming of Obesity and Birth Weight
During the last few years, both experimental and clinical research studies have highlighted the crucial role of the intrauterine environment in the subsequent growth and development of the individual. Fetal under-nutrition, caused mainly by poor or unbalanced maternal nutrition, suboptimal body composition, excessive physical or psychosocial stress or placental insufficiency, may increase the biological vulnerability of the fetus to later development of obesity, MetS and CVD [56]. Based on epidemiologic research studies, which revealed an association between low birth weight and components of the MetS and CVD in adulthood, Barker and colleagues, developed the “fetal origins of adult disease hypothesis” [57]. This was explained by the “thrifty phenotype hypothesis” [58] according to which the fetus in a poor intrauterine environment maximizes the conservation of resources by altering its metabolism. However, this adaptive alteration, which is useful in a deprived environment becomes maladaptive in plentiful conditions. A much broader approach, the “predictive adaptive response”, was later suggested by Gluckman and Hanson, extending the fetal adaptive response to environmental stimuli beyond energy use and conservation [59, 60]: predictive adjustments of the fetus include physiological and behavioral changes in body and organ size, neuroendocrine activity and aspects of behavior in order to survive.
There are further effects of poor maternal diet on later health, and emerging evidence highlights the important role of maternal obesity, excessive weight gain during pregnancy and gestational diabetes as variables that influence disease risk in the next generation [61–63]. Epidemiologic studies have demonstrated that the relation between birth weight and later disease is U-shaped: similarly to low birth weight, large birth weight is also associated with greater disease risk [64]. Maternal BMI has been positively correlated with total and abdominal adiposity, and with hepatic lipid content in infancy across the entire range of maternal BMI [65•]. Maternal gestational diabetes, diabetes and glycosuria are all associated with higher offspring fasting glucose and insulin concentrations in adolescence [66], whereas maternal gestational diabetes was associated with increased cardiovascular risk markers in childhood [67•]. Pregnancy weight gain that exceeds the medical guidelines is also associated with higher incidence of obesity in the neonatal period and adolescence [68, 69]. In maternal obesity, an up-regulation of specific placental amino acid transporter isoforms might contribute to fetal overgrowth. This effect may be mediated by activation of insulin/IGF-I and mTOR signaling pathways, which are positive regulators of placental amino acid transporters [70].
The concept of fetal programming of obesity also supports the notion that exposure to excessive or deficient nutrition before birth alters the development of the adipocytes, resulting in a permanent increase in the capacity to form new foam cells in the adipose tissue, or to store lipids in existing adipocytes. Adipogenesis, the process of adipose development, occurs mainly during late fetal and early postnatal life. This process is highly sensitive to environmental conditions during this time frame. Based on experimental studies, changes in gene expression within visceral adipocytes before birth influence the subsequent properties of subcutaneous adipose tissue as it continues to develop from stem cells throughout life. Factors secreted by these “programmed” adipocytes promote pre-adipocyte differentiation in other depots, resulting in further increase in adipose tissue [71].
Epigenetic processes in early development have been implicated in the origins of obesity. In a recent study, researchers examined the methylation status of CpGs in the promoters of candidate genes in DNA extracted from umbilical cord tissue obtained at birth in children who were later assessed for adiposity at the age of 9 years. Maternal carbohydrate consumption during the first trimester of pregnancy was inversely correlated with methylation levels at a single CpG in the RXRA gene in umbilical cord tissue and, in turn, was associated with the child’s adiposity at ages 6 and 9 years of age [72•].
Stress and Pediatric Obesity
The experience of chronic and/or severe stress during childhood and adolescence may have permanent effects on growth, metabolism, reproduction, and immune and cardiovascular functions [73]. Both neuroendocrine and behavioral mechanisms contribute to the development of obesity and metabolic alterations in stressed individuals [74]. In normal conditions, activation of the stress system, consisting of the hypothalamic–pituitary–adrenal axis and the locus caeruleus/norepinephrine system in the brainstem, results in adaptive biologic changes that help maintain homeostasis [75]. In contrast, the experience of acute severe or chronic stress, real or perceived, that exceeds a certain threashold, may lead to chronically excessive and prolonged hyper- or hypo-activation of the stress system with a variety of somatic and psychological consequences [76•].
Thus, chronic dysregulation of stress mediators, as evidenced, in most cases, by elevated peripheral cortisol and/or catecholamine concentrations, may result in insulin hypersecretion and growth hormone and sex steroid hyposecretion, leading to the accumulation of fat in visceral adipose tissue. Visceral adiposity is further associated with carbohydrate intolerance, dyslipidemia, arterial hypertension (components of the MetS), loss of muscle (sarcopenia) and osteoporosis [73–75, 76•]. Furthermore, recent evidence on the effects of the circadian CLOCK system on the sensitivity of peripheral tissues to cortisol suggests that even mild elevations of cortisol concentrations, for instance the slight chronic elevation of evening cortisol associated with stress and depression, may contribute to the development of central obesity and the MetS [77•, 78].
Associations between chronic stress and pediatric obesity have been defined in studies examining children and adolescents with emotional and behavioral problems and disorders in relation to weight gain [74]. Anxiety and depression represent the most typical behavioral phenotypes of chronic stress in humans. Several studies have shown that overweight and obese children have a higher prevalence of symptoms of anxiety and depression [79, 80], and that children with anxiety disorders or depression, especially females, may have a higher BMI than comparison groups [81, 82]. In addition, stress-related disorders, such as anxiety and mood disorders, are associated with a chronically abnormal secretion of cortisol and catecholamines, which may lead to the development of obesity and obesity-related manifestations [83, 84].
We have demonstrated an increased prevalence of anxiety and depressive symptoms in obese children and that trait anxiety is associated positively with salivary cortisol concentrations determined at noon [80]. Furthermore, overall salivary cortisol concentrations were increased in children with anxiety or depressive symptomatology compared to obese children without any affective morbidity and to those with anxiety and depression co-morbidity, suggesting that symptoms of emotional stress may further contribute to obesity and the adverse metabolic profile of stress in obese children. Several molecules also contribute to the link between stress and obesity: glucocorticoids induce insulin and leptin secretion and contribute to the “leptin resistance” that characterizes obesity. Cortisol elevations are also accompanied by increased NPY secretion and disruption of the balance of food intake regulation. Insulin and leptin have also been shown to play important roles in the regulation of central pathways related to food reward and uptake [85].
In addition to cortisol and catecholamine dysregulation, a number of behavioral parameters also contribute to the development of obesity in chronically stressed individuals, such as poor adherence to self-care activities, sedentary habits with decreased motivation for activity, lack of sleep and disturbed eating behaviors, such as emotional eating and consumption of comfort foods [86]. The clinical implications of research studies linking stress to pediatric obesity extend beyond the understanding of pathophysiologic pathways. Screening for symptoms of stress in obese children and incorporating stress management techniques in comprehensive weight programs is of utmost importance in obesity prevention and intervention strategies [87, 88].
Childhood Obesity and Pubertal Development
During the last decades, a secular trend towards earlier puberty has taken place in relation to modern life style. The parallel worldwide increase in the prevalence of childhood obesity led researchers to investigate the associations between BMI, body composition and the timing and tempo of puberty. Evidence has shown an association between early weight gain and advanced pubertal development in both sexes, more clearly in girls. Furthermore, earlier menarche in girls is associated with increased risk of adult obesity, diabetes type 2 and breast cancer. Pathways linking early adiposity to timing of puberty include genetic and epigenetic factors in relation to the pathophysiologic role of the adipokines [89•, 90]. Higher birth weight and weight gain until the age of 5 years, have also been associated to earlier sexual maturation in girls [91].
In a study investigating hormonal concentrations in relation to sexual maturation, earlier menarche was associated with greater body weight, height, and BMI at age 8 years [92]. Before adjustment for BMI, earlier menarche was associated with higher insulin-like growth factor (IGF)-1, androstenedione, dehydroepiandrosterone-sulphate (DHEAS), leptin, and fasting insulin concentrations, and with lower IGF binding protein-1 and sex-hormone binding globulin concentrations at age 8 years, whereas after adjustment for BMI at age 8 years, only IGF-I, androstenedione, and DHEAS were associated with earlier menarche.
Maternal pre-pregnancy weight was also associated with the timing of menarche in daughters. This association was not mediated by the daughter’s birth weight, prepubertal BMI or maternal weight gain during pregnancy [93].
Genetic Factors
In the last few decades, the role of genetic factors in the regulation of energy and body fat has been established, while monogenic disorders of obesity resulting from disruption of the leptin-melanocortin pathways have been described. In these disorders, early onset of severe obesity is the predominant feature [94]. Congenital leptin deficiency, leptin receptor deficiency, pro-opiomelanocortin deficiency, melanocortin-4 receptor deficiency and prohormone convertase-1 deficiency represent rare monogenic causes of early-onset childhood obesity [95, 96].
Pleiotropic syndromes with monogenic childhood obesity manifest typically in association with intellectual disabilities, organ-specific abnormalities and dysmorphic features. Such disorders include the syndromes of Bardet–Biedl, Albright’s hereditary osteodystrophy, Cohen and Almstrong syndromes, etc. Finally, the Prader–Willi syndrome is the most common syndromal cause of human obesity and is due to chromosomal rearrangements in the paternal chromosome 15.
The monogenic disorders of severe childhood obesity are rare, however, the identification of candidate children suffering from such disorders is a major challenge for pediatricians.
Conclusions
Beyond genetic vulnerability, cardiometabolic risk can be defined epigenetically very early in life. However, this risk can be modified by nutrition, life style, exposure to chronic stress and environmental exposures. Interventions to promote healthy behaviors should be targeted to mothers during the gestational period, to mother–child pairs in the first 2–3 years of life, as well as to adolescents-future mothers. Only such approaches will stop the vicious cycle of maternal–child–adolescent obesity.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance
Weiss R, Dziura J, Burgert TS, et al. Obesity and the metabolic syndrome in children and adolescents. N Engl J Med. 2004;350:2362–74.
Hannon TS, Rao G, Arslanian SA. Childhood obesity and type 2 diabetes mellitus. Pediatrics. 2005;116:473–80.
Kassi E, Pervanidou P, Kaltsas G, Chrousos G. Metabolic syndrome: definitions and controversies. BMC Med. 2011;9:48.
Reaven GM. Banting lecture, Role of insulin resistance in human disease. Diabetes. 1988;1988(37):1595–607.
Cook S, Weitzman M, Auinger P, et al. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med. 2003;157:821–7.
Duncan GE, Li SM, Zhou XH. Prevalence and trends of a metabolic syndrome phenotype among U.S. adolescents, 1999–2000. Diabetes Care. 2004;27:2438–43.
Pervanidou P, Kanaka-Gantenbein C, Chrousos GP. Assessment of metabolic profile in a clinical setting. Curr Opin Clin Nutr Metab Care. 2006;9:589–95.
Brambilla P, Lissau I, Flodmark CE, et al. Metabolic risk-factor clustering estimation in children: to draw a line across pediatric metabolic syndrome. Int J Obes (Lond). 2007;31:591–600.
Viner RM, Segal TY, Lichtarowicz-Krynska E, Hindmarsh P. Prevalence of the insulin resistance syndrome in obesity. Arch Dis Child. 2005;90:10–4.
Zimmet P, Alberti KG, Kaufman F, et al. IDF Consensus Group: the metabolic syndrome in children and adolescents—an IDF consensus report. Pediatr Diabetes. 2007;8:299–306.
Goodman E, Daniels SR, Meigs JB, Dolan LM. Instability in the diagnosis of metabolic syndrome in adolescents. Circulation. 2007;115:2316–22.
• Goodman E, Li C, Tu YK, et al. Stability of the factor structure of the metabolic syndrome across pubertal development: confirmatory factor analyses of three alternative models. J Pediatr. 2009;155:S5–8. This study examined the fit and stability of 3 alternative models of the metabolic syndrome’s factor structure across 3 developmental stages (pre-puberty, puberty and post-puberty) and found that the diagnosis of metabolic syndrome may vary by maturation stage in children. Thus, the concept of metabolic syndrome may not provide adequate fit in children and adolescents whereas a components-based approach to cardiovascular risk may be more appropriate for youth.
Trayhurn P. Endocrine and signalling role of adipose tissue: new perspectives on fat. Acta Physiol Scand. 2005;184(4):285–93.
Trayhurn P, Wood IS. Adipokines: inflammation and the pleiotropic role of white adipose tissue. Br J Nutr. 2004;92:347–55.
Weisberg SP, McCann D, Desai M, et al. Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003;112(12):1796–808.
Pervanidou P, Eleftheriades M, Papassotiriou I. Adipokines and anthropometry: childhood and adolescent obesity. In: Preedy R, editor. Handbook of anthropometry: physical measures of human form in health and disease. New York: Springer; 2012. p. 1221–36.
Zhang Y, Proenca R, Maffei M, et al. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372(6505):425–32.
Lago F, Dieguez C, Gómez-Reino J, Gualillo O. The emerging role of adipokines as mediators of inflammation and immune responses. Cytokine Growth Factor Rev. 2007;18(3–4):313–25.
Antunes H, Santos C, Carvalho S. Serum leptin levels in overweight children and adolescents. Br J Nutr. 2008;28:1–5.
Holm JC, Gamborg M, Kaas-Ibsen K, et al. Time course and determinants of leptin decline during weight loss in obese boys and girls. Int J Pediatr Obes. 2007;2:2–10.
• Zhang S, Liu X, Brickman WJ, et al. Association of plasma leptin concentrations with adiposity measurements in rural Chinese adolescents. J Clin Endocrinol Metab. 2009;94(9):3497–504. The study examined plasma leptin concentrations in relation to different adiposity measurements and showed similar associations of leptin with BMI, waist circumference (WC), fat mass index (FMI), and percentage body fat (%BF) in females. In males, %BF and FMI were more strongly associated with leptin levels than BMI and WC. The study shows that further investigation of leptin, as a biomarker of adiposity, is needed in adolescents.
Yoshinaga M, Sameshima K, Tanaka Y, et al. Adipokines and the prediction of the accumulation of cardio-vascular risk factors or the presence of metabolic syndrome in elementary school children. Circ J. 2008;72:1874–8.
Berg AH, Scherer PE. Adipose tissue, inflammation, and cardiovascular disease. Circ Res. 2005;96(9):939–49.
Araki S, Dobashi K, Kubo K, et al. High molecular weight, rather than total, adiponectin levels better reflect metabolic abnormalities associated with childhood obesity. J Clin Endocrinol Metab. 2006;91(12):5113–6.
Cambuli VM, Musiu MC, Incani M, et al. Assessment of adiponectin and leptin as biomarkers of positive metabolic outcomes after lifestyle intervention in overweight and obese children. J Clin Endocrinol Metab. 2008;93:3051–7.
Jeffery AN, Murphy MJ, Metcalf BS, et al. Adiponectin in childhood. Int J Pediatr Obes. 2008;3:130–40.
Winer JC, Zern TL, Taksali SE, et al. Adiponectin in childhood and adolescent obesity and its association with inflammatory markers and components of the metabolic syndrome. J Clin Endocrinol Metab. 2006;91(11):4415–23.
Gilardini L, McTernan PG, Girola A, et al. Adiponectin is a candidate marker of metabolic syndrome in obese children and adolescents. Atherosclerosis. 2006;189(2):401–7.
Bush NC, Darnell BE, Oster RA, et al. Adiponectin is lower among African Americans and is independently related to insulin sensitivity in children and adolescents. Diabetes. 2005;54:2772–8.
Butte NF, Comuzzie AG, Cai G, et al. Genetic and environmental factors influencing fasting serum adiponectin in Hispanic children. J Clin Endocrinol Metab. 2005;90:4170–6.
Kanaka-Gantenbein C, Margeli A, Pervanidou P, et al. Retinol- binding protein 4 and lipocalin-2 in childhood and adolescent obesity: when children are not just “small adults”. Clin Chem. 2008;54(7):1176–82.
Bozaoglu K, Bolton K, McMillan J, et al. Chemerin is a novel adipokine associated with obesity and metabolic syndrome. Endocrinology. 2007;148:4687–94.
• Landgraf K, Friebe D, Ullrich T et al. Chemerin as a mediator between obesity and vascular inflammation in children. J Clin Endocrinol Metab. 2012;97(4):E556–564. The chemoattractant protein chemerin is associated with obesity and inflammatory and endothelial activation markers and may be a molecular link between increasing adiposity and an early atherogenic risk profile in obese children.
Bridger T. Childhood obesity and cardiovascular disease. Paediatr Child Health. 2009;14:177–82.
Berenson GS, Srinivasan SR SR, Bao W, et al. Association between multiple cardiovascular risk factors and atherosclerosis in children and young adults. The Bogalusa Heart Study. N Engl J Med. 1998;338:1650.
Morrison JA, Glueck CJ, Wang P. Childhood risk factors predict cardiovascular disease, impaired fasting glucose plus type 2 diabetes mellitus, and high blood pressure 26 years later at a mean age of 38 years: the Princeton-lipid research clinics follow-up study. Metabolism. 2012;61(4):531–41.
McMahan CA, Gidding SS, Malcom GT, et al. Pathobiological Determinants of Atherosclerosis in Youth Research Group. Pathobiological determinants of atherosclerosis in youth risk scores are associated with early and advanced atherosclerosis. Pediatrics. 2006;118:1447–55.
Montero D, Walther G, Perez-Martin A, et al. Endothelial dysfunction, inflammation, and oxidative stress in obese children and adolescents: markers and effect of lifestyle intervention. Obes Rev. 2012;13(5):441–55.
Woo KS, Chook P, Yu CW, et al. Overweight in children is associated with arterial endothelial dysfunction and intima-media thickening. Int J Obes Relat Metab Disord. 2004;28:852–7.
Reinehr T, Wunsch R, de Sousa G, Toschke AM. Relationship between metabolic syndrome definitions for children and adolescents and intima-media thickness. Atherosclerosis. 2008;199:193–200.
Fang J, Zhang JP, Luo CX, et al. Carotid Intima-media thickness in childhood and adolescent obesity relations to abdominal obesity, high triglyceride level and insulin resistance. Int J Med Sci. 2010;7:278–83.
Huang K, Zou CC, Yang XZ, et al. Carotid intima-media thickness and serum endothelial marker levels in obese children with metabolic syndrome. Arch Pediatr Adolesc Med. 2010;164:846–51.
Zalesin KC, Franklin BA, Miller WM, et al. Impact of obesity on cardiovascular disease. Endocrinol Metab Clin N Am. 2008;37:663–84.
Hanevold C, Waller J, Daniels S, et al. The effects of obesity, gender, and ethnic group on left ventricular hypertrophy and geometry in hypertensive children: a collaborative study of the International Pediatric Hypertension Association. Pediatrics. 2004;113:328.
Misra A. Risk factors for atherosclerosis in young individuals. J Cardiovasc Risk. 2000;7:215.
Daniels SR. Cardiovascular disease risk factors and atherosclerosis in children and adolescents. Curr Atheroscler Rep. 2001;3:479.
DeBoer MD. Obesity, systemic inflammation, and increased risk for cardiovascular disease and diabetes among adolescents: a need for screening tools to target interventions. Nutrition. 2013;29(2):379–86.
Danenberg HD, Szalai AJ, Swaminathan RV, et al. Increased thrombosis after arterial injury in human C-reactive protein—transgenic mice. Circulation. 2003;108:512–5.
Siervo M, Ruggiero D, Sorice R, et al. Angiogenesis and biomarkers of cardiovascular risk in adults with metabolic syndrome. J Intern Med. 2010;268:338–47.
• Siervo M, Ruggiero D, Sorice R et al. Body mass index is directly associated with biomarkers of angiogenesis and inflammation in children and adolescents. Nutrition. 2012;28:262–66. The study examines associations of BMI with plasma hs-CRP, VEGF, hs-TnT and NT proBNP, biomarkers that provide the physiopathologic progression of cardiovascular risk in obese children and adolescents.
Pervanidou P, Akalestos A, Bastaki D et al. Increased circulating high sensitivity-troponin (hs-TnT) concentrations in children and adolescents with obesity and the metabolic syndrome: a marker for early cardiac damage? Metabolism. 2012. doi:10.1016/j.metabol.2012.09.012.
Wallace TW, Abdullah SM, Drazner MH, et al. Prevalence and determinants of troponin T elevation in the general population. Circulation. 2006;113:1958–65.
Apple FS, Collinson PO. IFCC task force on clinical applications of cardiac biomarkers. Analytical characteristics of high-sensitivity cardiac troponin assays. Clin Chem. 2012;58:54–61.
Pervanidou P, Akalestos A, Sakka S, et al. Gender dimorphic associations between N-terminal pro-brain natriuretic peptide, body mass index and blood pressure in children and adolescents. Horm Res Paediatr. 2010;73(5):341–8.
Pervanidou P, Margeli A, Akalestos A, et al. Associations between circulating N-terminal pro-brain natriuretic peptide (NT-proBNP) and adiponectin concentrations depend on obesity level in female adolescents: gender dimorphic findings. Horm Metab Res. 2009;41(11):829–33.
Eleftheriades M, Pervanidou P, Chrousos GP. Fetal stress. In: George F, editors. Encyclopedia of stress, 2nd edn. Oxford: Oxford Academic Press; 2007. p. 46–51.
Barker DJ. The fetal origins of diseases of old age. Eur J Clin Nutr. 1992;46(3):S3–59.
Hales CN, Barker DJ. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia. 1992;35(7):595–601.
Gluckman PD, Hanson MA. Living with the past: evolution, development, and patterns of disease. Science. 2004;305(5691):1733–6.
Gluckman PD, Hanson MA. The developmental origins of the metabolic syndrome. Trends Endocrinol Metab. 2004;15(4):183–7.
Weng SF, Redsell SA, Swift JA, et al. Systematic review and meta-analyses of risk factors for childhood overweight identifiable during infancy. Arch Dis Child. 2012;97(12):1019–26.
Mamun AA, O’Callaghan MJ, Williams GM, Najman JM. Change in maternal body mass index is associated with offspring body mass index: a 21-year prospective study. Eur J Nutr. 2012. doi:10.1007/s00394-012-0465-7.
Hanson MA, Gluckman PD, Ma RC, et al. Early life opportunities for prevention of diabetes in low and middle income countries. BMC Public Health. 2012;12(1):1025.
Barouki R, Gluckman PD, Grandjean P, et al. Developmental origins of non-communicable disease: implications for research and public health. Environ Health. 2012;27(11):42.
• Modi N, Murgasova D, Ruager-Martin R et al. The influence of maternal body mass index on infant adiposity and hepatic lipid content. Pediatr Res. 2011;70(3):287–91. This study reveals maternal BMI, across the entire range, to positively correlate with total and abdominal adiposity, as assessed by whole body MR imaging, of the offspring and with hepatic lipid content, as assessed by HMR spectroscopy, in infancy. The study supports alterations related to pediatric adiposity starting in utero.
Patel S, Fraser A, Davey Smith G, et al. Associations of gestational diabetes, existing diabetes, and glycosuria with offspring obesity and cardiometabolic outcomes. Diabetes Care. 2012;35(1):63–71.
• Krishnaveni GV, Veena SR, Hill JC et al. Intrauterine exposure to maternal diabetes is associated with higher adiposity and insulin resistance and clustering of cardiovascular risk markers in Indian children. Diabetes Care. 2010;33(2):402–04. The study supports maternal gestational diabetes to increase diabetes and cardiovascular risk markers in the offspring over genetic factors.
Laitinen J, Jääskeläinen A, Hartikainen AL, et al. Maternal weight gain during the first half of pregnancy and offspring obesity at 16 years: a prospective cohort study. BJOG. 2012;119(6):716–23.
Ferraro ZM, Barrowman N, Prud’homme D, et al. Excessive gestational weight gain predicts large for gestational age neonates independent of maternal body mass index. J Matern Fetal Neonatal Med. 2012;25(5):538–42.
Jansson N, Rosario FJ, Gaccioli F, et al. Activation of placental mTOR signaling and amino acid transporters in obese women giving birth to large babies. J Clin Endocrinol Metab. 2012;98(1):105–13.
Muhlhausler B, Smith SR. Early-life origins of metabolic dysfunction: role of the adipocyte. Trends Endocrinol Metab. 2009;20(2):51–7.
• Godfrey KM, Sheppard A, Gluckman PD et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes. 2011;60(5):1528–34. The study shows maternal diet during pregnancy associated with methylation levels at a single CpG in the RXRA gene in umbilical cord tissue and, in turn, associated with child’s adiposity at ages 6 and 9. The study supports epigenetic processes in early development implicated in pediatric obesity.
Charmandari E, Kino T, Souvatzoglou E, Chrousos GP. Pediatric stress: hormonal mediators and human development. Horm Res. 2003;59(4):161–79.
Pervanidou P, Chrousos GP. Stress and obesity/metabolic syndrome in childhood and adolescence. Int J Pediatr Obes. 2011;6(1):21–8.
Charmandari E, Tsigos C, Chrousos G. Endocrinology of the stress response. Annu Rev Physiol. 2005;67:259–84.
• Chrousos GP. Stress and disorders of the stress system. Nat Rev Endocrinol. 2009;5(7):374–81. Review article updating the concepts of stress and stress system disorders.
• Nader N, Chrousos GP, Kino T: Interactions of the circadian CLOCK system and the HPA axis. Trends Endocrinol Metab. 2010;21(5):277–86. Article summarizing evidence on the circadian CLOCK system and its effects on the sensitivity of peripheral tissues to cortisol. The findings suggest that even mild elevations of cortisol concentrations may contribute to the development of central obesity and the metabolic syndrome.
Charmandari E, Chrousos GP, Lambrou GI, et al. Peripheral CLOCK regulates target-tissue glucocorticoid receptor transcriptional activity in a circadian fashion in man. PLoS ONE. 2011;6(9):e25612.
Onyike CU, Crum RM, Lee HB, et al. Is obesity associated with major depression? Results from the Third National Health and Nutrition Examination Survey. Am J Epidemiol. 2003;158(12):1139–47.
Pervanidou P, Bastaki D, Chouliaras G, et al. Cortisol profiles, anxiety and depressive symptomatology, and body mass index in a clinical population of obese children. Stress. 2013;16(1):34–43.
Anderson SE, Cohen P, Naumova EN, et al. Association of depression and anxiety disorders with weight change in a prospective community-based study of children followed up into adulthood. Arch Pediatr Adolesc Med. 2006;160(3):285–91.
Noll JG, Zeller MH, Trickett PK, et al. Obesity risk for female victims of childhood sexual abuse: a prospective study. Pediatrics. 2007;120(1):e61–7.
Pervanidou P. Biology of post-traumatic stress disorder in childhood and adolescence. J Neuroendocrinol. 2008;20(5):632–8.
Pervanidou P, Kolaitis G, Charitaki S, et al. The natural history of neuroendocrine changes in pediatric posttraumatic stress disorder (PTSD) after motor vehicle accidents: progressive divergence of noradrenaline and cortisol concentrations over time. Biol Psychiatry. 2007;62(10):1095–102.
Figlewicz DP. Adiposity signals and food reward: expanding the CNS roles of insulin and leptin. Am J Physiol Regul Integr Comp Physiol. 2003;284:R882–92.
Pervanidou P, Chrousos GP. Metabolic consequences of stress during childhood and adolescence. Metabolism. 2012;61(5):611–9.
Bathrellou E, Yannakoulia M, Papanikolaou K, et al. Development of a Multi-Disciplinary Intervention for the Treatment of Childhood Obesity Based on Cognitive Behavioral Therapy. Child Fam Behav Therapy. 2010;32:34–50.
Bathrellou E, Yannakoulia M, Papanikolaou K, et al. Parental involvement does not augment the effectiveness of an intense behavioral program for the treatment of childhood obesity. Hormones (Athens). 2010;9(2):171–5.
• Wagner IV, Sabin MA, Pfäffle RW et al. Effects of obesity on human sexual development. Nat Rev Endocrinol 2012;8(4):246–54. Review article discussing the associations between BMI and body composition in children with timing and tempo of puberty. Pathophysiologic pathways such as the genetic and epigenetic factors and the adipokines are discussed.
Ahmed ML, Ong KK, Dunger DB. Childhood obesity and the timing of puberty. Trends Endocrinol Metab. 2009;20(5):237–432.
Wang Y, Dinse GE, Rogan WJ. Birth weight, early weight gain and pubertal maturation: a longitudinal study. Pediatr Obes. 2012;7(2):101–9.
Thankamony A, Ong KK, Ahmed ML. Higher levels of IGF-I and adrenal androgens at age 8 years are associated with earlier age at menarche in girls. J Clin Endocrinol Metab. 2012;97(5):E786–90.
Deardorff J, Berry-Millett R, Rehkopf D. Maternal pre-pregnancy BMI, gestational weight gain, and age at menarche in daughters. Matern Child Health J. 2012. doi:10.1007/s10995-012-1139-z.
Farooqi IS. Genetic, molecular and physiological insights into human obesity. Eur J Clin Invest. 2011;41(4):451–5.
Farooqi IS, O’Rahilly S. Leptin: a pivotal regulator of human energy homeostasis. Am J Clin Nutr. 2009;89(3):980S–4S.
Farooqi IS, O’Rahilly S. Mutations in ligands and receptors of the leptin-melanocortin pathway that lead to obesity. Nat Clin Pract Endocrinol Metab. 2008;4(10):569–77.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Pervanidou, P., Charmandari, E. & Chrousos, G.P. Endocrine Aspects of Childhood Obesity. Curr Pediatr Rep 1, 109–117 (2013). https://doi.org/10.1007/s40124-013-0011-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40124-013-0011-y