
Abstract
Introduction
To evaluate the short-term efficacy of cyclosporine A (CsA)-0.1% cationic emulsion (CE) in patients with dry eye disease (DED) and mitigation of the inflammatory flares triggered by desiccating stress environments.
Methods
A single-center non-randomized clinical trial was performed at a tertiary care setting. Twenty patients with DED treated with CsA 0.1% CE were exposed to a normal controlled environment (NCE) (23 °C, 50% relative humidity) and an adverse controlled environment (ACE) (23 °C, 10% relative humidity, 0.43 m/s localized airflow) during baseline and the 1- and 3-month visits. Patients underwent the following evaluations: conjunctival hyperemia and staining, corneal fluorescein staining (CFS) using the Oxford and Cornea and Contact Lens Research Unit (CCLRU) scale, meibomian gland (MG) secretion quality, Dry Eye Questionnaire-5, Symptom Assessment in Dry Eye (SANDE II), and Change in Dry Eye Symptoms Questionnaire. Multivariate models were adjusted for statistical analysis.
Results
Nineteen women and one man (mean age, 58.9 ± 12.3 years) completed the study. All symptom questionnaires, CFS, conjunctival hyperemia and staining, and MG secretion quality improved (p ≤ 0.003) with 1 month of treatment; improvements were maintained after 3 months (p ≤ 0.02), except for SANDE II (p ≥ 0.07). The CFS worsening (total CCLRU) after baseline ACE exposure (from 8.6 to 10.1) was higher, although not significant (p = 0.64), compared with 1 month (from 5.4 to 5.8) and 3 months (from 5.0 to 5.9) after treatment.
Conclusion
Topical CsA-0.1% CE improved DED signs and symptoms after 1 month of treatment under controlled environmental conditions. Future studies should confirm the benefit of CsA-0.1% CE in desiccating stress environments.
Trial Registration
ClinicalTrials.gov identifier, NCT04492878.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
The literature is scarce regarding the efficacy of topical treatment with cyclosporine (CsA) 0.1% cationic emulsion (CE) (Ikervis) for reducing signs and symptoms after 1 and 3 months of use in patients with dry eye disease with severe keratitis. |
Patients with chronic dry eye disease (DED) suffer from episodic flares that are usually associated with exacerbation of DED signs and symptoms. These flares can be triggered by the exposition to adverse environmental conditions (e.g., air-conditioning office buildings). |
What was learned from the study? |
Topical CsA 0.1% CE can improve signs and symptoms by 1 month of treatment in patients with moderate DED who previously did not benefit from tear substitute therapy. This improvement continues 3 months after starting the treatment. |
Further studies are needed to show that topical CsA 0.1% CE could provide a prophylactic effect against aggravation of DED signs and symptoms when patients are exposed to desiccating stress environments. |
Expositions to controlled environments can help to minimize potential confounding factors in DED clinical trials, which could be useful for assessing new treatments for DED more reliably. |
Introduction
Dry eye disease (DED) is one of the most prevalent ocular diseases and affects 5–50% of the population depending on the diagnosis criteria [1]. This multifactorial inflammatory disorder is characterized by loss of homeostasis and instability of the tear film accompanied by ocular surface damage and symptoms [1].
The external environment is one of the most important triggers of DED. Adverse environmental conditions such as high temperature, airflow, or low relative humidity exacerbate DED signs and symptoms [2,3,4,5]. Environmental chambers allow re-creation of specific controlled environmental conditions in clinical studies and evaluation of individual responses to different stimuli in a standardized way [3, 6].
Artificial tears and lubricants ameliorate DED symptoms and signs only in the short term and do not address the pathogenic inflammatory mechanism of the disease [7]. Topical corticosteroids improve DED signs and symptoms by ameliorating the inflammatory process [8], but their known potential adverse effects such as intraocular pressure elevation or cataract development may prevent long-term use [9]. Long-term topical anti-inflammatory drugs for DED, such as cyclosporine A (CsA) and lifitegrast ophthalmic solution (Xiidra, Novartis, Basel, Switzerland), are also commercially available. Only topical CsA (multiple formulations), available in most countries, is safe for long-term use [10]. CsA is an anti-inflammatory and immunosuppressant drug that reduces the inflammatory status of the ocular surface [11,12,13] and improves signs and symptoms of DED [14, 15].
In 2015, the European Medicines Agency approved CsA 0.1% cationic emulsion (CE) (Ikervis, Santen Oy, Tampere, Finland) to treat severe keratitis in patients with DED refractory to treatment with tear substitutes [16]. CsA 0.1% CE is safe and effective for reducing DED signs and symptoms after 6 and 12 months of use [17,18,19,20]. However, little evidence is available from high-quality clinical trials that assessed the safety and efficacy of CsA 0.1% CE during shorter follow-up periods [17, 18]. This information should be valuable for ophthalmologists because it could prevent patients from using other concomitant medications until the effectiveness of CsA has been determined. In addition, outcomes of DED clinical trials can be biased because of environment-related conditions [5], especially in large sample phase III multicenter studies. Controlling the environmental conditions when evaluating patients can help to better assess the efficacy of any DED therapy. Therefore, the purpose of the current study was to evaluate the efficacy of CsA 0.1% CE on the signs and symptoms of DED after 1 and 3 months of therapy in patients with moderate to severe disease under controlled environmental conditions. The study also assessed the potential prophylactic benefits of CsA 0.1% CE to reduce inflammatory worsening in patients exposed to controlled adverse environmental conditions.
Methods
This was a single-center, open-label, non-controlled, phase IV clinical trial that explored the short-term efficacy of CsA 0.1% CE and its response after exposure to a controlled adverse indoor environment. The Ethics Committee of the Valladolid University Clinical Hospital (Valladolid, Spain) and the Spanish Drugs and Health Products Administration (https://www.aemps.gob.es/) (EudraCT number: 2019-000982-19) approved the trial. It was registered at the US National Institutes of Health (ClinicalTrials.gov, ID: NCT04492878). It was conducted at the Institute of Applied Ophthalmobiology, University of Valladolid, Valladolid, Spain. The study followed the tenets of the Declaration of Helsinki and the Good Clinical Practices.
Study Design
Patients completed four visits (inclusion, baseline, and follow-up visits at 1 and 3 months). All visits except the inclusion visit were performed at the Controlled Environment Laboratory (CELab, Vision R&D, Valladolid, Spain). Participants were evaluated first after a 30-min exposure to a normal controlled environment (NCE) (23 °C temperature and 50% relative humidity). They then were evaluated after a 2-h exposure to an adverse controlled environment (ACE) (23 °C temperature, 10% relative humidity, and localized 0.43 m/s airflow). Clinical evaluations were performed at each visit (Fig. 1).

Sequence of clinical evaluations. ACE adverse controlled environment, BCVA best corrected visual acuity, CDES-Q Change in Dry Eye Symptoms Questionnaire, DEQ Dry Eye Questionnaire, MG Meibomian gland, NCE normal controlled environment, SANDE Symptom Assessment in Dry Eye, TBUT tear film break-up time, VAS visual analog scale
During the inclusion visit, the nature of the study, associated potential risks and benefits, and visit schedule were explained and informed consent was obtained from the patients. Inclusion and exclusion criteria were checked, and if the criteria were met, the baseline visit was scheduled.
The baseline visit was performed 7 ± 1 days from the inclusion visit. Baseline measurements were obtained in the NCE and ACE. The inclusion/exclusion criteria were re-evaluated, and subjects who did not meet the established criteria were excluded. The included patients received the study treatment, i.e., one drop of CsA 0.1% CE in each eye every day until the end of the study. The follow-up visits followed the same methodology as at the baseline visit.
Subjects were instructed not to use any eye drops in the 2 h before the visits. Patients were asked about adherence to the protocol before beginning the follow-up visits; the vials used until that time point were collected and counted.
Patient Selection
The inclusion criteria included age of least 18 years; moderate or worse DED defined as an Oxford scale corneal fluorescein staining (CFS) ≥ 2 bilaterally and a dry eye questionnaire (DEQ)-5 score exceeding six points; use of artificial tears four or more times daily; and when concomitant medication was used that could affect the ocular surface, patients had to have been using it at least 3 months before the inclusion visit and keep using it according to the same schedule for the study duration.
Patients were excluded if they had any ocular disease except DED, a history of eye surgery or trauma that could affect corneal sensitivity or tear film distribution within 6 months before inclusion in the study, used any ocular medication for anomalies other than DED, and used any ocular medication for DED other than artificial tears during the previous month such as steroids or during the previous 3 months such as cyclosporine or tacrolimus. Patients also were excluded if they had any uncontrolled systemic disease that may affect the eyes, used any systemic treatment that could affect the DED during the last 3 months before the inclusion visit, underwent occlusion of the lacrimal punctum within the month before the study, wore contact lens during the month before the study or during its duration, or were pregnant or breastfeeding.
Clinical Evaluation
Both eyes of all patients were clinically evaluated. The following parameters were evaluated following the schedule shown in Fig. 1.
Ocular Symptoms
The presence of DED symptoms was assessed using the DEQ-5. The questionnaire score ranges from 0 to 22, and the cut-off value to detect DED is a score of 6 or higher [21].
The Symptom Assessment in Dry Eye (SANDE) II questionnaire was used to compare symptoms among study visits and between both environmental conditions. This instrument records the frequency and intensity of dry/irritated eyes, and the score ranges from − 50 to 50 [22].
Change in Dry Eye Symptoms Questionnaire (CDES-Q), which ranges from − 100 to 100 [23], also was used to assess changes.
For the SANDE II and CDES-Q, negative values corresponded to a worsening of symptoms and positive values to an improvement.
Finally, the visual analog scale (VAS) ranging from 0 to 100 evaluated satisfaction with the study treatment [24].
Visual Acuity
The best-corrected visual acuity (BCVA) was measured monocularly (Topcon Corp. Ltd., Tokyo, Japan) at 4 m using a logarithm of the minimum angle of resolution (logMAR) scale with 100% contrast.
Ocular Surface Evaluation (I)
A slit-lamp (SL-D7, Topcon Corp.) examination was performed to assess the ocular surface. Bulbar and tarsal hyperemia was evaluated based on the Cornea and Contact Lens Research Unit (CCLRU) scale [25].
Tear Film Break-up Time (TBUT)
The TBUT was measured after the instillation of 5 μl of 2% sodium fluorescein into the inferior fornix. A cobalt blue filter (Topcon Corp.) and a yellow Wratten no. 12 filter (Eastman Kodak, Rochester, NY, USA) were used. Three measurements were taken, and the average was recorded.
CFS
After 2 min of fluorescein instillation, the CFS was evaluated using the Oxford (0–5) and CCLRU (0–4 extent score for each of the 5 corneal areas) scales [25, 26]. The total CCLRU (extent) score was calculated as the sum of each score obtained in the five corneal areas.
Lissamine Green Conjunctival Staining
Staining was evaluated with the Oxford scale using lissamine green strips (GreenGlo, HUB Pharmaceuticals LLC, Rancho Cucamonga, CA, USA) wet with 25 μl sodium chloride and applied to the inferior fornix [26].
Meibomian Glands (MGs) Assessment
The expressibility and quality of secretion of the MG were measured on a 0–3 scale [27].
Outcome Measures
The primary outcome measure was a significant reduction in CFS after 1 and/or 3 months of treatment. The secondary outcome measures were significant differences in the worsening of clinical signs and/or symptoms after exposure to ACE at the 1- and 3-month follow-up visits compared with baseline.
Safety outcome measures were assessed by recording adverse events, BCVA, and slit-lamp findings.
Sample Size and Statistical Analysis
The calculation indicated that 25 patients were needed to detect a minimum change of 1 point in the CFS score (Oxford scale) considering a 20% loss (α = 0.05; p = 0.90).
A PhD licensed statistician (I.F.) performed the statistical analysis using the R statistical package version 4.0.4 (R Core Team. Foundation for Statistical Computing, Vienna, Austria (available at https://www.R-project.org/). To evaluate the effect of CsA 0.1% CE treatment and exposure to ACE and their interaction in quantitative variables, linear mixed effects models were adjusted. In addition, scores from both eyes also were considered for analysis. The required model assumptions (normality, homoelasticity, linearity, and lack of outliers) were checked. Post-hoc Tukey’s method for multiple comparisons adjustment was performed. Cumulative link mixed models were used to assess the effect of CsA 0.1% CE treatment and exposure to ACE (and their interaction) in non-parametric variables. Odds ratio (OR) calculations were obtained to characterize the changes observed in non-parametric variables after treatment or ACE exposure.
Parametric variables are expressed as the mean ± standard deviation and non-parametric variables as the median and interquartile range (IQR). P ≤ 0.05 was considered statistically significant.
Results
Baseline Characteristics
Twenty-six patients with DED were recruited, three did not meet the inclusion criteria, two dropped out of the study because of lack of tolerability to treatment, and one left because of scheduling constraints. In addition, nine patients could not complete the 1-month follow-up visit because of the COVID-19 pandemic lockdown, but they were contacted by phone to complete the DED questionnaires.
Twenty patients with DED (19 women, 1 man) (age 58.9 ± 12.3; range 35–77 years) completed the study. Four had Sjögren syndrome-related DED, and the others had no extraocular involvement. At the inclusion visit, the mean BCVA was 0.10 ± 0.21 (range − 0.20/0.85) logMAR in the right eye and 0.04 ± 0.11 (range − 0.12/0.28) in the left eye. The mean DEQ-5 score was 14.8 ± 3.3 points (range 7.0–19.0), and the median CFS (Oxford scale) was 3 (IQR, 1) bilaterally.
Efficacy Analysis
The primary endpoint was achieved as the CFS (Oxford scale) decreased significantly (p < 0.001) after 1 and 3 months of CsA 0.1% CE treatment compared to baseline in the NCE (Fig. 2). No significant (p = 0.69) differences were seen between both eyes. After 1 and 3 months of treatment, the cumulative link mixed model estimated that the ORs for a 1-point increase in the Oxford scale were 0.02 (95% confidence interval [CI], 0.006–0.04) and 0.06 (95% CI 0.03–0.13), respectively.

Corneal fluorescein staining (Oxford scale) under a normal controlled environment at baseline and after 1 and 3 months of topical cyclosporine 0.1% cationic emulsion. Both eyes were considered for statistical analysis. The squares represent medians, and vertical bars represent the 25th and 75th percentiles. OD, right eye; OS, left eye. ***p < 0.0001
Regarding CFS as measured with the CCLRU scale (extent score), the linear mixed effects model showed that CsA 0.1% CE treatment had a significant (p < 0.0001) positive effect on the CFS. The estimated marginal means for the total CCLRU (extent score) provided by the statistical model for baseline, and the 1- and 3-month visits were 9.25 (95% CI 8.05–10.45), 6.20 (95% CI 4.90–7.50), and 5.85 (95% CI 4.65–7.10), respectively (range 0–20). The differences in the marginal means of the total CCLRU scale between baseline and the 1-month (3.05, 95% CI 2.05–4.05) and 3-month (3.40, 95% CI 2.60–4.15) follow-up visits were both significant (p < 0.0001).
Secondary Outcome Measures
Regarding ocular symptoms, the linear mixed effects model showed that values obtained for all questionnaires (DEQ-5, SANDE II, and CDES-Q) significantly (p ≤ 0.003) improved after 1 month of CsA 0.1% CE treatment (Table 1). In addition, the DEQ-5 values (baseline vs. 3-month visit) improved significantly (p < 0.0001) after the 3 months of treatment (Table 1). The CDES-Q scores showed improvement (p = 0.02) 3 months after treatment compared with the 1-month visit. The SANDE II also showed a marked tendency for improvement in the frequency and intensity items (p = 0.08 and p = 0.07, respectively). After ACE exposure, the linear mixed effects model showed that DED symptoms significantly (p < 0.0001) worsened as measured with the SANDE II (frequency and intensity) and CDES-Q (Table 1). Finally, patients reported a high level of satisfaction with the treatment based on the VAS score (mean ≥ 75 units), which was similar for both follow-up visits (Table 1).
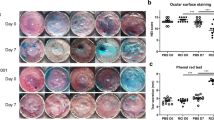
The outcomes of the remaining clinical signs evaluated are shown in Table 2. A significant (p ≤ 0.03) positive effect of CsA 0.1% CE treatment was seen in conjunctival bulbar and tarsal hyperemia, corneal (Oxford and CCLRU scales) and conjunctival staining, and quality of MG secretion. In contrast, exposure to the ACE had a significant (p ≤ 0.02) negative effect on bulbar hyperemia and CFS (Oxford and CCLRU scales). Specifically, the cumulative link mixed model showed that the ORs for an increase of 1 point in the bulbar hyperemia score after ACE exposure during the 1- and 3-month visits were 0.74 (95% CI 0.03–0.15) and 0.19 (95% CI 0.09–0.38), respectively. Regarding CFS, the OR of a 1-point increase in the Oxford scale score after ACE exposure was 2.46 (95% CI 1.35–4.47). Regarding the total CCLRU scale (extent score), the linear mixed effects model showed that exposure to ACE increased the estimated marginal means by 1.0 point (95% CI 0.4–1.5). Regarding the Oxford and CCLRU scales, the interactions between CsA 0.1% CE treatment and environmental exposures were not significant (p = 0.26 and p = 0.64, respectively). No significant (p ≥ 0.15) differences were seen in the TBUT or MG expressibility among study visits or between environmental conditions. No significant (p ≥ 0.06) differences were seen between both eyes for any clinical sign.
Safety Analysis
No significant differences were seen in the BCVA among the study visits or between both exposures. Sixteen of the 20 study patients reported mild adverse reactions after treatment instillation, i.e., ocular discomfort, itching, or stinging, with the last the most frequent. These symptoms resolved within a few minutes after instillation.
Discussion
Treatment strategies for DED should follow staged management depending on the disease severity [28, 29]. Anti-inflammatory medications are included in a stepwise approach. They are also very useful in chronic DED to ameliorate flares triggered by desiccating stress environments [30]. CsA is an immunomodulatory drug with anti-inflammatory benefits, and its topical administration is recommended for DED management [29]. The efficacy of topical CsA has been demonstrated without causing severe side effects [31]; however, the tolerability during instillation should be fully addressed. Recently, a new CsA formulation, CsA 0.1% CE (Ikervis), has resulted in improved DED signs and symptoms 6 and 12 months after treatment [17,18,19,20]. However, data from clinical trials assessing the safety and efficacy of CsA 0.1% CE after 1 month of treatment are scarce. In addition, the prophylactic effect of CsA 0.1% CE against flares due to environmental stress, which are very common in chronic DED [32], has not been tested. The present study provides not only such relevant information but also data obtained under controlled environmental conditions. Thus, reliability of the outcomes is higher because this methodology reduces bias from weather-related conditions [5, 33], in contrast to previous CsA 0.1% CE clinical trials in which environmental conditions were not controlled.
Regarding the primary outcome measure of this clinical trial, CFS significantly improved by 1 point (median values: 3 [1] vs. 2 [1]) as measured with the Oxford scale (0–5) after 1 month of CsA 0.1% CE use, and the effect was maintained out to the 3-months visit (Fig. 1). The OR showed that the likelihood of a 1-point increase in the CFS score (Oxford scale) was 50.0 (1/0.02) and 16.6 (1/0.06) times lower after 1 and 3 months of treatment, respectively. The CFS was evaluated using the CCLRU scale (extent score), in which the five corneal areas were assessed individually and could provide more precise measures of corneal integrity. We also wanted to characterize these possible changes in CFS using a more detailed scale instead of only using the Oxford scale that assesses the cornea. In the present study, assessment of the CFS using the total CCLRU scale also showed improved corneal integrity after CsA 0.1% CE. Regarding DED symptoms, the DEQ-5 scores improved 1 and 3 months after treatment compared with baseline. SANDE II (frequency and intensity) and CDES-Q scores improved after 1 month of treatment; however, further symptom relief was not reported during the 3-months visit compared with 1 month (Table 1, NCE).
Several questionnaires were used to assess the conventional efficacy of the treatment to ameliorate DED over 3 months and the possible symptom changes after ACE exposure. The DEQ-5 is a questionnaire recommended by TFOS DEWS II diagnostic methodology report [34]. However, because this questionnaire asks for the symptoms during the last month, it could not be used to assess changes before and after the ACE exposure. Instead, the SANDE II and CDES-Q were designed specifically to evaluate the changes in DED symptoms [22, 23].
The SANSIKA study [17], a phase III clinical trial that assessed the effect of CsA 0.1% CE in severe DED compared with vehicle, also reported significant CFS improvements using a modified Oxford scale (0–5) after 3 and 6 months of treatment. However, the reduction in CFS after 1-month treatment was not significant from a statistical viewpoint [17]. In the SANSIKA study, the severity of the DED in patients recruited regarding the CFS score was higher than in the present study. It is possible that keratitis can take longer to improve in the presence of more severe DED, and patients might obtain only mild benefits from the protective effect of the vehicle and need more time for the DED signs to improve until the effect of CsA 0.1% showed clinical advancements. Previous authors have reported improved signs and symptoms in mild and moderate DED as soon as 2 weeks after treatment using a compound similar to the CsA 0.1% CE vehicle [35]. Thus, this might be a reason for the slightly different findings between the SANSIKA study and the present study. In fact, Geerling et al. [36] reported significant improvements in the CFS after 1 month of treatment with CsA 0.1% CE in less severe DED (Mean Oxford scale, baseline, 2.56 vs. 1 month, 1.77) in a prospective multicenter study from routine clinical practice. Another important reason to observe CFS improvement after 1 month of treatment in the present study is that the patients were always evaluated under the same environmental conditions. Thus, more consistent results should be expected during the visits when assessing ocular surface parameters, because the environmental conditions might not have biased the outcomes as occurs in other studies. The SICCANOVE study was another phase III clinical trial that assessed the efficacy and safety of CsA 0.1% CE in DED [18]. The authors used a modified Oxford scale (0–7) to assess the CFS and reported that the treatment group had lower CFS score (− 0.77, CsA 0.1% CE) than the vehicle group (− 0.52, CsA 0.1% CE) during the 1-month follow-up visit. Differences in the CFS between the CsA 0.1% CE and the vehicle (CE-CKC) were clinically negligible. The reason might be that the vehicle can act alone as a tear substitute as previously observed [17], and patients with non-severe DED are even more likely to obtain benefits from the drug (CsA 0.1%) and the vehicle (CE).
Regarding the safety outcomes, no changes in BCVA or marked slit-lamp findings were observed. However, most patients experienced discomfort immediately after CsA instillation, which resolved within a few minutes, except for two patients whose tolerability was much lower and caused them to leave the study. In fact, the most commonly reported ocular adverse event in the literature for CsA treatments was mild instillation site pain or eye irritation [17,18,19,20]. This drug-related ocular adverse event is usually well tolerated in most patients with DED; thus, study discontinuation due to this secondary effect was observed in only 9.3% of the recruited patients in the previous CsA 0.1% CE clinical trials [37]. In the present study, two patients recruited discontinued CsA 0.1% CE treatment, which is a similar proportion to that in previous clinical trials [38]. Geerling et al. [36] also assessed the tolerability of CsA 0.1% CE in routine clinical practice and reported that only 6.6% of the participants discontinued the treatment due to poor local tolerance.
Regarding the secondary outcome measures of the present study, we assessed the DED signs and symptoms after ACE exposure because most of the population are exposed daily to adverse outdoor and indoor environmental conditions (i.e., auto climate control cars, airplane cabins, office buildings) [38]. It is well known that patients with chronic DED have episodic flares that are usually associated with exacerbation of DED signs and symptoms [32]. The flares can be triggered using environmental chambers to produce desiccating stress environments [38]. However, they also can be limited if patients were previously treated using drugs having a prophylactic effect [30, 39]. In the present study, the worsening of CFS after ACE exposure during baseline was higher (CCLRU scale from 8.6 to 10.1) than the one observed after CsA 0.1% CE treatment during the 1-month (from 5.4 to 5.8) and 3-month (from 5.0 to 5.9) visits (Table 2). However, the interaction between treatment and environmental exposures was not significant (p = 0.64). A larger sample size could have helped to observe significant differences in CFS before and after treatment; however the sample size was calculated based on the primary outcome measure. Regarding the DED symptoms, although questionnaire scores significantly improved 1 and 3 months after treatment (p ≤ 0.002), the worsening reported by patients after ACE was not significant for any visit (p ≥ 0.74). Thus, further larger studies should confirm that topical CsA 0.1% CE also might prevent symptom worsening provoked by adverse environments.
The main limitation of this clinical trial was the lack of a vehicle-treated control group. However, the effects of the vehicle in DED have been reported widely during previous large-sample clinical trials, the SANSIKA and SICCANOVE studies [17,18,19,20]. The aims of the present study were to provide new more reliable data describing the early short-term efficacy of CsA 0.1% CE controlling the environmental conditions to reduce bias and to assess the ability of CsA 0.1% CE to ameliorate flares when patients with DED are exposed to adverse conditions. Thus, there was no need to include a control group using the CsA 0.1% CE vehicle because an adequate statistical analysis can be performed using multivariate models that consider the clinical parameters assessed during all visits and during both environmental conditions. Another limitation was that the study followed an open-label unmasked design. It was decided this way because it was already proven that CsA 0.1% CE was safe and effective after 6 months of use [17,18,19,20], and the goal of the present study was different from a pivotal phase III clinical trial aiming to achieve regulatory agency approval. Besides, the investigator who performed the qualitative assessments was masked to the data corresponding to previous visits; consequently, bias associated with the design was likely to be low. Finally, another limitation is that the study started before the COVID-19 pandemic lockdown, and several participants could not complete the 1-month visit. Nonetheless, data from the rest of the participants was collected and properly analyzed from a statistical viewpoint.
Conclusion
In conclusion, the present study showed that topical CsA 0.1% CE can improve signs and symptoms by 1 month of treatment in patients with moderate DED who previously did not benefit from tear substitute therapy. This improvement in the signs and symptoms of ocular surface damage continued 3 months after starting treatment. Larger studies should corroborate that topical CsA 0.1% CE can protect patients from exacerbation of DED signs and symptoms when exposed to desiccating stress environments.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Craig JP, Nelson JD, Azar DT, et al. TFOS DEWS II report executive summary. Ocul Surf. 2017;15:802–12.
López-Miguel A, Tesón M, Martín-Montañez V, et al. Dry eye exacerbation in patients exposed to desiccating stress under controlled environmental conditions. Am J Ophthalmol. 2014;157:788–98.
Martín-Montañez V, Enríquez-de-Salamanca A, López-De La Rosa A, et al. Effect of environmental conditions on the concentration of tear inflammatory mediators during contact lens wear. Cornea. 2016;35:1192–8.
Gupta SK, Gupta V, Joshi S, Tandon R. Subclinically dry eyes in urban Delhi: an impact of air pollution? Ophthalmologica. 2002;216:368–71.
Van Setten G, Labetoulle M, Baudouin C, Rolando M. Evidence of seasonality and effects of psychrometry in dry eye disease. Acta Ophthalmol. 2016;94:499–506.
Tesón M, González-García MJ, López-Miguel A, et al. Influence of a controlled environment simulating an in-flight airplane cabin on dry eye disease. Invest Ophthalmol Vis Sci. 2013;54:2093–9.
Lee SY, Tong L. Lipid-containing lubricants for dry eye: a systematic review. Optom Vis Sci. 2012;89:1654–61.
Pflugfelder SC, Maskin SL, Anderson B, et al. A randomized, double-masked, placebo-controlled, multicenter comparison of loteprednol etabonate ophthalmic suspension, 0.5%, and placebo for treatment of keratoconjunctivitis sicca in patients with delayed tear clearance. Am J Ophthalmol. 2004;138:444–57.
Marsh P, Pflugfelder SC. Topical nonpreserved methylprednisolone therapy for keratoconjunctivitis sicca in Sjögren syndrome. Ophthalmology. 1999;106:811–6.
Barber LD, Pflugfelder SC, Tauber J, Foulks GN. Phase III safety evaluation of cyclosporine 0.1% ophthalmic emulsion administered twice daily to dry eye disease patients for up to 3 years. Ophthalmology. 2005;112:1790–4.
Matsuda S, Koyasu S. Mechanisms of action of cyclosporine. Immunopharmacology. 2000;47:119–25.
Kunert KS, Tisdale AS, Stern ME, Smith JA, Gipson IK. Analysis of topical cyclosporine treatment of patients with dry eye syndrome: effect on conjunctival lymphocytes. Arch Ophthalmol. 2000;118:1489–96.
Turner K, Pflugfelder SC, Ji Z, Feuer WJ, Stern M, Reis BL. Interleukin-6 levels in the conjunctival epithelium of patients with dry eye disease treated with cyclosporine ophthalmic emulsion. Cornea. 2000;19:492–6.
Baiza-Duran L, Medrano-Palafox J, Hernandez-Quintela E, Lozano-Alcazar J, Alaniz-de la OJ. A comparative clinical trial of the efficacy of two different aqueous solutions of cyclosporine for the treatment of moderate-to-severe dry eye syndrome. Br J Ophthalmol. 2010;94:1312–5.
Sall K, Stevenson OD, Mundorf TK, Reis BL. Two multicenter, randomized studies of the efficacy and safety of cyclosporine ophthalmic emulsion in moderate to severe dry eye disease. CsA Phase 3 Study Group. Ophthalmology. 2000;107:631–9.
Eroglu YI. A comparative review of Haute Autorité de Santé and National Institute for health and care excellence health technology assessments of Ikervis® to treat severe keratitis in adult patients with dry eye disease which has not improved despite treatment with tear substitutes. J Mark Access Health Policy. 2017;5:1336043.
Leonardi A, Van Setten G, Amrane M, et al. Efficacy and safety of 0.1% cyclosporine A cationic emulsion in the treatment of severe dry eye disease: a multicenter randomized trial. Eur J Ophthalmol. 2016;26:287–96.
Baudouin C, Figueiredo FC, Messmer EM, et al. A randomized study of the efficacy and safety of 0.1% cyclosporine A cationic emulsion in treatment of moderate to severe dry eye. Eur J Ophthalmol. 2017;27:520–30.
Baudouin C, de la Maza MS, Amrane M, et al. One-Year Efficacy and safety of 0.1% cyclosporine a cationic emulsion in the treatment of severe dry eye disease. Eur J Ophthalmol. 2017;27:678–85.
Pisella PJ, Labetoulle M, Doan S, et al. Topical ocular 0.1% cyclosporine A cationic emulsion in dry eye disease patients with severe keratitis: experience through the French early-access program. Clin Ophthalmol. 2018;12:289–99.
Chalmers RL, Begley CG, Caffery B. Validation of the 5-Item Dry Eye Questionnaire (DEQ-5): discimination across self-assessed severity and aqueous tear deficient dry eye diagnosis. Cont Lens Anterior Eye. 2010;33:55–60.
Schaumberg DA, Gulati A, Mathers WD, et al. Development and validation of a short global dry eye symptom index. Ocul Surf. 2007;5:50–7.
Pinto-Fraga J, Calonge M, Enríquez-de-Salamanca A, Fernández I, González-García MJ, Steven P. Development of a questionnaire for detecting changes in dry eye disease-related symptoms. Eye Contact Lens. 2021;47:8–14.
Aitken RC. Measurement of feelings using visual analogue scales. Proc R Soc Med. 1969;62:989–93.
Terry RL, Schnider CM, Holden BA, et al. CCLRU standards for success of daily and extended wear contact lenses. Optom Vis Sci. 1993;70:234–43.
Bron AJ, Evans VE, Smith JA. Grading of corneal and conjunctival staining in the context of other dry eye tests. Cornea. 2003;22:640–50.
Tomlinson A, Bron AJ, Korb DR, et al. The international workshop on meibomian gland dysfunction: report of the diagnosis subcommittee. Invest Ophthalmol Vis Sci. 2011;52:2006–49.
Geerling G, Tauber J, Baudouin C, et al. The international workshop on meibomian gland dysfunction: report of the subcommittee on management and treatment of meibomian gland dysfunction. Invest Ophthalmol Vis Sci. 2011;52:2050–64.
Jones L, Downie LE, Korb D, et al. TFOS DEWS II management and therapy report. Ocul Surf. 2017;15:575–628.
Pinto-Fraga J, López-Miguel A, González-García MJ, et al. Topical fluorometholone protects the ocular surface of dry eye patients from desiccating stress: a randomized controlled clinical trial. Ophthalmology. 2016;123:141–53.
Wan KH, Chen LJ, Young AL. Efficacy and safety of topical 0.05% cyclosporine eye drops in the treatment of dry eye syndrome: a systematic review and meta-analysis. Ocul Surf. 2015;13:213–25.
Perez VL, Stern ME, Pflugfelder SC. Inflammatory basis for dry eye disease flares. Exp Eye Res. 2020;201: 108294.
Tesón M, López-Miguel A, Neves H, Calonge M, González-García MJ, González-Méijome JM. Influence of climate on clinical diagnostic dry eye tests: pilot study. Optom Vis Sci. 2015;92:284–9.
Wolffsohn JS, Arita R, Chalmers R, et al. TFOS DEWS II diagnostic methodology report. Ocul Surf. 2017;15:539–74.
Fogagnolo P, Quisisana C, Caretti A, et al. Efficacy and safety of VisuEvo® and Cationorm® for the treatment of evaporative and non-evaporative dry eye disease: a multicenter, double-blind, cross-over, randomized clinical trial. Clin Ophthalmol. 2020;14:1651–63.
Geerling G, Hamada S, Trocmé S, et al. Real-world effectiveness, tolerability and safety of cyclosporine a 0.1% cationic emulsion in severe keratitis and dry eye treatment. Ophthalmol Ther. 2022;11:1101–7.
Leonardi A, Messmer EM, Labetoulle M, et al. Efficacy and safety of 0.1% ciclosporin A cationic emulsion in dry eye disease: a pooled analysis of two double-masked, randomised, vehicle-controlled phase III clinical studies. Br J Ophthalmol. 2019;103:125–31.
Calonge M, Pinto-Fraga J, González-García MJ, et al. Effects of the external environment on dry eye disease. Int Ophthalmol Clin. 2017;57:23–40.
Moore QL, De Paiva CS, Pflugfelder SC. Effects of dry eye therapies on environmentally induced ocular surface disease. Am J Ophthalmol. 2015;160:135–42.
Acknowledgements
We thank the participants for their involvement in the study.
Medical Writing, Editorial, and Other Assistance.
Santen SA reviewed the manuscript for scientific and medical accuracy but had no input into the content. This assistance was funded by Santen SA.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Funding
The study was partially supported by investigator-initiated study grants from Santen SA (Geneva, Switzerland). No funding or sponsorship was received for the publication of this article.
Author information
Authors and Affiliations
Contributions
All authors contributed to the study conception and design. Material preparation, data collection and analysis were performed by Laura Valencia-Nieto, José Pinto-Fraga, Marta Blanco-Vázquez, Itziar Fernández, Carmen García-Vázquez and Amalia Enríquez de Salamanca. The first draft of the manuscript was written by Laura Valencia-Nieto, Alberto López-Miguel, Itziar Fernández and María J. González-García, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
No conflicting relationship exists for any author. Disclosures of Dr. Margarita Calonge are the following: Research/clinical trials contracts, consultantships,advisory boards and/or lectures for Novartis, Santen Pharmaceutical, Johnson & Johnson, Horus Pharma, Fidia Farmaceutica, UrsaPharm GmbH, and Thea Laboratories. Alberto Lopez Miguel is an Editorial Board member of Ophthalmology and Therapy. Alberto Lopez Miguel was not involved in the selection of peer reviewers for the manuscript nor any of the subsequent editorial decisions. The remaining authors have no relationship to disclose.
Ethical Approval
The Ethics Committee of the Valladolid University Clinical Hospital (Valladolid, Spain) and the Spanish Drugs and Health Products Administration (https://www.aemps.gob.es/) (EudraCT number: 2019-000982-19) approved the trial. It was registered at the US National Institutes of Health (ClinicalTrials.gov, ID: NCT04492878). It was conducted at the Institute of Applied Ophthalmobiology, University of Valladolid, Valladolid, Spain. The study followed the tenets of the Declaration of Helsinki and the Good Clinical Practices.
Additional information
Prior Presentation: The outcomes of this study were partially presented at the 2022 Association for Research in Vision and Ophthalmology Annual Meeting, May 1–4, (Denver, CO, USA).
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Valencia-Nieto, L., Pinto-Fraga, J., Blanco-Vázquez, M. et al. Short-Term Efficacy of Ophthalmic Cyclosporine: A 0.1% Cationic Emulsion in Dry Eye Patients Assessed Under Controlled Environment. Ophthalmol Ther 13, 1197–1210 (2024). https://doi.org/10.1007/s40123-024-00906-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-024-00906-1