
Abstract
Introduction
To assess the safety and efficacy of repeated intravitreal injections of RC28-E, a novel bispecific antibody that simultaneously binds vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF) in patients with neovascular age-related macular degeneration (AMD).
This was a prospective, multicenter, open-label clinical trial; 37 patients with choroidal neovascularization secondary to AMD and best-corrected visual acuity (BCVA) letter scores between 73 and 34 were enrolled.
Methods
Treatment regimens consisted of a 3-month loading phase and a pro re nata (PRN) maintenance phase. This study included three treatment groups: the 0.5, 1.0, and 2.0 mg RC28-E groups, with escalating doses ranging from 0.5 to 2.0 mg. Patients were evaluated monthly for 48 weeks.
Safety was assessed based on ocular and systemic adverse events (AEs), pharmacokinetic characteristics, and the presence of anti-RC28-E antibodies. Efficacy was assessed using the mean change in BCVA and central subfield thickness (CST) from baseline to week 48.
Results
Most AEs were mild or moderate. The most common AE was a minor injection-related subconjunctival hemorrhage (16.2%). The AEs did not increase with dose or repeated injections. At week 48, mean improvements in BCVA from baseline in the 0.5, 1.0, and 2.0 mg groups were 6.1 ± 8.3, 9.9 ± 10.7, and 7.6 ± 9.38 letters, respectively; mean reductions in CST in the three groups were 112.1 ± 160.5, 175.1 ± 212.4, and 128.7 ± 145.8 μm, respectively. The serum RC28-E concentrations in 95% of the patients were below the quantification limit of the assay. No significant change from baseline was observed in the mean plasma concentrations of VEGF or FGF over the 48 weeks of treatment. Pre-treatment antibodies to RC28-E were detected in 1 of the 37 patients. Antibodies to RC28-E were detected in two patients after dosing with RC28-E for 48 weeks.
Conclusion
RC28-E was well tolerated and exhibited an overall favorable safety profile with evidence of improvements in BCVA and anatomical parameters.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Age-related macular degeneration (AMD) causes progressive loss of central vision. Vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF, also known as FGF-2) are important regulators of macular angiogenesis. RC28-E simultaneously blocks both VEGF-A and FGF-2. |
RC28-E may promote vascular stability, reduce neovascularization, and improve durability. The clinical activity of RC28-E should be further investigated in a phase 1b trial. |
What was learned from the study? |
In this prospective, multicenter, non-randomized, open-label, sequential multiple ascending dose study. Most adverse events (AEs) were mild or moderate. Best-corrected visual acuity (BCVA) improvements and central subfield thickness (CST) reductions were observed from baseline to week 48. |
These results proved RC28-E was well tolerated and exhibited an overall favorable safety profile with evidence of improvements in BCVA and anatomical parameters. |
Introduction
Age-related macular degeneration (AMD) affects the macular region of the retina and causes progressive loss of central vision [1]. Most patients with AMD have a dry form of the disease, but severe vision loss occurs most frequently in patients who develop choroidal neovascularization (CNV) [2]. Anti-vascular endothelial growth factor (VEGF) therapy is currently the most effective first-line treatment for neovascular AMD [3]. A key challenge with currently available anti-VEGF treatments is the need for frequent monitoring and injections to maintain initial gains in vision [4]. Novel targets beyond the VEGF pathway might offer longer durability, reduce treatment burden, and improve outcomes compared to currently available therapies.
Basic fibroblast growth factor (bFGF, also known as FGF-2) has also been shown to be an important regulator of angiogenesis. It promotes degradation of the vessel basement membrane and stimulates the survival, proliferation, migration, and differentiation of endothelial cells [5]. Moreover, FGF-2 signaling has been shown to interact with VEGF signaling, and upregulation of FGF-2 may partially account for the development of VEGF resistance [6]. Therefore, selective neutralization of both VEGF and FGF-2 may further promote vascular stability, reduce neovascularization, and improve durability compared with therapies targeting the VEGF pathway alone.
RC28-E is a novel dual-decoy receptor immunoglobulin G (IgG) 1 Fc-fusion protein that simultaneously blocks both VEGF-A and FGF-2. The RC28-E structure is composed of the second binding domain of VEGF receptor 1 (VEGFR1), the third domain of VEGF receptor 2 (VEGFR2), and the second third of FGF receptor 1 (FGFR1) fused to the Fc region of human IgG [7]. Preclinical studies have demonstrated that RC28-E exhibits improved bioactivity in vitro and more significant efficacy than other VEGF antagonists in vivo [6, 8]. Primate studies have shown that RC28-E largely attenuates CNV formation and vascular leakage in retinal injury models and has a favorable safety profile [6, 9]. The clinical activity of RC28-E was initially demonstrated in a 6-week, sequential, single ascending dose phase 1 study (identifier NCT03777254) in patients with neovascular AMD. Intravitreal injection of 0.25–2.0 mg of RC28-E was well tolerated, with no evidence of ocular inflammation. At 6 weeks, treatment with RC28-E resulted in an improvement in the mean best-corrected visual acuity (BCVA) of 5.6 letters for all groups combined, which correlated with anatomical improvement.
Based on these encouraging results from previous studies, a multiple ascending-dose phase 1b study was designed to investigate the safety and efficacy of repeated doses of RC28-E. The results of this study are presented herein.
Methods
Ethical Considerations
This study was conducted in accordance with the tenets of the Declaration of Helsinki, China’s good clinical practice regulations, and applicable institutional regulatory requirements. Institutional review board and health authority approvals were obtained. Written informed consent was obtained from each study participant before protocol-related procedures were performed. This study was registered at www.clinicaltrials.gov (identifier: NCT04270669).
Study Design
This study was a prospective, multicenter (nine sites in China), non-randomized, open-label, sequential multiple ascending dose study to investigate the safety, efficacy, pharmacokinetics, and immunogenicity of RC28-E in patients with neovascular AMD. This study was conducted from April 2020 to December 2021. The primary objectives of this study were to assess the ocular and systemic safety and tolerability of multiple doses of RC28-E and to assess the effect of intravitreal injection of RC28-E on BCVA. A key secondary objective was to assess the effect of RC28-E on central subfield thickness (CST). The major eligibility criteria included age ≥ 50 years, a confirmed diagnosis of neovascular AMD in the study eye (one eye per patient), BCVA letter scores between 73 and 34, evidence of leakage resulting from CNV, evidence of intraretinal fluid or subretinal fluid, and treatment-naïve or the last intravitreal anti-VEGF treatment ≥ 3 months prior to enrollment. The key exclusion criteria were significant subfoveal atrophy or scarring, CNV resulting from causes other than AMD, vitreous hemorrhage in the preceding 2 months, active ocular inflammation or infection, previous photodynamic or laser therapy in the previous 3 months, and previous intravitreal corticosteroid injection or device implantation within the previous 6 months. Subjects were enrolled sequentially into three dose-escalating groups, and the central randomized system for clinical trials (DAS for IWRS, provided by Beijing BioVoice Technology Co., Ltd.) was used to assign subject numbers.
Intervention
This study included three treatment groups: 0.5, 1.0, and 2.0 mg RC28-E groups. The intravitreal injection volume was 0.05 ml for 0.5 mg, 1.0 mg, and 2.0 mg RC28-E doses. A dose-escalation program was only used when patients in the higher dose group received their second injection, since a single ascending-dose study has been performed previously.
After a minimum of three patients in the lower dose group completed their second dose, and a favorable safety and tolerability assessment was achieved, dose escalation was allowed to proceed in a different group of patients. Dose escalation was guided by the occurrence of drug-related ocular dose-limiting toxicity, described as (1) intraocular inflammation, uveitis, or vitreitis, defined as a change of two units on standard grading scales, (2) sustained elevation of intraocular pressure (IOP), or (3) loss of more than 15 letters of visual acuity.
Initially, all patients were administered three doses at monthly intervals. After the 3-month loading phase, patients were evaluated monthly and retreated as needed (pro re nata [PRN]) with their assigned dose. Retreatment decisions were based on the investigator’s evaluation of BCVA, optical coherence tomography (OCT), and other ophthalmic examination results. Patients were re-treated if any of the following criteria were met compared with the last visit: (1) BCVA decreased by five or more letters and subfoveal fluid was present, (2) CST increased to 100 μm or more, (3) persistent retinal fluid based on a review of OCT scans, (4) new macular hemorrhage, or (5) new active CNV.
Endpoints and Assessments
The 48-week assessment measured visual and anatomical changes, including BCVA, IOP measurements, slit-lamp examinations, indirect ophthalmoscopy, fundus photography, OCT, fundus fluorescence angiography, and indocyanine green angiography. Study visits were scheduled every 28 ± 7 days. Additionally, safety was monitored 7 ± 3 days after the first and second injections.
Safety was assessed based on ocular and systemic adverse events (AEs), clinical laboratory tests, vital signs, and ophthalmic examinations, grouped by System Organ Class, identified using their Preferred Term (Medical Dictionary for Regulatory Activities, version 24.1), and graded from 1 to 5 according to National Institutes of Health Common Terminology Criteria for Adverse Events(version 5.0). The primary efficacy endpoint was the mean change in BCVA from baseline at 48 weeks. Certified examiners assessed BCVA using the Early Treatment Diabetic Retinopathy Study (ETDRS) protocol with a starting distance of 4 m.
Secondary endpoints included the change in BCVA at 12 weeks, mean changes in CST (based on OCT results) at 12 and 48 weeks, and mean number of injections over time. Spectral-domain OCT scans were acquired during each visit by trained personnel. The same OCT instrument used at baseline was used throughout the study. CST was defined as the distance between the inner limiting membrane and the inner border of the retinal pigment epithelium (RPE)/choriocapillaris complex, including any subretinal fluid and the thickness of any observable choroidal neovascular membrane or scar tissue in the central 1 mm of the posterior pole scan.
Statistical Analyses
Safety and tolerability analyses were performed on the safety analysis set, which included all the enrolled patients who received the study treatment. Efficacy analyses were performed on the full analysis set, which included all patients who underwent baseline and at least one post-baseline assessments. Formal hypothesis testing and formal sample size calculation were not conducted in this study. All safety, efficacy, pharmacokinetic, and immunogenicity parameters are reported using descriptive statistics.
Results
Patient Population and Baseline Characteristics
Thirty-seven patients (0.5 mg group, n = 12; 1.0 mg group, n = 13; 2.0 mg group, n = 12) were enrolled in the study and received treatment. Of these, 29 (78.4%) completed the 48-week study period (0.5 mg group, 58.3%; 1.0 mg group, 84.6%; 2.0 mg group, 91.7%). The reasons why the eight patients exited before the final visit included subject’s requests (n = 6) and inability to attend visits (n = 2).
Baseline characteristics were comparable across the treatment groups (Table 1). Across all groups, the proportion of CNV lesion types were as follows: predominantly classic (10.8%), minimally classic (18.9%), occult (24.3%), and polypoidal choroidal vasculopathy (PCV) (45.9%). Patients with previous anti-VEGF treatment comprised 73% (27/37) of the study population.
Safety and Tolerability
Safety data were available for all patients for at least 28 days after the final dose. Multiple intravitreal administrations of RC28-E were well tolerated up to 13 monthly doses of 2 mg in the 48-week study period. There was no dose-limiting toxicity, and the maximum tolerated dose was not identified.
Treatment-emergent adverse events (TEAEs) were comparable between groups (Table 2). The AEs did not increase with dose escalation or repeated injections. Most AEs were mild or moderate and disappeared with or without treatment.
Ocular Safety
Ocular TEAEs were reported in 43.2% of the patients in the combined treatment groups. Most ocular AEs were related to the injection procedure, including subconjunctival hemorrhage (16.2%), dry eye (5.4%), transient increased intraocular pressure (5.4%), corneal epithelial defect (2.7%), and vitreous floaters (2.7%). The most common ocular AE was injection-related subconjunctival hemorrhage (16.2%); all instances were of mild intensity. No serious ocular AEs, clinically significant ocular inflammation, or endophthalmitis was reported in any study eyes during the entire study period.
Systemic Safety
Systemic TEAEs were reported in 78.4% of patients in the combined treatment groups.
Electrocardiogram results were mildly abnormal in two patients during the treatment period. Supraventricular extrasystole was reported in one patient in the 1.0 mg group, and an abnormal T wave was reported in one patient in the 2.0 mg group. Neither abnormality was considered related to the study drug. No cardiovascular or cerebrovascular events such as heart failure, stroke, or arterial thrombosis occurred during the study.
Serious systemic AEs were observed in three patients, including one case of leiomyoma of the uterus (0.5 mg group) and two cases of intestinal polyposis (both in the 1.0 mg group). All serious AEs were judged to be unrelated to the study drug by the principal investigators.
Pharmacokinetics and Immunogenicity
Serum concentrations of RC28-E were below the quantification limit of 1.56 ng/ml in 95% (35/37) of patients. Meanwhile, no significant change from baseline was observed in the mean plasma concentrations of VEGF or FGF over the 48 weeks of treatment.
Pre-treatment antibodies to RC28-E were detected in 1 of the 37 patients. After dosing with RC28-E for 48 weeks, antibodies against RC28-E were detected in 2 of the 37 patients. There were no differences in safety or efficacy between patients with and without immunoreactivity.
Best-Corrected Visual Acuity
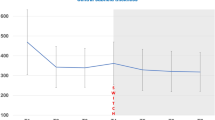
All RC28-E treatment groups showed improvement in visual acuity at weeks 12 and 48. The changes in mean BCVA over time for each group are shown in Fig. 1. Most improvements occurred during the loading phase in the first 12 weeks. The mean changes in BCVA from baseline at week 12 were 4.5 ± 4.8 letters for the 0.5 mg group, 10.8 ± 8.3 letters for the 1.0 mg group, and 7.2 ± 9.2 letters for the 2.0 mg group. The improvements gained during the loading phase were maintained or increased during the study period. At week 48, mean improvements in BCVA from baseline in the 0.5, 1.0, and 2.0 mg groups were 6.1 ± 8.3, 9.9 ± 10.7, and 7.6 ± 9.4 letters, respectively.

Mean change in BCVA from baseline at week 48 for patients in the 0.5 mg (n = 12), 1.0 mg (n = 13), and 2.0 mg groups (n = 12). BCVA best-corrected visual acuity; ETDRS Early Treatment Diabetic Retinopathy Study
Central Subfield Thickness
Changes in the mean CST over time for each group are shown in Fig. 2. The mean decrease in CST at week 12 was 14.0 ± 111.4 μm for the 0.5 mg group, 61.7 ± 98.6 μm for the 1.0 mg group, and 96.7 ± 122.7 μm for the 2.0 mg group. Patients in the 2.0 mg group showed the greatest reduction in CST after the loading phase.

Mean change in CST from baseline at week 48 for patients in the 0.5 mg (n = 12), 1.0 mg (n = 13), and 2.0 mg groups (n = 12). The CST was measured using optical coherence tomography. CST central subfield thickness
The improvement in CST observed at week 12 was maintained until week 48 after PRN administration of RC28-E. At week 48, mean reductions in CST from baseline in the 0.5, 1.0, and 2.0 mg groups were 112.1 ± 160.5, 175.1 ± 212.4, and 128.7 ± 145.8 μm, respectively.
Number of Injections
The mean number of RC28-E injections after 48 weeks were 8.8 ± 3.9 (0.5 mg group), 9.6 ± 3.7 (1.0 mg group), and 9.0 ± 3.1 (2.0 mg group). During the PRN dosing phase, the mean number of injections in the 0.5, 1.0, and 2.0 mg groups were 6.5 ± 3.5, 7.9 ± 2.0, and 6.5 ± 2.5, respectively.
Discussion
We report the outcomes of a phase 1b trial of the safety and efficacy of the novel bispecific antibody RC28-E in anti-VEGF-exposed or treatment-naïve patients with neovascular AMD. Multiple dose-escalating intravitreal injections of RC28-E were well tolerated when administered at doses of 0.5–2.0 mg and as frequently as every 4 weeks for 1 year. Most ocular AEs were mild and caused by the intravitreal injection procedure. Subconjunctival hemorrhage was the most common AE (16.2%). It was painless, reversible, and usually resolved without treatment. A corneal epithelial defect occurred in one patient in the 0.5 mg group and was considered to be related to the injection procedure. The lesion was punctate, limited to the lower part of the cornea, and recovered without treatment. Most of the systemic AEs reported in this study were due to the medical history and concomitant conditions of the enrolled elderly patients. All serious AEs were systemic, and none were related to the study drug. The overall safety profile of RC28-E was similar to that previously reported for other intravitreal anti-VEGF agents. Due to the limited number of participants in this study, a larger randomized trial is needed to accurately assess the true incidence of ocular and systemic AEs.
Visual and anatomical improvements were observed in all dosing groups. Both the BCVA and CST improved more rapidly in the 1.0 mg and 2.0 mg group than in the 0.5 mg group. Most of the change from baseline observed at week 48 was already evident after the first three monthly injections in the 1.0 and 2.0 mg groups. Furthermore, the efficacy achieved during the loading phase was maintained at the end of the study with PRN dosing. Despite the similar average changes in BCVA and CST between the 1.0 and 2.0 mg groups, the discrete degree of data was higher in the 1.0 mg group. An evident discrete value of CST was observed in one patient in the 1.0 mg group. The CST of this patient was reduced by 742 μm from baseline at week 48, which was a much greater reduction than that of all the other enrolled patients.
All patients in the study followed PRN dosing during the maintenance phase, and injections were given when certain retreatment criteria were met. The most common reason for retreatment was the presence of persistent fluid on OCT examination. Notably, the fluid incorporated all types of retinal fluid, including intraretinal, subretinal, and sub-RPE fluids. The mean number of injections was 8.8 for the 0.5 mg group, 9.6 for the 1.0 mg group, and 9.0 for the 2.0 mg group. The high dropout rate in the 0.5 mg group (41.7%) contributed to its low injection numbers. The number of injections also appeared to be lower in the 2.0 mg RC28-E group, but there were too few patients in each group to allow any definitive conclusions.
In this study, 73% of the patients had previously received anti-VEGF agents, suggesting that the study likely included a large number of patients who failed to control their disease with other marketed anti-VEGF therapies. Additionally, 45.9% of included patients had PCV, which is a subtype of AMD that is particularly prevalent in Asian populations. In a clinic-based case series of Asian patients with neovascular AMD, the proportion of PCV patients ranged from 20 to 60% [10, 11], which is much higher than that in Western patients. Disparities in the clinical features, natural history, and response to anti-VEGF agents between patients with PCV and typical AMD have been reported [12, 13]. The results of this study suggest that intravitreal RC28-E monotherapy can achieve visual gains in both PCV and non-PCV patients.
Anti-VEGF treatment is currently the standard of care for patients with neovascular AMD. Preclinical and clinical research suggests that targeting additional proangiogenic pathways could potentially improve disease control [4, 14, 15]. RC28-E simultaneously binds to VEGF and FGF-2 with a high affinity and selectivity. In theory, it has the potential to confer additional treatment benefits through an enhanced anti-VEGF effect [16] and prevention of retinal fibrosis [17, 18]. However, much remains to be learned regarding the exact role of FGF-2 in retinal fibrotic scarring, and the actual clinical benefits of this novel bispecific antibody.
There were two limitations of this study. First, the sample size of our study is limited. Subsequent studies would be carried out to further investigate the safety and efficacy of RC28-E. Second, only Chinese patients were enrolled. Thus, caution should be paid when these results were interpreted for patients with other ethnicities.
Conclusion
In summary, RC28-E was well tolerated and exhibited an overall favorable safety profile, with evidence of improvements in BCVA and CST in patients with neovascular AMD. The results of this study provide support for the continued investigation of RC28-E in larger phase III trials.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available because RC28-E has not been released to market, and we need to further analyze the raw data for research objectives.
References
Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. 2018;392:1147–59.
Brown DM, Heier JS, Ciulla T, et al. Primary endpoint results of a phase II study of vascular endothelial growth factor trap-eye in wet age-related macular degeneration. Ophthalmology. 2011;118:1089–97.
Flaxel CJ, Adelman RA, Bailey ST, et al. Age-related macular degeneration preferred practice pattern®. Ophthalmology. 2020;127:P1–65.
Heier JS, Khanani AM, Quezada Ruiz C, et al. Efficacy, durability, and safety of intravitreal faricimab up to every 16 weeks for neovascular age-related macular degeneration (TENAYA and LUCERNE): two randomised, double-masked, phase 3, non-inferiority trials. Lancet. 2022;399:729–40.
Cross MJ, Claesson-Welsh L. FGF and VEGF function in angiogenesis: signalling pathways, biological responses and therapeutic inhibition. Trends Pharmacol Sci. 2001;22:201–7.
Jiang J, Xu K, Wang L, et al. Pharmacology study of a chimeric decoy receptor trap fusion protein on retina neovascularization by dual blockage of VEGF and FGF-2. Eur J Pharm Sci. 2018;121:251–9.
Li D, Xie K, Zhang L, et al. Dual blockade of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (FGF-2) exhibits potent anti-angiogenic effects. Cancer Lett. 2016;377:164–73.
Yang QH, Zhang Y, Jiang J, et al. Protective effects of a novel drug RC28-E blocking both VEGF and FGF2 on early diabetic rat retina. Int J Ophthalmol. 2018;11:935–44.
Jiang J, Wang L, Kou X, et al. In vivo characterization of RC28-E, a fusion protein targeting VEGF and bFGF: pharmacokinetics and ocular distribution in primates. Exp Eye Res. 2020;190: 107823.
Cheung CMG, Lai TYY, Ruamviboonsuk P, et al. Polypoidal choroidal vasculopathy: definition, pathogenesis, diagnosis, and management. Ophthalmology. 2018;125:708–24.
Li Y, You QS, Wei WB, et al. Polypoidal choroidal vasculopathy in adult chinese: the Beijing Eye Study. Ophthalmology. 2014;121:2290–1.
Wong CW, Yanagi Y, Lee WK, et al. Age-related macular degeneration and polypoidal choroidal vasculopathy in Asians. Prog Retin Eye Res. 2016;53:107–39.
Koh A, Lai TYY, Takahashi K, et al. Efficacy and safety of ranibizumab with or without verteporfin photodynamic therapy for polypoidal choroidal vasculopathy: a randomized clinical trial. JAMA Ophthalmol. 2017;135:1206–13.
Chakravarthy U, Bailey C, Brown D, et al. Phase I trial of anti-vascular endothelial growth factor/anti-angiopoietin 2 bispecific antibody RG7716 for neovascular age-related macular degeneration. Ophthalmol Retina. 2017;1:474–85.
Jaffe GJ, Ciulla TA, Ciardella AP, et al. Dual antagonism of PDGF and VEGF in neovascular age-related macular degeneration: a phase IIb, multicenter, randomized controlled trial. Ophthalmology. 2017;124:224–34.
Malabanan KP, Kanellakis P, Bobik A, Khachigian LM. Activation transcription factor-4 induced by fibroblast growth factor-2 regulates vascular endothelial growth factor-A transcription in vascular smooth muscle cells and mediates intimal thickening in rat arteries following balloon injury. Circ Res. 2008;103:378–87.
Matsuda Y, Nonaka Y, Futakawa S, et al. Anti-angiogenic and anti-scarring dual action of an anti-fibroblast growth factor 2 aptamer in animal models of retinal disease. Mol Ther Nucleic Acids. 2019;17:819–28.
Nakamura Y. Multiple therapeutic applications of RBM-007, an anti-FGF2 aptamer. Cells. 2021;10:1617.
Acknowledgements
We would like to thank all the participants of the study.
Funding
This work, including the journal’s Rapid Service fee, was supported by RemeGen Co., Ltd.
Author information
Authors and Affiliations
Contributions
Concept and design and supervision: Hong Dai, Jianmin Fang, Wenxiang Wang. Analysis and interpretation of data, drafting and revision of manuscript: Yingyi Lu , Xiaobing Yu, Lin Li, He Chen, Yifan Zhang. Drafting of the manuscript, analysis, or interpretation of data: Yingyi Lu, Xiaobing Yu. Leading the trial at different centers: Youxin Chen, Chan Wu, Qin Jiang, Shaoping Ha, Dan Zhu, Yanlong Bi, Xiaoling Liu,Han Zhang, Zhuo Li. All authors have read the final manuscript and reached an agreement.
Corresponding authors
Ethics declarations
Conflict of Interest
Wenxiang Wang, Lin Li, He Chen, Yifan Zhang are employees of RemeGen Co., Ltd., Yingyi Lu, Xiaobing Yu, Youxin Chen, Chan Wu, Qin Jiang, Shaoping Ha, Dan Zhu, Yanlong Bi, Xiaoling Liu, Han Zhang, Zhuo Li, Hong Dai, Jianmin Fang has nothing to disclose.
Ethical Approval
This study was conducted in accordance with the tenets of the Declaration of Helsinki, China’s good clinical practice regulations, and applicable institutional regulatory requirements. Institutional review board and health authority approvals were obtained. Written informed consent was obtained from each study participant before protocol-related procedures were performed.
Additional information
Prior Presentation: Previously presented as an oral presentation at the World Ophthalmology Congress 2022 (Online, September 9–12).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lu, Y., Yu, X., Chen, Y. et al. Safety and Efficacy of Multiple Escalating Doses of RC28-E for Neovascular Age-Related Macular Degeneration: A Phase 1b Trial. Ophthalmol Ther 13, 2405–2415 (2024). https://doi.org/10.1007/s40123-024-00994-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-024-00994-z