
Abstract
The port delivery system (PDS) of anti-VEGF therapy provides continuous delivery of ranibizumab (RBZ). In October of 2021, the American Food and Drug Administration (FDA) approved the PDS with RBZ as a treatment option for neovascular age-related macular degeneration (nAMD). As the field of PDS with RBZ is progressing rapidly, this narrative review provides a much-needed overview of existing clinical trials as well as ongoing and upcoming trials investigating PDS with RBZ. The phase 2 LADDER trial reported that the mean time to first refill with RBZ PDS 100 mg/ml was 15.8 months (80% CI 12.1–20.6), and pharmacokinetic profiling revealed a sustained concentration of RBZ in serum and aqueous humor. Later, the phase 3 ARCHWAY trial reported that PDS with RBZ (100 mg/ml) refilled every 24 weeks was non-inferior to monthly intravitreal injection (IVI) with RBZ (0.5 mg) in patients with nAMD over 9 months and 2 years. However, patients with PDS had a higher rate of adverse events including vitreous hemorrhage and endophthalmitis. Patients indicate high treatment satisfaction with both PDS and IVI, but the lower number of treatments with PDS was reported as a preferred choice. Several ongoing and future clinical trials, of which details are discussed in this paper, are further exploring the potentials of PDS with RBZ. We conclude that the PDS provides continuous deliverance of RBZ and that clinical efficacy levels are non-inferior to IVI therapy for nAMD. Yet, a higher rate of adverse events remains a concerning detail for widespread implementation. Future studies are warranted to better understand which patients may benefit best from this treatment approach, if long-term efficacy can be sustained, and if safety of PDS can be further improved.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The port delivery system with ranibizumab has shown sustained deliverance and non-inferiority. | |
The treatment approach is approved for treating neovascular age-related macular degeneration. | |
Patients seem to prefer the port delivery system with ranibizumab over intravitreal injections. | |
The port delivery system shows a higher rate of adverse events than intravitreal injections. | |
Ongoing and future trials seek to further investigate the port delivery system with ranibizumab. |
Introduction
Age-related macular degeneration (AMD) is a leading cause of blindness in high-income countries [1]. Globally, the prevalence is estimated to be 8.7%, and by 2040, 288 million people are anticipated to be suffering from the disease, which will have a large burden on healthcare systems [2].
Non-neovascular or ‘dry AMD’ is the most common subtype of AMD while neovascular AMD (nAMD) or ‘wet AMD’ is the primary cause of vision loss. nAMD is an advanced stage of AMD and is represented by the development of macular neovascularization [3], which pathophysiologically is linked to ageing [4,5,6], environmental factors [6,7,8], genetic disposition [3, 9], and para-inflammation [10,11,12,13]. Growth factors released from the retinal pigment epithelium, including vascular endothelial growth factor (VEGF), play a crucial role in mediating the pathogenesis of AMD due to proangiogenic abilities. The formation of vulnerable and leaky new blood vessels can result in vision loss due to retinal edema, retinal hemorrhages, and fibrosis [14].
Ranibizumab (RBZ) (Lucentis, Genentech, Inc., South San Francisco, CA, USA) is a humanized monoclonal antibody fragment that targets VEGF-A. As a result, the downstream signaling pathway is negatively affected through inactivation of vascular endothelial growth factor receptors 1 and 2, which promote angiogenesis, leakage of vessels, and inflammation [14]. In 2006, RBZ was introduced for the treatment of nAMD following approval from the Food and Drug Administration (FDA) based on the pivotal ANCHOR and MARINA trials [15, 16]. Anti-VEGF treatment has led to substantial reductions in blindness [17, 18], stabilized vision [19], and has provided an important vision-related daily function in the majority of the patients [20]. However, intravitreal injection (IVI)-based therapy can significantly burden patients and may result in discontinuation of anti-VEGF therapy [21,22,23].
The port delivery system implant (PDS) (Susvimo, Genentech, Inc., South San Francisco, CA, USA) with RBZ may potentially mitigate the burden associated with IVI-based therapy. The implant contains a reservoir filled with RBZ and is implanted permanently into the superotemporal quadrant of the sclera [24]. The mechanism allows for a controlled and sustained RBZ deliverance [25,26,27] and refill through a self-sealing septum [28]. RBZ is transferred from the implant to the vitreous cavity by passive diffusion and the diffusion rate depends on the starting concentration [25, 27].
The FDA approved the PDS with RBZ for patients with nAMD with response to at least two anti-VEGF treatments in October 2021 (https://www.fda.gov/media/158153/download) (accessed July 3, 2023) after the phase 2 LADDER trial [27] and the phase 3 ARCHWAY trial [26]. Since this FDA approval, new and exploring research has been conducted and interesting future clinical trials are in the pipeline. We are conducting this narrative review due to rapid progressions in the field of PDS. Our objective was to evaluate prior studies and future outlooks of PDS with RBZ.
Methods
We searched the databases PubMed, Web of Science, the Cochrane Central, and ClinicalTrials.gov for publications and protocols on the port delivery system with ranibizumab. The database searches were conducted between November 2022 and July 2023. During the review process, a Genentech letter accessed on October 3, concerning a voluntary recall of the Susvimo implant was added. References were reviewed for relevance for the narrative structure of this review. We only considered articles written in English. This narrative review is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Clinical Trials
The phase 1 trial supported by Genentech, Inc., South San Francisco, CA, USA, presented initial PDS results. PDS implants with RBZ (10 mg/ml) were implanted in 20 treatment-naïve patients with nAMD. With ocular adverse events (AE) as the primary endpoint, 19 patients (95%) experienced hyperemia, five patients (25%) experienced vitreous hemorrhage, and four patients (20%) experienced hyphemia. Four cases of serious ocular adverse events (SAE) were reported including two eyes with vitreous hemorrhage and one eye with endophthalmitis. The authors also found a steady mean RBZ serum concentration after 8 weeks (n = 10) staying above the lower limit of quantification (15 pg/ml). Furthermore, there was a mean refill rate of 4.1 injections per patient (95% CI 2.9–5.3) and a median time to first refill of 3 months (interquartile range 2–3 months). The authors found an increase in visual acuity over 1 year [29].
The LADDER Trial
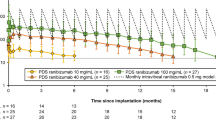
The phase 2 LADDER trial, supported by Genentech, Inc., South San Francisco, CA, USA, launched in 2015 (NCT02510794 [30]) [27, 31] as a randomized, multicenter, active-treatment controlled clinical trial. The trial aimed at evaluating efficacy and safety of PDS in patients with nAMD (n = 220). Patients were randomly allocated to four arms as 3:3:3:2 to receive PDS with RBZ 10 mg/ml, 40 mg/ml, or 100 mg/ml after a pro re nata-regime or monthly IVI with 0.5 mg/ml RBZ, respectively [27]. Inclusion criteria were (1) ≥ 50 years of age, (2) previous responsiveness to anti-VEGF treatment, (3) previous treatment with 2–9 anti-VEGF treatments in the study eye, and (4) nAMD diagnosis in the study eye within 9 months prior to start. Anti-VEGF responsiveness was defined as (1) a decrease in central foveal thickness (CFT) of minimum 50 µm or (2) a stabilized or improved visual acuity after initiating anti-VEGF treatment [27].
Principal endpoint was time to first refill when last enrolled patient completed the month 9 visit. The median time to first refill for the PDS arms with 10 mg/ml (n = 58), 40 mg/ml (n = 62), and 100 mg/ml (n = 59) was 8.7 months (80% CI 6.9–9.0), 13.0 months (80% CI 11.8–24.6), and 15.8 months (80% CI 12.1–20.6), respectively [31]. Patients were assessed monthly and received implant refill if (1) CFT increased, (2) best-corrected visual acuity (BCVA) decreased, or (3) macular hemorrhage occurred. For patients in PDS-based therapy receiving 10, 40, and 100 mg/ml percentages without refill after 1 year were 28.9, 56.0, and 59.4%, respectively.
Secondary endpoints comprised change in CFT and BCVA measured using the Early Treatment Diabetic Retinopathy Study Chart (ETDRS). Month 22 was the mean time on the study and here the mean change in BCVA from baseline was − 4.6 ETDRS letters, - 2.3 ETDRS letters, + 2.9 ETDRS letters, and + 2.7 ETDRS letters for PDS 10, 40, 100 mg/ml, and monthly IVI, respectively. Furthermore, at month 22, 57.7%, 80.0%, 87.5%, and 88.9% of patients receiving the PDS 10, 40, 100 mg/ml, and monthly IVI maintained their baseline visual acuity, respectively [31]. CFT increase was the most frequent reason for refill [31]. At month 22, the CFT measured from the internal limiting membrane to retinal pigment epithelium was for patients receiving PDS-based therapy 10, 40, and 100 mg/ml, and monthly IVI-based therapy − 0.7, − 20.9, − 4.0, and − 10.9 µm, respectively.
Overall, from LADDER it was reported that patients receiving PDS-based therapy had more AE compared to patients receiving IVI-based therapy. Within the first month from implantation (37 days), 89.9% of PDS-treated patients and 9.8% of IVI-treated patients experienced AE. Beyond the first month, 68.2% of patients receiving PDS-based therapy and 63.4% of patients receiving IVI-based therapy experienced AE [31], highlighting the importance of post-implantation monitoring. From the results section reported in the study protocol (NCT02510794), conjunctival hemorrhage was at the completion of the study the most frequent non-serious adverse event across all arms with 70.69, 70.97, 62.71, and 19.51% of patients receiving 10, 40, and 100 mg/ml, and IVI with RBZ 0.5 mg, respectively. At completion of the study, 9.5% of patients in PDS-based therapy had experienced SAE events in contrast to no patients receiving monthly IVI-based therapy. While LADDER did not report a definition of SAE, the most common SAE reported was vitreous hemorrhage. A high rate of vitreous hemorrhage was observed prior to May 2016 in 11/22 (50%) of the already-investigated patients receiving PDS-based therapy leading to a pause of the trial. Following surgical procedure optimization [32], the rate of vitreous hemorrhage in the PDS arm dropped to 8/157 (5.1%) after May 2016 [31]. Three patients receiving PDS-based therapy experienced endophthalmitis in contrast to none of the patients receiving IVI-based therapy.
Serum samples were collected in all patients and aqueous humor samples in PDS-treated patients. Analyses were conducted in the pharmaco-kinetic evaluable population comprising patients in PDS-based therapy but with no IVI-treatment in the fellow eye, supplemental RBZ treatment, or bevacizumab 9 months prior to randomization. For patients receiving PDS-based therapy with RBZ (100 mg/ml), the mean serum concentration was 160 pg/ml through month 9 and thus in the range expected with monthly IVI with RBZ (0.5 mg) of 130–2220 pg/ml. Through month 16, the mean serum concentration was 51 pg/ml and thus remained above the lower limit of quantification (15 pg/ml), indicating a steady serum concentration of RBZ. For patients in PDS-based therapy (100 mg/ml), the mean maximum concentration was roughly 1080 pg/ml at implantation and above the maximum concentration found in other PDS arms [33]. The serum concentration was approximately correlated to the aqueous humor concentration for all PDS arms (10, 40, and 100 mg/ml) [33]. In vitro findings conducted prior to LADDER suggested the PDS could deliver a continuous and predictable supply of RBZ. Drug release depended on the starting concentration of RBZ and in the PDS arm loaded with 100 mg/ml the release rate was 16.69 µg/day at 3.5 days and 4.16 µg/day at 6 months, thus showing a continuous release of RBZ over 6 months [25]. An overview of the primary findings from LADDER is found in Table 1.
The ARCHWAY Trial
The phase 3 ARCHWAY trial [26] (NCT03677934 [34]), supported by Genentech, Inc., South San Francisco, CA, USA, was an open-label, randomized, non-inferiority trial with the purpose to evaluate safety and efficacy of PDS with RBZ for treatment of nAMD. A total of 418 participants were enrolled and randomized 3:2 to receive PDS with RBZ (100 mg/ml) every 24 weeks (n = 251) or IVI (0.5 mg) monthly (n = 167). For patients allocated to the PDS arm, 248 received treatment and in the IVI arm, 167 received treatment. For enrolment, patients should (1) be ≥ 50 years of age, (2) have nAMD lesions located to the macula diagnosed within the preceding 9 months, (3) have a minimum of three anti-VEGF treatments (RBZ, bevacizumab, or aflibercept) within the preceding 6 months, and (4) respond to treatment both visually and anatomically. PDS-treated patients were allowed supplemental IVI-based therapy if a decrease in BCVA or an increase in central retinal thickness was observed.
The principal endpoint was evaluation of change in BCVA assessed on the Early Treatment Diabetic Retinopathy Study (ETDRS) chart averaged at week 36 and 40 from baseline. Change in adjusted mean from baseline averaged through week 36 and week 40 was + 0.2 (SE; 0.5) and + 0.5 (SE; 0.6) among PDS-treated patients and IVI-treated patients, respectively. Non-inferiority (± 4.5 ETDRS letters) was demonstrated for patients receiving the PDS with RBZ (100 mg/ml). The difference in adjusted means was − 0.3 letters (95% CI − 1.7 to 1.1 ETDRS letters).
Secondary endpoints included both visual and anatomical outcomes of BCVA and center point thickness (CPT). Of the total patients, 98.4% (n = 246) did not require supplemental IVI therapy before first refill at 24 weeks. Statistically corresponding CPT values between the two treatment arms were found through week 40.
The study reported more PDS-treated patients (n = 248) than IVI-treated patients (n = 167) experiencing both AE (94.4 and 35.5%) and SAE (5.6 and 1.2%) through week 40. SAE was defined as fatality, life threat, prolonged hospitalization, persistent or significant disability, congenital birth defect in infant born to study participant, or significant medical event judged by an investigator. Thirteen patients in PDS-based therapy experienced vitreous hemorrhage (5.2%) of which 12 were during the first month and one case was serious. Four patients (1.6%) experienced endophthalmitis, all occurring outside the first month. In comparison, four (2.4%) patients receiving IVI-based therapy experienced vitreous hemorrhage and no experienced endophthalmitis. In vitro findings conducted prior to the ARCHWAY trial suggested continuous and quantifiable release rates of RBZ when assessing the PDS with RBZ (100 mg/ml) of 17.97, 4.44, and 2.45 µg/day at 3.5 days, 6 months, and 9 months, respectively [25]. An overview of the ARCHWAY trial is found in Table 2.
An ARCHWAY extension, supported by Genentech, Inc., South San Francisco, CA, USA [35] reported results from approximately 2 years. The final population for analysis comprised 415 patients receiving PDS-based therapy (n = 248) and IVI-based therapy (n = 167). Overall, results were consistent with the primary analysis of ARCHWAY [26]. It was reported that for patients receiving PDS-based therapy 24 discontinued treatment and for patients receiving IVI-based therapy 13 discontinued treatment. Reasons for discontinuation included AE, withdrawal, and death.
The primary endpoint was identical to the ARCHWAY trial. Secondary endpoints included mean change in BCVA averaged over weeks 60 and 64 and by request from the European Medicines Agency also over weeks 44–48 and 88–92. For the PDS-treated patients, adjusted mean change from baseline through weeks 44 to 48, weeks 60 to 64, and weeks 88 to 92 was 0.0 letters (SE 0.5 letters), − 0.4 letters (SE 0.57 letters), and − 1.1 letters (SE 0.61 letters), respectively. For the IVI-treated patients receiving monthly RBZ, the mean change from baseline through weeks 44–48, weeks 60–64, and weeks 88–92 was 0.2 letters (SE, 0.62 letters), − 0.8 letters (SE, 0.69 letters), and − 0.5 letters (SE, 0.75). Difference between treatment arms were − 0.2 letters (95% CI − 1.8 to 1.3 letters), 0.4 letters (95% CI − 1.4 to 2.1 letters), and − 0.6 letters (95% CI − 2.5 to 1.3), respectively. After nearly 2 years, the authors found that patients receiving PDS-based therapy with RBZ (100 mg/ml) showed non-inferiority compared to patients receiving monthly IVI-based therapy with RBZ (0.5 mg) with a non-inferiority margin of ± 3.9 ETDRS letters. However, the non-inferiority test was not prespecified. 98.4% did not need supplemental IVI therapy within the first 24 weeks [26] and 94.7% did not need supplemental IVI therapy during the last 24 weeks (weeks 73–96) [35]. The difference in change from baseline in CPT was11.3 µm (95% CI − 0.1 to 22.6) and did not differ statistically significantly between treatment arms through week 96.
Consistent, with the primary ARCHWAY analysis, the rate of AE and SAE was higher among patients receiving PDS-based therapy compared to patients receiving IVI-based therapy. At week 96, 22 patients in PDS-based therapy (8.9%) had experienced SAE in contrast to four patients in IVI-based therapy (2.4%). At week 96, 15 patients receiving PDS-based therapy had experienced vitreous hemorrhage in contrast to six patients receiving IVI-based therapy. Four patients treated with PDS experienced endophthalmitis and one patient in IVI-based therapy experienced endophthalmitis.
For the pharmaco-kinetic evaluable population of patients receiving PDS-based therapy (100 mg/ml), a serum concentration of RBZ in the same range as for patients receiving IVI-based therapy with RBZ (0.5 mg) was maintained through week 96. The study stated that the PDS with RBZ (100 mg/ml) can maintain a steady concentration through 96 weeks with a fixed refill interval of 24 weeks. An overview of the primary findings from the ARCHWAY extension is found in Table 3.
Patient Preferences
Chang et al. [36] evaluated treatment satisfaction among patients in PDS- and IVI-based treatment from LADDER and ARCHWAY by using the Macular Disease Treatment Satisfaction Questionnaire (MacTSQ) [37]. The MacTSQ comprises two sub-scale scores (range, 0–36 points) and a total score (range, 0–72 points). A sub-scale score of 30 and an overall score of 60 indicated ‘high treatment satisfaction’.
On patients from the ARCHWAY trial, the MacTSQ was evaluated at baseline and week 40 [36]. At baseline, both the PDS-treated patients (n = 248) and the IVI-treated patients (n = 167) showed high satisfaction in total and subscale scores. At week 40, the mean total score for the PDS-treated patients (n = 237) and the IVI-treated patients (n = 159) were 68.0 (95% CI 67.4–68.6) and 66.1 (95% CI 64.9–67.3) with a difference of 1.9 (95% CI 0.7–3.1). In addition, both groups had subscale scores above 30 and high overall satisfaction was found across both arms with and without SAE. In patients from the LADDER trial, high treatment satisfaction was found in both treatment arms through month 9 using the MacTSQ [36].
Preference was evaluated only in PDS-treated patients from ARCHWAY by using the validated PDS Patient Preference Questionnaire (PPPQ). The validation study was presented as a conference abstract [38]. A total of 234 patients who received a minimum of three IVI treatments prior to baseline and completed the PPPQ were included in the final analysis; 218 preferred the PDS (93%) and 172 (73%) had a ‘very strong’ preference for the PDS. Reasons for preferring PDS were ‘fewer treatments’, ‘less discomfort’, and ‘less nervousness’ [36]. Of 222 patients without AE, 209 (94%) preferred the PDS. Of 12 patients with a SAE, nine (75%) preferred the PDS.
Cost Analyses
By using data from LADDER [27] and ARCHWAY [26], Sood et al. [39] conducted cost analyses. The authors examined the number of IVI treatments needed to break even with the PDS both with and without refills. Furthermore, total cost over 1 and 5 years for patients receiving PDS with RBZ refilled every 6 months against IVI-based therapy one time per month (monthly) or one time every 2 months (bimonthly). The ARCHWAY trial reported that 98% of the PDS-treated patients did not require supplemental IVI therapy before the first refill at 24 weeks [26] and therefore a single refill the first year and nine refills over 5 years were assumed [39]. Finally, authors conducted scenario analyses assuming pro re nata dosing based on data from the LADDER trial [31] and real-world data from the American Academy of Ophthalmology Intelligent Research in Sight Registry. For pro re nata dosing, it was assumed that 40.6% of the PDS-treated patients required a refill inside year 1 and 3.8 refills over 5 years as the median time to first refill was 15.8 months [31].
To break even with implantation and baseline medication of PDS with RBZ, 6.4 (SD, 0.8) RBZ injections, 5.5 (SD, 0.7) aflibercept injections, or 34.5 (4.2) bevacizumab injections were needed. When adding one refill 10.8 (SD, 1.3) RBZ injections, 9.3 (SD, 1.1) aflibercept injections, or 58.1 (SD, 7.1) bevacizumab injections were needed.
The 1-year mean cost of PDS with RBZ refilled every 6 months was $21,017 (SD, $2,101). In comparison, bimonthly IVI-based therapy with aflibercept was $7.658 cheaper (p = 0.006) and monthly IVI-based therapy with bevacizumab was $16,732 cheaper (p = < 0.001). Monthly IVI-based therapy with aflibercept was $5,702 more costly (p = 0.04). No statistical difference was found when compared to monthly IVI-based therapy with RBZ.
The 5-year mean cost of PDS with RBZ refilled every 6 months was $89,218 (SD, $8,922). In comparison, monthly IVI-based therapy with RBZ was $25,581 more costly (p = 0.04). Monthly IVI-based therapy with aflibercept was $44,374 more costly (p = 0.01). Monthly IVI-based therapy with bevacizumab was $67,793 cheaper (p = < 0.001). Bimonthly IVI-based therapy with aflibercept was $22,422 cheaper (p = 0.03).
In scenario analyses over 1 year, the PDS with RBZ was more costly whether refilled as needed or as fixed every 6 months when compared to data based on real-world use of anti-VEGF. As an exception, when administered as needed no statically significant difference was found comparing the PDS with RBZ against a real-world regime of IVI-based therapy with aflibercept over 1 year. PDS with RBZ administered as-needed was cheaper than monthly IVI-based treatment with RBZ and aflibercept, respectively.
In scenario analyses over a 5-year period, PDS with RBZ administered as-needed was $15,129 cheaper (p = 0.03) than IVI-based treatment with RBZ and $23,097 cheaper (p = 0.01) than IVI-based treatment with aflibercept administered after a real-world regime, respectively. In contrast, IVI-based treatment with bevacizumab following a real-world regime was $35,136 cheaper (p = > 0.001) than PDS with RBZ administered as-needed. PDS with RBZ administered at a fixed 6-month interval was statistically significantly more expensive than RBZ, aflibercept and bevacizumab administered after a real-world regime.
Future Outlooks
An overview of the studies presented in this section can be found in Table 4.
The PAVILION trial, sponsored by Hoffmann La-Roche (NCT04503551 [40]) (accessed June 19, 2023) is a randomized, multicenter, phase 3 clinical trial investigating efficacy and safety in patients suffering from moderately severe to severe non-proliferative diabetic retinopathy (DR) without center-involved diabetic macular edema (DME) (central subfield thickness ≥ 325 µm). Prior to implantation of the PDS with RBZ (100 mg/ml) refilled every 36 weeks, patients receiving PDS-based therapy receive two IVI with RBZ. The comparator arm will be monitored monthly before implantation with the PDS device with refill every 36 weeks. The comparator arm is eligible for supplemental IVI treatment. The primary outcome is rate of patients with ≥ 2 step improvement on the ETDRS-Chart-Diabetic Retinopathy Severity Scale (ETDRS-DRSS) 52 weeks from baseline. Among others, secondary endpoints include evaluation of DR status, rate of patients who develop sight-threatening complications (proliferative DR, anterior segment neovascularization, or center-involving DME), visually, anatomically, and pharmacokinetically outcomes. The PAVILION trial was launched in 2020 and is estimated to end in 2024.
The PAGODA trial, sponsored by Hoffmann La-Roche (NCT04108156 [41]) (accessed June 20, 2023) is a randomized, multicenter, phase 3, clinical trial investigating efficacy, safety, and pharmacokinetics of the PDS in patients suffering from DR with DME (central subfield thickness ≥ 325 µm). The treatment arm receiving PDS will be loaded monthly with IVI containing RBZ prior to implantation and hereafter refill of the implant will occur every 24 weeks. The comparator arm will receive monthly IVI with RBZ (0.5 mg) and hereafter implantation of the PDS device with refill every 24 weeks. The primary endpoint is change in BCVA over weeks 60 and 64 from baseline using the ETDRS chart. Secondary endpoints include visual outcomes, anatomical outcomes, and patient preference in patients who receive PDS-based therapy in one eye and IVI-based therapy in the other. Quality of life is assessed in all patients. Furthermore, adverse events and pharmacokinetics are investigated. PAGODA was launched in 2019 and is estimated to end in 2024.
A randomized, multicenter, phase 3, clinical trial is conducted in Chinese patients diagnosed with nAMD within 9 months from baseline and who have shown prior responsiveness to anti-VEGF treatment (NCT05562947 [42]) (accessed July 14, 2023). The study is much like ARCHWAY [26] and investigates PDS with RBZ (100 mg/ml) refilled every 24 weeks against monthly IVI-based therapy with RBZ (0.5 mg). The primary endpoint is change in BCVA from baseline averaged over weeks 36 and 40. The study is sponsored by Hoffmann La-Roche and is expected to launch in 2023 and end in 2029.
The DIAGRID-trial, sponsored by Hoffmann La-Roche (NCT05126966 [43]) (accessed June 20, 2023) is a randomized, multicenter, phase 3b study. The aim is to evaluate efficacy and safety of the PDS with RBZ (100 mg/ml) refilled every 36 weeks compared to IVI-based therapy with aflibercept (2 mg) administered as a treat-and-extend regime in patients suffering from nAMD and with prior responsiveness to anti-VEGF. The primary endpoints will be change in BCVA between baseline and week 80 assessed on the ETDRS chart and treatment frequency until week 80. The study is now suspended because Roche/Genentech has paused new implantations as the implants from commercial supply did not meet the filed specifications for the intended use in the clinical studies. A Genentech letter from October 2022 explained a voluntary recall of the Susvimo Ocular Implant and Insertion Tool Assembly including the Susvimo drug vial and initial fill needle. The recall was due to dislodgement problems. (https://www.gene.com/download/pdf/Susvimo_DHCP_Important_Prescribing_Information_2022-10-18.pdf), (accessed on October 3, 2023). The letter advises that patients without septum dislodgement can continue treatment after careful examination of the vitreous cavity has been conducted. For patients with septum dislodgement, the physicians are advised to stop performing refill procedures and to discuss removal of the implant after providing information on potential benefits and risks of keeping versus explanting the implant. Patients can be started in IVI-based therapy with anti-VEGF. The DIAGRID trial is estimated to launch in 2023 and end in 2026.
The VELODROME trial, sponsored by Hoffmann La-Roche (NCT04657289 [44]) (accessed June 20, 2023, is a randomized, multicenter, active-comparison, phase 3b study. The aim is to evaluate the efficacy, safety, and pharmacokinetics of the PDS with RBZ (100 mg/ml) refilled every 36 weeks compared to the PDS with RBZ (100 mg/ml) refilled every 24 weeks in patients with nAMD. Patients must have received at least three treatments prior to baseline, show responsiveness to anti-VEGF, and be diagnosed within 9 months from baseline. The primary endpoint is BCVA change from baseline averaged over weeks 68 and 72. BCVA is assessed with the ETDRS chart. Secondary endpoints comprise visual outcomes, anatomical outcomes, patient preferences, pharmacokinetics, and safety. The VELODROME trial launched in 2021 and is estimated to end in 2026.
The PORTAL trial, sponsored by Hoffmann La-Roche, (NCT03683251 [45]) (accessed June 20, 2023) is an open-label, multicenter trial investigating tolerability and long-term safety of PDS with RBZ (100 mg/ml) in patients with nAMD. Patient recruitment is ongoing, and investigators estimate 1000 patients to be enrolled. Patients who have completed either LADDER [27], ARCHWAY [26], or VELODROME [44] are eligible for inclusion. The primary endpoints investigate the incidence, severity, and duration of ocular and non-ocular AE (up to week 240). The secondary endpoints investigate visual and anatomical outcomes (up to week 240). Upon study entry patients will receive the PDS with RBZ (100 mg/ml) with refill every 24 weeks. However, patients from VELODROME allocated to receive a refill at 36 weeks will continue to do so. Patients from LADDER and ARCHWAY will switch to refill every 12 weeks at weeks 168 and 144, respectively. The PORTAL trial launched in 2018 and is estimated to end in 2026.
Presented as a conference abstract, interim analyses of PORTAL at 48 months showed mean BCVA change from baseline was 0.1 ETDRS letters (95% CI − 6.6 to 6.8) and 2.3 ETDRS letters (95% CI − 9.4 to 14.1) for patients who received the PDS with RBZ (100 mg/ml) (n = 31) and IVI-based therapy with RBZ (0.5 mg) (n = 15), respectively; 92% of patients who in LADDER received IVI-based therapy now preferred the PDS device [46]. In pooled data (n = 555) over a mean time of 111 weeks, 11.4% experienced cataracts, 6.1% vitreous hemorrhage, and 2.0% endophthalmitis [46].
The ongoing VOYAGER study, sponsored by Hoffmann La-Roche (NCT05476926 [47]) (accessed June 23, 2023) is a prospective, multinational, cohort study collecting long-term real-world data in patients who receive treatment with either the PDS with RBZ or faricimab over 5 years. The aim is to assess the efficacy, safety, and clinical insights. Investigators estimate 5000 patients to be included. The primary endpoint is change in BCVA from baseline to 1 year using the local method for obtaining BCVA and then converted to ETDRS letters. Secondary endpoints run over 5 years and include visual and safety outcomes. The VOYAGER study was launched in 2022 and is expected to end in 2027.
The ongoing BELVEDERE trial, sponsored by Genentech, Inc., (NCT04853251 [48]) (accessed June 23, 2023) is a phase 4, multicenter, open-label trial. The objective is to investigate the treatment response of patients with nAMD receiving the PDS with RBZ (100 mg/ml) refilled every 24 weeks. Patients must have previously been treated with anti-VEGF other than RBZ. The primary endpoint is BCVA at week 40 measured using an ETDRS chart. Secondary endpoints comprise vision, safety, and anatomical outcomes including mean change in center point thickness from baseline to week 52, percentage of participants with and without subretinal and/or intraretinal fluid over week 52, among others. Originally, 200 patients were estimated but currently 58 patients are estimated. The BELVEDERE trial was launched in 2021 and is expected to end in 2024.
Discussion
In summary, clinical trials find that PDS have potential to continuously deliver RBZ with a median time to first refill of 15.8 months [31] and with a fixed refill time of 24 weeks the PDS with RBZ (100 mg/ml) may be non-inferior to fixed-dose monthly IVI-based therapy with RBZ [26]. More AEs were found in patients in PDS-based therapy compared to patients in IVI-based therapy, which highlights the need for careful consideration of the safety profile when considering which patients to recommend for PDS. However, patients seem to prefer PDS over IVI, although it should be underscored that both PDS and IVI lead to high treatment satisfaction [36]. Ongoing and future trials are in the pipeline for further investigations on the PDS with RBZ.
LADDER (n = 218) was limited and, notably, underpowered in detecting a difference between the arms in the PDS-treated patients receiving 100 and 40 mg/ml [31] as reported by the authors. Also, PAVILION [40], the Chinese trial [42], and BELVEDERE [48] estimate smaller sample sizes of 174, 68, and 58 patients, respectively. Hence, potentially reducing the reproducibility when extrapolated to a larger population. Further trials with larger sample sizes are warranted. In contrast, the ongoing PORTAL trial [45] and VOYAGER study [47] estimate 1000 and 5000 patients for enrolment, respectively.
Patients from LADDER and ARCHWAY were required to show responsiveness to two prior IVI treatments [26, 31] potentially reducing generalizability. Likewise, VELODROME [44], DIAGRID [43], and BELVEDERE [48] also require prior responsiveness among participants. In contrast, the phase 1 study [29] and the ongoing PAVILION [40] included patients who were treatment-naïve in the study eye. Non-treatment-naïve patients receiving treatment with anti-VEGF prior to baseline might hypothetically present with a higher visual acuity at baseline than treatment-naïve patients. A potential ceiling effect should be considered, which could possibly result in poorer gain in visual acuity among eyes with higher baseline visual acuity while treatment-naïve patients could instead potentially experience larger gains in visual acuity [49,50,51]. It may be advantageous to conduct further research on the PDS in treatment-naïve patients suffering from nAMD and DME prior to real-world implementation to learn about its significance. Additionally, a direct comparison of treatment-naïve patients against prior responders could be an interesting approach. The upcoming trial conducted in Chinese patients also requires prior responsiveness [42]. However, considering ethnic differences in subtypes of nAMD [52,53,54,55] results of the trial conducted specifically in a Chinese population may, to a certain extent, differ from the ARCHWAY trial conducted primarily in white Americans [26]. Importantly, other ongoing trials including PORTAL [45], VELODROME [44], and VOYAGER [47] are conducted in a multi-center and an international fashion. More future studies of the PDS with RBZ on multi-ethnic populations may improve generalizability.
Contrary to other clinical studies summarized in this review, the ongoing VELODROME trial [44] will compare refill every 36 weeks to refill every 24 weeks in patients with nAMD (estimated n = 442). This might potentially help determine an optimal refill interval for PDS with RBZ. However, as already mentioned, patients must show prior responsiveness to anti-VEGF treatment potentially limiting generalizability. In addition, a control group receiving IVI-based therapy with RBZ for active comparison would be intriguing. Interestingly, DIAGRID compares PDS with RBZ to IVI-based therapy with aflibercept. However, as already mentioned, the trial is for now suspended as the implants from commercial supply did not meet the filed specifications of the intended use [43]. A letter issued October 2022 from Genentech states that 33 cases of septum dislodgement was reported in approximately 1419 patients across clinical trials assessing the PDS with RBZ emphasizing the awareness of physicians assessing patients implanted with the PDS. The Genentech letter also states, however, that no cases of septum dislodgement were reported from implants used in commercial supply. Perhaps the problem may for now mainly be of scientific and not public concern. However, time will tell if patients and physicians will have any concerns using the PDS with RBZ in future clinical and scientific practice. (https://www.gene.com/download/pdf/Susvimo_DHCP_Important_Prescribing_Information_2022-10-18.pdf) (accessed October 3, 2023).
Because the PDS with RBZ is intended as a permanent solution, clinical trials assessing long-term efficacy and safety are crucial. The total study duration of LADDER was 38 months with the mean study time of 22.1 months for patients receiving PDS-based therapy and 21.7 months for patients receiving IVI-based therapy [31]. ARCHWAY had a duration of only 9 months [26], highlighting the need for the ARCHWAY extension [35], the PORTAL trial [45], and the VOYAGER study [47] for the assessment of the PDS with RBZ over extended periods of time. The results from the ARCHWAY extension [35] were consistent with the primary analysis [26]. However, 39 patients from the treatment arm receiving IVI-based therapy missed at least one treatment due to the corona virus pandemic, potentially introducing bias. In contrast, no patients receiving PDS-based therapy missed a refill due to the corona virus pandemic [35]. Patients enrolled in PORTAL are not randomized but allocated based on the trial they came from [45], which can also be a limitation. Likewise, patients and study personnel were not masked to treatment-arm randomization in both LADDER and ARCHWAY due to differences in treatment administration between arms resulting in potential risk of bias.
In the LADDER trial, the implant procedure was altered and optimized following a high rate of vitreous hemorrhage leading to unequal intervention before and after May 2016 in patients receiving PDS-based treatment [31], potentially adding bias. Both LADDER [31] and ARCHWAY [26] indicate more AE among patients receiving PDS-based therapy than patients receiving IVI-based therapy. Along with a high frequency of vitreous hemorrhage, the US label for PDS with RBZ warns against an association with a threefold higher rate of endophthalmitis compared to monthly IVI-based therapy. This emphasizes the viability of close monitoring and early detection. Ericksen et al. [56] investigated monitoring of the PDS through non-invasive high-resolution images in six eyes and with no control group. Through qualitative assessments, the authors found a thinning of the tenons capsule and overlying conjunctiva over the corners of the implant flange. Given the study design, conclusions cannot be drawn. Yet, the study may be a step in detecting PDS-related complications earlier. Future larger-scale studies with quantitative monitoring would be intriguing.
Many complications appeared within 1 month from implantation in the ARCHWAY and LADDER trials [26, 31] and it is possible that surgery may be responsible. For example, vitreous hemorrhage dropped after procedure optimization in the LADDER trial [31] and in ARCHWAY authors stated that most AE cases were linked to the implantation procedure [26]. Moreover, in the ARCHWAY trial, it was discussed that three out of four cases of endophthalmitis were associated with conjunctival retraction and suggested these cases could be mitigated through a more careful handling of the conjunctiva and tenons capsule [26]. This emphasizes the significance of surgeons. However, perhaps proper patient education and post-surgical self-care could also play a role in mitigating AE and SAE. For clinicians, simulation-based virtual reality provides a safe training space. VRmagic GmbH and Genentech Inc. developed two virtual reality training systems for the training in implantation and refill of the PDS device for the ARCHWAY trial [57, 58]. Results from other modalities like simulation-based cataract surgery and vitreoretinal surgery have been interesting. Yet, they must also be interpreted carefully due to risk of bias and low grading of evidence certainty [59,60,61]. Hence, the need for future quality trials to provide construct validity and transfer of skills for the implantation procedure of the PDS must be stressed. Importantly, ongoing trials are also assessing safety as endpoint and for that matter, large observational studies like the VOYAGER study [47] will be of interest.
Cost is a significant parameter to consider when evaluating novel treatment approaches. The cost analyses by Sood et al. [39] were thoroughly conducted with model input based on significant clinical trials. However, analyses were made upon assumptions, which should be considered when interpreting the results. Compared to a real-world regime of IVI-based therapy, the PDS with RBZ may offer cost-savings after 5 years. Perhaps, this could limit the use of the PDS with RBZ and increase the preference for alternative treatment approaches. Moreover, in a comment by Patel and Sternberg [62], it was argued that while PDS may reduce number of IVI treatments, monitoring of the implant and potential fellow-eye disease may increase in-clinic visiting frequency [62]. Additionally, utilization of bevacizumab should be considered [62] as bevacizumab potentially offers immense cost savings [39], and has demonstrated non-inferiority to RBZ when using similar methods of administration [63]. However, it is mentionable that bevacizumab is only used off-label in the treatment of nAMD. Still, future trials assessing PDS with RBZ against IVI-based therapy with bevacizumab could be intriguing. Preferably, future trials should investigate IVI-based regimes other than monthly injections which patients in general do not really adhere to in a real-world setting. Yet, in vitro findings notably suggest aflibercept and bevacizumab are sub-optimal compared to RBZ for a sustained drug deliverance with the PDS [64].
Patient involvement may also offer invaluable feedback. While PDS-treated patients and IVI-treated patients from the ARCHWAY trial both had high overall treatment satisfaction, the PDS-based therapy was preferred over IVI-based therapy [36]. However, the PPPQ used for assessing treatment preference was evaluated in only 11 patients from the LADDER trial [38] making the PPPQ more uncertain. The study by Chang et al. [36] received criticism for not connecting visual acuity to patient satisfaction and preference [65]. Additionally, since treat-and-extend regimes better reflect real-world AMD treatment than monthly IVI treatment, it was questioned if treatment satisfaction would differ [65]. Out of 234 patients, 172 (73.5%) preferred the PDS despite AE [36]. Perhaps this indicates a risk willingness. However, assumedly, patients participating in the trial would be more willing to undergo surgery despite a risk of potential complications. Preference is also being evaluated in both VELODROME among patients with nAMD [44] and in PAGODA [41] among patients with DME.
Globally, DME is a visual thread [66] but real-world data suggest lower anti-VEGF IVI frequency than in clinical trials [67, 68]. This highlights the usefulness of the PAGODA and PAVILION trials investigating the PDS in DR-patients with and without DME, respectively. PAGODA expects 634 patients while PAVILION expects only 174 patients. Duration between the trials is almost similar and comparable. However, PAVILION uses treatment-naïve patients whereas PAGODA does not. Moreover, PAVILION evaluates patients on the ETDRS-DDRS scale as outcome measurement while PAGODA evaluates BCVA on an ETDRS chart [40, 41]. Nevertheless, the results can hopefully expand our knowledge of the PDS with RBZ for the treatment of patients suffering from DME.
Conclusions
The PDS is a permanent drug reservoir implanted into the sclera to allow for a sustained delivery of RBZ potentially able to mitigate the treatment burden of IVI. The PDS may continuously deliver RBZ (100 mg/ml) with a mean time to first refill of 15.8 months and may be non-inferior to monthly IVI-based therapy. Patients seem to prefer the PDS with RBZ over monthly IVI-based therapy. However, limitations also apply to the PDS including cost and AE comprising vitreous hemorrhage and endophthalmitis. Yet, the PDS with RBZ remains an intriguing treatment approach. To gain comprehensive understanding of the PDS, further large future studies are needed, exploring long-term efficacy, safety, satisfaction, preference, and cost in real-world settings. Also, while ethnicity may be an important aspect for differences in subtypes of AMD the importance of investigating the PDS with RBZ in different populations must be stressed. Moreover, anti-VEGF drugs other than RBZ including bevacizumab and aflibercept might be useful to investigate in clinical trials to get a comprehensive understanding of potential benefits and disadvantages. Fortunately, ongoing and future trials will further investigate this treatment approach.
References
Bourne RR, Jonas JB, Flaxman SR, et al. Prevalence and causes of vision loss in high-income countries and in Eastern and Central Europe: 1990–2010. Br J Ophthalmol. 2014;98(5):629–38.
Wong WL, Su X, Li X, et al. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–16.
Fleckenstein M, Keenan TDL, Guymer RH, et al. Age-related macular degeneration. Nat Rev Dis Primers. 2021;7(1):31.
Subhi Y, Nielsen MK, Molbech CR, et al. T-cell differentiation and CD56+ levels in polypoidal choroidal vasculopathy and neovascular age-related macular degeneration. Aging (Albany NY). 2017;9(11):2436–52.
Subhi Y, Forshaw T, Sørensen TL. Macular thickness and volume in the elderly: a systematic review. Ageing Res Rev. 2016;29:42–9.
Harris J, Subhi Y, Sørensen TL. Effect of aging and lifestyle on photoreceptors and retinal pigment epithelium: cross-sectional study in a healthy Danish population. Pathobiol Aging Age Relat Dis. 2017;7(1):1398016.
Knudtson MD, Klein R, Klein BE. Physical activity and the 15-year cumulative incidence of age-related macular degeneration: the Beaver Dam Eye Study. Br J Ophthalmol. 2006;90(12):1461–3.
Detaram HD, Joachim N, Liew G, et al. Smoking and treatment outcomes of neovascular age-related macular degeneration over 12 months. Br J Ophthalmol. 2020;104(7):893–8.
Nielsen MK, Subhi Y, Molbech CR, Grønskov K, Sørensen TL. Distribution of risk alleles in patients with age-related macular degeneration. Dan Med J. 2020;67(3).
Nahavandipour A, Krogh Nielsen M, Sørensen TL, Subhi Y. Systemic levels of interleukin-6 in patients with age-related macular degeneration: a systematic review and meta-analysis. Acta Ophthalmol. 2020;98(5):434–44.
Subhi Y, Krogh Nielsen M, Molbech CR, et al. Plasma markers of chronic low-grade inflammation in polypoidal choroidal vasculopathy and neovascular age-related macular degeneration. Acta Ophthalmol. 2019;97(1):99–106.
Rozing MP, Durhuus JA, Krogh Nielsen M, et al. Age-related macular degeneration: a two-level model hypothesis. Prog Retin Eye Res. 2020;76: 100825.
Holtz JK, Thinggaard BS, Grauslund J, Subhi Y. Association between oral metformin use and the risk of age-related macular degeneration: a systematic review with meta-analysis. Acta Ophthalmol. 2023;101(6):595–605.
Heloterä H, Kaarniranta K. A linkage between angiogenesis and inflammation in neovascular age-related macular degeneration. Cells. 2022;11(21):3453.
Rosenfeld PJ, Brown DM, Heier JS, et al. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–31.
Brown DM, Kaiser PK, Michels M, et al. Ranibizumab versus verteporfin for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1432–44.
Bloch SB, Larsen M, Munch IC. Incidence of legal blindness from age-related macular degeneration in Denmark: year 2000 to 2010. Am J Ophthalmol. 2012;153(2):209-13.e2.
Finger RP, Daien V, Eldem BM, et al. Anti-vascular endothelial growth factor in neovascular age-related macular degeneration—a systematic review of the impact of anti-VEGF on patient outcomes and healthcare systems. BMC Ophthalmol. 2020;20(1):294.
Brynskov T, Munch IC, Larsen TM, Erngaard L, Sørensen TL. Real-world 10-year experiences with intravitreal treatment with ranibizumab and aflibercept for neovascular age-related macular degeneration. Acta Ophthalmol. 2020;98(2):132–8.
Ferløv Baselius NJ, Brynskov T, Falk MK, Sørensen TL, Subhi Y. Driving vision in patients with neovascular AMD in anti-VEGF treatment. Acta Ophthalmol. 2021;99(8):e1360–5.
Spooner KL, Mhlanga CT, Hong TH, Broadhead GK, Chang AA. The burden of neovascular age-related macular degeneration: a patient’s perspective. Clin Ophthalmol. 2018;12:2483–91.
Prenner JL, Halperin LS, Rycroft C, Hogue S, Williams Liu Z, Seibert R. Disease burden in the treatment of age-related macular degeneration: findings from a time-and-motion study. Am J Ophthalmol. 2015;160(4):725-31.e1.
Subhi Y, Sørensen TL. Neovascular age-related macular degeneration in the very old (≥90 years): epidemiology, adherence to treatment, and comparison of efficacy. J Ophthalmol. 2017;2017:7194927.
Pieramici DJ, Wieland MR, Stewart JM, et al. Implant insertion procedure of the port delivery system with ranibizumab: overview and clinical pearls. Ophthalmic Surg Lasers Imaging Retina. 2022;53(5):249–56.
Yohe S, Maass KF, Horvath J, Rea J, Barteselli G, Ranade SV. In-vitro characterization of ranibizumab release from the port delivery system. J Control Release. 2022;345:101–7.
Holekamp NM, Campochiaro PA, Chang MA, et al. Archway randomized phase 3 trial of the port delivery system with ranibizumab for neovascular age-related macular degeneration. Ophthalmology. 2022;129(3):295–307.
Campochiaro PA, Marcus DM, Awh CC, et al. The port delivery system with ranibizumab for neovascular age-related macular degeneration: results from the randomized phase 2 ladder clinical trial. Ophthalmology. 2019;126(8):1141–54.
Khanani AM, Graff JM, Marcus DM, et al. Refill-exchange procedure of the port delivery system with ranibizumab: overview and clinical trial experience. Ophthalmic Surg Lasers Imaging Retina. 2022;53(5):257–65.
Loewenstein A, Laganovska G, Bressler NM, et al. Phase 1 Clinical study of the port delivery system with ranibizumab for continuous treatment of neovascular age-related macular degeneration. Investig Ophthalmol Visual Sci. 2020;61(7).
Genentech I. Study of the efficacy and safety of the ranibizumab port delivery system for sustained delivery of ranibizumab in patients with subfoveal neovascular age-related macular degeneration. ClinicalTrials.gov: NCT02510794. Accessed March 30, 2023. https://classic.clinicaltrials.gov/show/NCT02510794.
Khanani AM, Callanan D, Dreyer R, et al. End-of-study results for the ladder phase 2 trial of the port delivery system with ranibizumab for neovascular age-related macular degeneration. Ophthalmol Retina. 2021;5(8):775–87.
Bantseev V, Schuetz C, Booler HS, et al. Evaluation of surgical factors affecting vitreous hemorrhage following port delivery system with ranibizumab implant insertion in a minipig model. Retina. 2020;40(8):1520–8.
Wykoff CC, Campochiaro PA, Pieramici DJ, et al. Pharmacokinetics of the port delivery system with ranibizumab in the ladder phase 2 trial for neovascular age-related macular degeneration. Ophthalmol Ther. 2022;11(5):1705–17.
Roche H-L. A phase III study to evaluate the port delivery system with ranibizumab compared with monthly ranibizumab injections in participants with wet age-related macular degeneration. ClinicalTrials.gov: NCT03677934. Accessed April 19, 2023. https://classic.clinicaltrials.gov/show/NCT03677934.
Regillo C, Berger B, Brooks L, et al. Archway phase 3 trial of the port delivery system with ranibizumab for neovascular age-related macular degeneration 2-year results. Ophthalmology. 2023. https://doi.org/10.1016/j.ophtha.2023.02.024.
Chang MA, Kapre A, Kaufman D, et al. Patient preference and treatment satisfaction with a port delivery system for ranibizumab vs intravitreal injections in patients with neovascular age-related macular degeneration: a randomized clinical trial. JAMA Ophthalmol. 2022;140(8):771–8.
Mitchell J, Bradley C. Design and development of the MacTSQ measure of satisfaction with treatment for macular conditions used within the IVAN trial. J Patient Rep Outcomes. 2018. https://doi.org/10.1186/s41687-018-0031-z.
Tschosik E, Kapre A, Ferrara D, Chang M. Content validity of the port delivery system with ranibizumab patient preference questionnaire. Investig Ophthalmol Visual Sci. 2019;60(9).
Sood S, Mandell J, Watane A, Friedman S, Parikh R. Cost of ranibizumab port delivery system vs intravitreal injections for patients with neovascular age-related macular degeneration. JAMA Ophthalmol. 2022;140(7):716–23.
Roche H-L. A Multicenter, randomized study in participants with diabetic retinopathy without center-involved diabetic macular edema to evaluate the efficacy, safety, and pharmacokinetics of ranibizumab delivered via the port delivery system relative to the comparator Arm. ClinicalTrials.gov: NCT04503551. Accessed June 19, 2023. https://classic.clinicaltrials.gov/show/NCT04503551.
Roche H-L. This study will evaluate the efficacy, safety, and pharmacokinetics of the port delivery system with ranibizumab in participants with diabetic macular edema compared with intravitreal ranibizumab. ClinicalTrials.gov: NCT04108156. Accessed June 20, 2023. https://classic.clinicaltrials.gov/show/NCT04108156.
Roche H-L. A study of the efficacy, safety, and pharmacokinetics of the port delivery system with ranibizumab in Chinese patients with neovascular age-related macular degeneration. ClinicalTrials.gov: NCT05562947. Accessed July 14, 2023. https://classic.clinicaltrials.gov/show/NCT05562947.
Roche H-L. A study of the effectiveness and safety of a 36-week refill regimen for the port delivery system with ranibizumab vs aflibercept treat and extend in subjects with neovascular age-related macular degeneration. ClinicalTrials.gov: NCT05126966. Accessed June 20, 2023. https://classic.clinicaltrials.gov/show/NCT05126966.
Roche H-L. A study of the efficacy, safety, and pharmacokinetics of a 36-week refill regimen for the port delivery system with ranibizumab in patients with neovascular age-related macular degeneration (velodrome). ClinicalTrials.gov: NCT04657289. Accessed June 20, 2023. https://classic.clinicaltrials.gov/show/NCT04657289.
Roche H-L. Extension study for the port delivery system with ranibizumab (portal). ClinicalTrials.gov: NCT03683251. Accessed June 20, 2023. https://classic.clinicaltrials.gov/show/NCT03683251.
Haug S, Callaway N, DeGraaf S, et al. Interim analysis of the portal extension trial evaluating the long-term safety and efficacy of the port delivery system with ranibizumab (PDS) in neovascular age-related macular degeneration (nAMD). Investig Ophthalmol Vis Sci. 2022;63(7).
Roche H-L. A Real-world study to gain clinical insights into Roche ophthalmology products. ClinicalTrials.gov: NCT05476926. Accessed June 23, 2023. https://classic.clinicaltrials.gov/show/NCT05476926.
Genentech I. A study of the response to treatment after transition to the port delivery system with ranibizumab Susvimo (ranibizumab injection) in patients with neovascular age-related macular degeneration previously treated with intravitreal agents other than ranibizumab. ClinicalTrials.gov: NCT04853251. Accessed June 23, 2023. https://classic.clinicaltrials.gov/show/NCT04853251.
Zhu M, Chew JK, Broadhead GK, et al. Intravitreal ranibizumab for neovascular age-related macular degeneration in clinical practice: five-year treatment outcomes. Graefes Arch Clin Exp Ophthalmol. 2015;253(8):1217–25.
The neovascular age-related macular degeneration database. multicenter study of 92 976 ranibizumab injections: report 1: visual acuity. Ophthalmology. 2014;121(5):1092–101.
Williams TA, Blyth CP. Outcome of ranibizumab treatment in neovascular age-related macula degeneration in eyes with baseline visual acuity better than 6/12. Eye (Lond). 2011;25(12):1617–21.
Wong CW, Wong TY, Cheung CM. Polypoidal choroidal vasculopathy in Asians. J Clin Med. 2015;4(5):782–821.
Matsumoto H, Hoshino J, Mukai R, Nakamura K, Kishi S, Akiyama H. Clinical characteristics and pachychoroid incidence in Japanese patients with neovascular age-related macular degeneration. Sci Rep. 2022;12(1):4492.
van Dijk EHC, Holtz JK, Sirks MJ, et al. European prevalence of polypoidal choroidal vasculopathy: a systematic review, meta-analysis, and forecasting study. J Clin Med. 2022;11(16):4766.
Lorentzen TD, Subhi Y, Sørensen TL. Prevalence of polypoidal choroidal vasculopathy in white patients with exudative age-related macular degeneration: systematic review and meta-analysis. Retina. 2018;38(12):2363–71.
Ericksen CJ, Christensen CA, Berger B, Gune S, Nielsen JS. Implantation site of a port delivery system with ranibizumab: anterior segment optical coherence tomography evaluation. J Vitreoret Dis. 2022;6(5):347–50.
Pieramici DJ, Heimann F, Brassard R, Barteselli G, Ranade S. Virtual reality becomes a reality for ophthalmologic surgical clinical trials. Transl Vis Sci Technol. 2020;9(7):1.
Heimann F, Barteselli G, Brand A, et al. A custom virtual reality training solution for ophthalmologic surgical clinical trials. Adv Simul (Lond). 2021;6(1):12.
Lin JC, Yu Z, Scott IU, Greenberg PB. Virtual reality training for cataract surgery operating performance in ophthalmology trainees. Cochrane Database Syst Rev. 2021;12(12):Cd014953.
Carr L, McKechnie T, Hatamnejad A, Chan J, Beattie A. Effectiveness of the Eyesi Surgical Simulator for ophthalmology trainees: systematic review and meta-analysis. Can J Ophthalmol. 2023. https://doi.org/10.1016/j.jcjo.2023.03.014.
Rasmussen RC, Grauslund J, Vergmann AS. Simulation training in vitreoretinal surgery: a systematic review. BMC Ophthalmol. 2019;19(1):90.
Patel S, Sternberg P Jr. Is there a cost benefit to the ranibizumab port delivery system? JAMA Ophthalmol. 2022;140(7):723–4.
Martin DF, Maguire MG, Ying GS, Grunwald JE, Fine SL, Jaffe GJ. Ranibizumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med. 2011;364(20):1897–908.
Chang DP, Burra S, Day ES, et al. Long-term stability of anti-vascular endothelial growth factor (a-VEGF) biologics under physiologically relevant conditions and its impact on the development of long-acting delivery systems. J Pharm Sci. 2021;110(2):860–70.
Beaulieu WT, Glassman AR. Patient-reported outcome measures in a clinical trial of the port delivery system with ranibizumab. JAMA Ophthalmol. 2022. https://doi.org/10.1001/jamaophthalmol.2022.2133.
Yau JW, Rogers SL, Kawasaki R, et al. Global prevalence and major risk factors of diabetic retinopathy. Diabetes Care. 2012;35(3):556–64.
Ciulla TA, Pollack JS, Williams DF. Visual acuity outcomes and anti-VEGF therapy intensity in diabetic macular oedema: a real-world analysis of 28 658 patient eyes. Br J Ophthalmol. 2021;105(2):216–21.
Van Aken E, Favreau M, Ramboer E, et al. Real-world outcomes in patients with diabetic macular edema treated long term with ranibizumab (VISION study). Clin Ophthalmol. 2020;14:4173–85.
Acknowledgements
Author Contribution
The idea for the manuscript was formulated by Simon Joel Lowater and Anna Stage Vergmann. Simon Joel Lowater searched databases, assessed articles for relevance, and prepared the original draft. Anna Stage Vergmann, Yousif Subhi, and Jakob Grauslund read, critically reviewed, and edited the draft through multiple rounds for intellectual content. The final version of the manuscript received approval from all authors before submission. All authors contributed to the creation of the manuscript with a substantial amount of work.
Funding
No funding or sponsorship was received for this review or publication of this article.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Conflict of Interest
Simon Joel Lowater does not have any conflicts of interest to declare. Anna Stage Vergmann and Yousif Subhi are editorial board members of Ophthalmology and Therapy. Jakob Grauslund is in advisory boards of Roche, Novartis, and Bayer. Yousif Subhi has received speakers fee from Bayer and Roche.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Lowater, S.J., Grauslund, J., Subhi, Y. et al. Clinical Trials and Future Outlooks of the Port Delivery System with Ranibizumab: A Narrative Review. Ophthalmol Ther 13, 51–69 (2024). https://doi.org/10.1007/s40123-023-00843-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-023-00843-5