
Abstract
Introduction
This trial aimed to compare the efficacy and safety between biosimilar QL1207 and the reference aflibercept for the treatment of neovascular age-related macular degeneration (nAMD).
Methods
This randomized, double-blind, phase 3 trial was conducted at 35 centers in China. Patients aged ≥ 50 years old with untreated subfoveal choroidal neovascularization secondary to nAMD and best-corrected visual acuity (BCVA) letter score of 73–34 were eligible. Patients were randomly assigned to receive intravitreous injections of QL1207 or aflibercept 2 mg (0.05 ml) in the study eye every 4 weeks for the first 3 months, followed by 2 mg every 8 weeks until week 48, stratified by baseline BCVA ≥ or < 45 letters. The primary endpoint was BCVA change from baseline at week 12. The equivalence margin was ± 5 letters. The safety, immunogenicity, pharmacokinetics (PK), and plasma vascular endothelial growth factor (VEGF) concentration were also evaluated.
Results
A total of 366 patients were enrolled (QL1207 group, n = 185; aflibercept group, n = 181) from Aug 2019 to Jan 2022 with comparable baseline characteristics. The least-squares mean difference in BCVA changes was − 1.1 letters (95% confidence interval − 3.0 to 0.7; P = 0.2275) between the two groups, within the equivalence margin. The incidences of treatment-emergent adverse events (TEAE; QL1207: 71.4% [132/185] vs. aflibercept: 71.8% [130/181]) and serious TEAE (QL1207: 14.1% [26] vs. aflibercept: 12.7% [23]) appeared comparable between treatment groups, and no new safety signal was found. Anti-drug antibody, PK profiles, and VEGF concentration were similar between the two groups.
Conclusions
QL1207 has equivalent efficacy to aflibercept for nAMD with similar safety profiles. It could be used as an alternative anti-VEGF agent for clinical practice.
Trial Registration
ClinicalTrials.gov: NCT05345236 (retrospectively registered on April 25, 2022); National Medical Products Administration of China: CTR20190937 (May 20, 2019).
Similar content being viewed by others

Why carry out this study? |
Neovascular age-related macular degeneration (nAMD) is one of the leading causes of blindness and visual impairment worldwide. Aflibercept, a fusion protein of vascular endothelial growth factor receptors-1, -2 and Fc portion of immunoglobulin G, is recommended for the treatment of nAMD. |
The similarity of biosimilar QL1207 and the reference aflibercept in patients with nAMD should be investigated in a phase 3 trial. |
What was learned from the study? |
In this randomized, double-blind, phase 3 trial, the difference in best-corrected visual acuity changes from baseline at week 12 (the primary endpoint) between the two groups was within the predefined equivalence margin. Secondary endpoints and safety profiles were also similar in the two groups. |
These results proved QL1207 has equivalent efficacy and similar safety profiles to aflibercept for nAMD. QL1207 is an option for nAMD. |
Introduction
Age-related macular degeneration (AMD) is one of major retinal diseases and a leading cause of blindness and visual impairment worldwide [1]. AMD may also increase the risk of cognitive impairment [2] and all-cause and cardiovascular mortality [3]. The number of people with AMD worldwide is estimated to be 288 million in 2040 [1]. Neovascular AMD (nAMD) is characterized by neovascularization. The pathogenesis of nAMD was not confirmed. However, it is considered to be associated with inflammation, oxidative stress, lipid metabolism, etc. [4]. Vascular endothelial growth factor (VEGF) also plays an important role in nAMD [5]. Thus, anti-VEGF treatment is now the standard treatment for nAMD [6,7,8].
Aflibercept (Eylea®, Bayer, Leverkusen, Germany; Regeneron, Tarrytown, NY, USA) is a fusion protein of VEGF receptor-1, -2, and Fc portion of immunoglobulin G [9]. The SIGHT study confirmed the superior efficacy of aflibercept over photodynamic therapy for nAMD [10]. The VIEW 1 and VIEW 2 studies indicated the similar efficacy and safety of aflibercept vs. anti-VEGF antibody ranibizumab in nAMD [11]. Aflibercept was recommended for nAMD treatment and approved in the USA, Europe, China, etc.
A biosimilar is a biological medicine that is highly similar to an existing approved biologic drug [12]. Biosimilars balance drug expenditure and efficacy, and are recognized in numerous countries [13]. QL1207 (Qilu pharmaceutical Co., Ltd., Jinan, China) is the first biosimilar of aflibercept in China, which contains a version of the active substance of the reference aflibercept. Preclinical studies indicated that QL1207 is comparable to the reference in terms of quality characteristics, in vitro and in vivo activity (data on file).
This phase 3 trial aimed to evaluate the equivalence of efficacy, similarity of safety, immunogenicity, and pharmacokinetics (PK) of QL1207 and aflibercept.
Methods
Study Design
This multicenter, randomized, double-blind, active-controlled, parallel-group phase 3 trial was conducted in 35 centers in China. The protocol (Supplementary Material) was approved by the ethics committee of each center (the approval number of the leading center Peking Union Medical College Hospital: KS2021031). The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All patients provided written informed consent before participation.
Eligibility Criteria
Key eligible criteria were (1) ≥ 50 years old, (2) with untreated subfoveal choroidal neovascularization (CNV) secondary to nAMD, (3) CNV comprising ≥ 50% of total lesion size, (4) total lesion area (including areas of CNV, blood, and scar) ≤ 12 disc areas, (5) best-corrected visual acuity (BCVA) letter score of 73–34 using Early Treatment Diabetic Retinopathy Study (ETDRS) charts in the study eye, and (6) no need to receive anti-VEGF treatment in the fellow eye. The key exclusion criteria were (1) subretinal or intraretinal hemorrhage comprising ≥ 50% of total lesion size or presence of subfoveal hemorrhage ≥ 1 disc area, (2) scar, fibrosis, atrophy that involved the center of the fovea, or subfoveal hard exudate, (3) CNV secondary to other reasons, (4) any ocular abnormality affecting visual acuity or macular examination in the study eye, and (5) previous intravitreal anti-VEGF or other nAMD treatment in the either eye. The full exclusion criteria were provided in Listing S1.
Eligibility was judged through medical history, ophthalmic and physical examinations, laboratory tests, optical coherence tomography (OCT), indocyanine green angiography (ICGA), fundus photography (FP), and fluorescein angiography (FA) before randomization during the screening period. OCT, ICGA, FP, and FA results were confirmed by the central reading centers.
Randomization, Masking, and Treatment
Patients were randomized 1:1 to receive intravitreal QL1207 or aflibercept 2 mg (0.05 ml) in the study eye every 4 weeks for the first 3 months, followed by 2 mg (0.05 ml) every 8 weeks until week 48 (eight doses in total). Randomization was stratified by BCVA ≥ or < 45 letters at baseline using Interactive Web Response System. Patients and assessors (including the central reading centers), but not investigators performing intravitreal injection, were blind to allocation.
Study Assessment
Patients were assessed every 4 weeks throughout week 52, using ETDRS chart for BCVA, intraocular pressure, slit lamp, and dilated fundus examination for safety by assessor. Central subfield retinal thickness (CRT; internal limiting membrane to retinal pigment epithelium) was evaluated using OCT by the central reading centers at weeks 4, 8, 12, 24, and 52, and by participating centers at the other timepoints. Treatment-emergent adverse events (TEAE), including TEAE in the study and fellow eyes, and non-ocular TEAE, were coded based on the Medical Dictionary for Regulatory Activities, version 24.1. FP and FA were performed at screening, weeks 12, 24, and 52. CNV leakage area was determined using FA by the central reading centers. Immunogenicity was evaluated by measuring the serum anti-drug antibody (ADA) before the first dose of study treatment and at weeks 4, 8, 12, 24, 36, and 52. ADA was analyzed based on electrochemiluminescence assay (Meso Scale Diagnostics, LLC, Rockville, MD, USA). Neutralizing antibody would be further tested using enzyme-linked immunosorbent assay if ADA is positive. Additional blood samples were obtained from patients volunteering for PK and VEGF analysis at the same time as ADA test and 4, 8, 12 h, 1, 2 days, and 1 and 2 weeks after the first dose of study treatment.
Study Endpoints
The primary endpoint was BCVA change from baseline at week 12. The secondary efficacy endpoints included CRT change from baseline at weeks 4, 8, 12, 24, and 52 and CNV leakage area change from baseline at weeks 12, 24, and 52. TEAE and immunogenicity were safety endpoints. Plasma drug concentration was PK endpoint. Plasma VEGF concentration was also analyzed.
Statistical Analysis
It is assumed that the difference in BCVA changes from baseline at week 12 between the two groups was 0. The pooled standard deviation (SD) of BCVA change from baseline at week 12 in the two groups was 11.6, calculated using meta-analysis of previous trials [11, 14,15,16,17]. Taking into account BCVA changes at week 12 for aflibercept and placebo in the previous studies [11, 14,15,16,17,18,19], and clinical experience, we set the equivalence margin at ± 5 in the present trial. A total of 282 (141 each group) patients were required for a two-sided significance level of 5% and a power of 90% with equivalence margin of ± 5. Considering a possible 20% dropout rate, a sample size of 354 (177 each group) patients were needed.
All statistical analyses were performed in SAS v9.4 (SAS Institute Inc., Cary, NC, USA). A two-sided P < 0.05 was considered statistically significant. The efficacy endpoints were evaluated in the full analysis set (FAS). The per-protocol set (PPS) was used for supplementary analysis. The FAS included all patients who received at least one dose of study drug and one efficacy assessment. The PPS included patients in the FAS without major protocol violation. Adverse events, immunogenicity, PK, and VEGF were summarized and analyzed in the safety set (SS), immunogenicity analysis set (IAS), pharmacokinetic analysis set (PKAS), and VEGF analysis set (VAS), including patients who received at least one dose of study drug and one corresponding post-baseline assessment.
Continuous data were presented as mean ± SD or median (interquartile range). Categorical data were presented as number (percentage). Continuous efficacy endpoints were analyzed using mixed model for repeated measures (MMRM). Missing data were assumed missing at random. Treatment group, visit, their interaction, and randomization strata were fixed effects. Within-subject variance–covariance structure of unstructured was used. If the model did not converge, the structure would be changed to compound symmetry. The results of MMRM were presented least-squares mean (LSM; standard error and/or 95% confidence intervals [CI]). For the primary endpoint, the equivalence margin was set at ± 5 letters. Predefined subgroup analysis on the primary endpoint was based on the randomization strata (BCVA ≥ or < 45 letters at baseline). Categorical efficacy endpoints were analyzed using Cochran–Mantel–Haenszel test adjusted by randomization strata.
Results
Patients
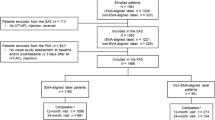
A total of 533 patients were screened from Aug 2019 to Jan 2022. A total of 366 were enrolled and randomly assigned to the QL1207 (n = 185) or aflibercept group (n = 181), all of whom were included in the FAS. The PPS included 319 patients (160 in the QL1207 and 159 in the aflibercept group). Finally, 321 patients (166 in the QL1207 and 155 in the aflibercept group) completed the study. The flowchart is shown in Fig. 1. The baseline characteristics were well balanced between the two groups in the FAS. The average ages were 67.4 ± 8.9 years in the QL1207 group and 67.1 ± 8.0 years in the aflibercept group. The mean BCVA were 56.1 ± 11.7 vs. 56.3 ± 11.8 letters, with 80.0% and 80.7% patient ≥ 45 letters. The mean CRT and CNV leakage areas were 428.2 ± 178.1 vs. 463.5 ± 193.3 μm and 3.99 ± 4.14 vs. 3.99 ± 4.13 mm2, respectively (Table 1).

Trial flow chart
Efficacy
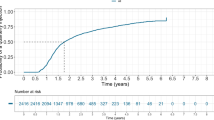
In the FAS, the mean BCVA changes from baseline at week 12 were 10.2 ± 8.8 letters in the QL1207 group and 11.0 ± 9.2 letters in the aflibercept group. The LSM changes in BCVA from baseline at week 12 were 10.4 (0.7) letters vs. 11.5 (0.7) letters in the two groups. The LSM difference was − 1.1 letters (95% CI − 3.0 to 0.7; P = 0.2275), within the equivalence margin of ± 5 (Fig. 2A and Table 2). Thus, the efficacy equivalence of QL1207 and the reference aflibercept is confirmed. Predefined subgroup analysis did not find significant differences in the primary endpoint between the two groups. The LSM differences were − 1.0 (95% CI − 2.9 to 1.0; P = 0.3292) and − 1.6 (95% CI − 6.8 to 3.6; P = 0.5441) in patients with BCVA ≥ and < 45 letters at baseline, respectively (Table 2). In the PPS, similar outcomes were achieved. The LSM difference of changes in BCVA at week 12 was also within the equivalence margin (− 1.2; 95% CI − 3.2 to 0.7; P = 0.2160; Fig. S1).

Least-squares mean (LSM) changes from baseline in A best-corrected visual acuity (BCVA), B central retinal thickness (CRT), and C choroidal neovascularization (CNV) leakage area in the full analysis set, analyzed using mixed model for repeated measures. Missing data were assumed missing at random. Treatment group, visit, their interaction, and randomization strata (BCVA ≥ or < 45 letters at baseline) were fixed effects. SE standard error, ETDRS Early Treatment Diabetic Retinopathy Study
For CRT, the LSM changes from baseline at week 52 were − 183.0 (12.1) μm vs. − 208.4 (12.4) μm, with an LSM difference of 25.4 μm (95% CI − 6.4 to 57.1, P = 0.1171). The LSM changes in CNV leakage area from baseline at week 52 were − 2.72 (0.26) vs. − 2.61 (0.26). The corresponding difference was − 0.11 mm2 (95% CI − 0.77 to 0.55, P = 0.7397). The changes at other timepoints were also similar in the two groups (Fig. 2B and C, and Table S1).
Safety
In total, 366 patients were included in the SS (185 in the QL1207 group and 181 in the aflibercept group). The times of study drug injection were similar between the two groups (7.5 ± 1.2 vs. 7.4 ± 1.4). The incidences of TEAE were comparable between the two groups (132 [71.4%] vs. 130 [71.8%]). TEAE in 11 (5.9%) and 15 (8.3%) patients were considered related to study drug in the two groups. Ocular TEAE in the study and fellow eyes, and non-ocular TEAE were reported in 52 (28.1%) vs. 54 (29.8%), 36 (19.5%) vs. 25 (13.8%), and 111 (60.0%) vs. 103 (56.9%) in the QL1207 and aflibercept groups, respectively. The most common ocular TEAE in the study eye were intraocular pressure increased (22 [11.9%] vs. 24 [13.3%]), conjunctival hemorrhage (11 [5.9%] vs. 11 [6.1%]), eye pain (5 [2.7%] vs. 3 [1.7%]), ocular hypertension (4 [2.2%] vs. 2 [1.1%]), cataract (3 [1.6%] vs. 3 [1.7%]), and conjunctivitis (3 [1.6%] vs. 3 [1.7%]). Most TEAE were mild or moderate. 12 (6.5%) and six (3.3%) patients experienced severe TEAE in the two groups. Serious TEAE were reported in 26 (14.1%) and 23 (12.7%) patients in the two groups, all of which were not related to study drugs. Ocular and non-ocular serious TEAE were also similar between the two groups. Two (1.1%) patients in the QL1207 group and four (2.2%) in the aflibercept group had treatment discontinuation due to TEAE. No TEAE leading to death was found (Table 3).
Immunogenicity and Pharmacokinetics
There were 365 patients in the IAS (184 for QL1207 and 181 for aflibercept). There were similar incidences of positive ADA between the two groups (13 [7.1%] in the QL1207 group and 14 [7.7%] in the aflibercept group). Positive Nab results were the same as those of ADA.
The PKAS and VAS included 54 (27 each) and 52 patients, respectively. Two patients had no blood sample for VEGF detection in the aflibercept group. Thus, they were excluded from the VAS. Plasma drug concentrations and PK parameters were comparable between the two groups, irrespective of free or bound state (Fig. S2 and Table S2). Further, similar VEGF levels were also detected in the two groups (Fig. S3).
Discussion
This is the first phase 3 trial of aflibercept biosimilar for nAMD. Anti-VEGF agents were the standard of care for nAMD recommended by guidelines [6,7,8]. Aflibercept, a fusion protein of VEGF receptor-1, -2 and Fc portion of immunoglobulin G, has high affinity to VEGF and potently blocks the VEGF-signaling pathway [9]. In the present study, the primary endpoint was met, indicating the efficacy equivalence of biosimilar QL1207 and the reference. Supplementary analysis in the PPS also supported this result. In addition, the similarity in safety profile, immunogenicity, and PK parameter was confirmed.
According to “Guidelines for Similarity Evaluation and Indication Extrapolation of Biosimilars” released by Center for Drug Evaluation of China National Medical Products Administration [20], the primary endpoint evaluation should be conducted before the efficacy plateau is reached, i.e., at a time point where differences between the two treatments could be best identified. In the case of aflibercept, which steeply increases BCVA during the first 4 months of treatment [10, 11], the highest sensitivity to detect differences in the primary endpoint BCVA would be within this time frame. Thus, BCVA change from baseline at week 12 was chosen as the primary endpoint to meet both regulatory and scientific needs. In the present study, after MMRM adjustment, the LSM changes in BCVA from baseline were 11.5 (0.7) letters at week 12 and 16.4 (0.9) at week 52 for aflibercept. In the SIGHT study [10], the mean BCVA change from baseline at week 12 was approximately 12 letters in the aflibercept group, and that at week 52 was 15.2 letters. In the integrated analysis of the VIEW 1 and VIEW 2 [11], corresponding results were approximately 7 and 8.4 letters, respectively. These results of aflibercept were consistent with those in the present study. At week 52, the LSM change in CRT from baseline in the present study for aflibercept (− 208.4 [12.4] μm) was comparable with the results previously reported (The SIGHT study: − 189.6 μm [10], The VIEW 1 study: − 129 μm, The VIEW 2 study: − 149 μm [11]). Due to different definitions and measurement methods, the changes in CNV leakage area or CNV lesion size cannot be compared directly among studies. However, the percentage reductions for aflibercept in the present study (leakage area: 65.4% [2.61/3.99]), SIGHT study (lesion size: 55.6% [1.007/1.812]) [10], VIEW 1 study (leakage area: 51.8% [3.4/6.57]), and VIEW 2 study (leakage area: 67.1% [5.2/7.75]) [11] were similar. The LSM changes in CRT and CNV leakage area at each timepoint in the QL1207 group were also comparable to aflibercept in this study.
The safety profile for aflibercept in this trial was consistent with previous data, with no new safety signal found [10, 11]. TEAE were also comparable between QL1207 and the reference. The most common ocular TEAE in the study eye were intraocular pressure increased, conjunctival hemorrhage, eye pain, ocular hypertension, cataract, and conjunctivitis. Most TEAE were mild or moderate, and not related to study drug. Only 14.1% patients in the QL1207 group and 12.7% in the aflibercept group suffered serious TEAE, all of which were non-study drug related. The incidences of treatment discontinuation were also low in the two groups (1.1% vs. 2.2%). Thus, QL1207 and aflibercept had similar safety profiles.
As proteins, immunogenic reaction of QL1207 and aflibercept was inevitable. It may lead to neutralization or hypersensitivity, affecting efficacy or safety [21]. Approximately 7% patients had ADA and Nab throughout week 52, indicating low and similar immunogenicity of the two drugs.
Previous studies demonstrated plasma free state concentrations at week 1 after single dose of intravitreal aflibercept 2 mg were 4.74–14.7 ng/ml [22]. The results of QL1207 and aflibercept in the present study were about 15 ng/ml. Systemic exposure to intravitreal QL1207 and aflibercept was dramatically lower than intravenous administration. Maximum drug concentrations (Cmax) of free and bound state QL1207 after single dose of intravitreal QL1207 2 mg were nearly 70 and 310 ng/ml, respectively, similar to aflibercept. In contrast, Cmax after intravenous administration of aflibercept 0.3 and 2 mg were 3.99 and 34.3 μg/ml in free state and 0.574 and 1.51 μg/ml in bound state [23]. It is suggested absorption of intravitreal anti-VEGF agent into systemic circulation is limited and not a safety concern.
There were two limitations of this study. Firstly, only Chinese patients were enrolled. Thus, caution should be paid when these results were interpreted for patients with other ethnicities. Secondly, OCT angiography (OCTA) was not implemented for CNV assessment because the use of OCTA in the clinical trial setting had not been validated during design of the present study. OCTA is a novel and noninvasive technique to show vasculature of the retina and choroid [24]. OCTA can be conducted as a part of multimodal imaging to provide more comprehensive data in future trials of nAMD.
Conclusions
This study indicated that QL1207 had potent efficacy, acceptable safety, immunogenicity, and PK profile similar to the aflibercept on the treatment of nAMD. The totality of evidence for QL1207 supports it is the first aflibercept biosimilar and an alternative option for patients with nAMD in China.
Data Availability
The datasets generated during and/or analyzed during the current study are not publicly available because we may further analyze the raw data for other research objectives.
References
Wong WL, Su X, Li X, Cheung CMG, Klein R, Cheng C-Y, Wong TY. Global prevalence of age-related macular degeneration and disease burden projection for 2020 and 2040: a systematic review and meta-analysis. Lancet Glob Health. 2014;2(2):e106–16. https://doi.org/10.1016/S2214-109X(13)70145-1.
Woo SJ, Park KH, Ahn J, et al. Cognitive impairment in age-related macular degeneration and geographic atrophy. Ophthalmology. 2012;119(10):2094–101. https://doi.org/10.1016/j.ophtha.2012.04.026.
Wang J, Xue Y, Thapa S, Wang L, Tang J, Ji K. Relation between age-related macular degeneration and cardiovascular events and mortality: a systematic review and meta-analysis. Biomed Res Int. 2016;2016:8212063. https://doi.org/10.1155/2016/8212063.
Ardeljan D, Chan C-C. Aging is not a disease: distinguishing age-related macular degeneration from aging. Prog Retin Eye Res. 2013;37:68–89. https://doi.org/10.1016/j.preteyeres.2013.07.003.
Mitchell P, Liew G, Gopinath B, Wong TY. Age-related macular degeneration. Lancet. 2018;392(10153):1147–59. https://doi.org/10.1016/S0140-6736(18)31550-2.
Schmidt-Erfurth U, Chong V, Loewenstein A, et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). Br J Ophthalmol. 2014;98(9):1144–67. https://doi.org/10.1136/bjophthalmol-2014-305702.
National Institute for Health and Care Excellence (NICE). NICE guideline [NG82]: age-related macular degeneration. Accessed May 25, 2022, https://www.nice.org.uk/guidance/ng82
American Academy of Ophthalmology Preferred Practice Pattern (PPP) Retina/Vitreous Committee. Age-Related Macular Degeneration PPP 2019. https://www.aao.org/preferred-practice-pattern/age-related-macular-degeneration-ppp
Holash J, Davis S, Papadopoulos N, et al. VEGF-Trap: a VEGF blocker with potent antitumor effects. Proc Natl Acad Sci USA. 2002;99(17):11393–8.
Li X, Chen Y, Zhang J, et al. Intravitreal aflibercept versus photodynamic therapy in Chinese patients with neovascular age-related macular degeneration: outcomes of the SIGHT study. J Ocul Pharmacol Ther. 2017;33(6):435–44. https://doi.org/10.1089/jop.2016.0071.
Heier JS, Brown DM, Chong V, et al. Intravitreal aflibercept (VEGF trap-eye) in wet age-related macular degeneration. Ophthalmology. 2012;119(12):2537–48. https://doi.org/10.1016/j.ophtha.2012.09.006.
Ishii-Watabe A, Kuwabara T. Biosimilarity assessment of biosimilar therapeutic monoclonal antibodies. Drug Metab Pharmacokinet. 2019;34(1):64–70. https://doi.org/10.1016/j.dmpk.2018.11.004.
Chandra A, Vanderpuye-Orgle J. Competition in the age of biosimilars. JAMA. 2015;314(3):225–6. https://doi.org/10.1001/jama.2015.6170.
Chang AA, Li H, Broadhead GK, Hong T, Schlub TE, Wijeyakumar W, Zhu M. Intravitreal aflibercept for treatment-resistant neovascular age-related macular degeneration. Ophthalmology. 2014;121(1):188–92. https://doi.org/10.1016/j.ophtha.2013.08.035.
Dugel PU, Jaffe GJ, Sallstig P, Warburton J, Weichselberger A, Wieland M, Singerman L. Brolucizumab versus aflibercept in participants with neovascular age-related macular degeneration: a randomized trial. Ophthalmology. 2017;124(9):1296–304. https://doi.org/10.1016/j.ophtha.2017.03.057.
Loewenstein A, Chang W, Chee C, et al. Evaluating the effect of ranibizumab and aflibercept on systemic vascular endothelial growth factor levels in neovascular age-related macular degeneration: the UNRAVEL study. Invest Ophthalmol Vis Sci. 2017;58(8):1198–1198.
Parvin P, Zola M, Dirani A, Ambresin A, Mantel I. Two-year outcome of an observe-and-plan regimen for neovascular age-related macular degeneration treated with Aflibercept. Graefes Arch Clin Exp Ophthalmol. 2017;255(11):2127–34. https://doi.org/10.1007/s00417-017-3762-2.
Rosenfeld PJ, Brown DM, Heier JS, Boyer DS, Kaiser PK, Chung CY, Kim RY. Ranibizumab for neovascular age-related macular degeneration. N Engl J Med. 2006;355(14):1419–31.
Regillo CD, Brown DM, Abraham P, Yue H, Ianchulev T, Schneider S, Shams N. Randomized, double-masked, sham-controlled trial of ranibizumab for neovascular age-related macular degeneration: PIER Study year 1. Am J Ophthalmol. 2008;145(2):239–48. https://doi.org/10.1016/j.ajo.2007.10.004.
Center for Drug Evaluation of China National Medical Products Administration (NMPA). Guideline for Similarity Evaluation and Indication Extrapolation of Biosimilars. Accessed Aug 10, 2023, https://www.cde.org.cn/main/news/viewInfoCommon/d92c6507a57bee9ccfc5baa1ee87fda9
Sauna ZE, Lagassé D, Pedras-Vasconcelos J, Golding B, Rosenberg AS. Evaluating and mitigating the immunogenicity of therapeutic proteins. Trends Biotechnol. 2018;36(10):1068–84. https://doi.org/10.1016/j.tibtech.2018.05.008.
Committee for Medicinal Products for Human Use (CHMP) of European Medicines Agency (EMA). Assessment report of aflibercept. 2015. Accessed May 25, 2022. https://www.ema.europa.eu/en/documents/variation-report/eylea-h-c-2392-ii-0021-epar-assessment-report-variation_en.pdf
Center for Drug Evaluation and Research (CDER) of the U.S. Food and Drug Administration (FDA). Clinical pharmacology and biopharmaceutics review(s) of aflibercept. 2012. Accessed May 25, 2022. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/125418Orig1s000ClinPharmR.pdf
Javed A, Khanna A, Palmer E, et al. Optical coherence tomography angiography: a review of the current literature. J Int Med Res. 2023;51(7):3000605231187933. https://doi.org/10.1177/03000605231187933.
Acknowledgements
The authors would like to thank all study participants.
Funding
This work was supported by Qilu Pharmaceutical Co., Ltd. (Jinan, China). The funding source also sponsored the Rapid Service Fee of the article.
Author information
Authors and Affiliations
Contributions
Jing Wang and Dr. Youxin Chen had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Concept and design: Hui Wang, Jing Wang, Feng Wang, and Youxin Chen. Acquisition, analysis, or interpretation of data: Bing Li, Ke Fan, Tonghe Zhang, Zhifeng Wu, Siming Zeng, Mingwei Zhao, Qian Ren, Dongping Zheng, Lifei Wang, Xiaoling Liu, Mei Han, Yanping Song, Jian Ye, Cheng Pei, Jinglin Yi, Xian Wang, Hui Peng, Hong Zhang, Zhanyu Zhou, Xiaoling Liang, Fangliang Yu, Miaoqin Wu, Chaopeng Li, Chunling Lei, Jilong Hao, Luosheng Tang, Huiping Yuan, Shanjun Cai, Qiuming Li, Jingxiang Zhong, Suyan Li, Lin Liu, Min Ke, Jing Wang, Hui Wang, Mengli Zhu, Zenghua Wang, Yang Yan, Feng Wang, and Youxin Chen. Drafting of the manuscript: Jing Wang and Mengli Zhu. Critical revision of the manuscript for important intellectual content: Bing Li, Ke Fan, Tonghe Zhang, Zhifeng Wu, Siming Zeng, Mingwei Zhao, Qian Ren, Dongping Zheng, Lifei Wang, Xiaoling Liu, Mei Han, Yanping Song, Jian Ye, Cheng Pei, Jinglin Yi, Xian Wang, Hui Peng, Hong Zhang, Zhanyu Zhou, Xiaoling Liang, Fangliang Yu, Miaoqin Wu, Chaopeng Li, Chunling Lei, Jilong Hao, Luosheng Tang, Huiping Yuan, Shanjun Cai, Qiuming Li, Jingxiang Zhong, Suyan Li, Lin Liu, Min Ke, Jing Wang, Hui Wang, Mengli Zhu, Zenghua Wang, Yang Yan, Feng Wang, and Youxin Chen. Statistical analysis: Yang Yan. Obtained funding: Hui Wang and Youxin Chen. Administrative, technical, or material support: Hui Wang, Jing Wang, Yang Yan, Zenghua Wang, and Mengli Zhu. Supervision: Feng Wang and Youxin Chen.
Corresponding authors
Ethics declarations
Conflict of Interest
Jing Wang, Hui Wang, Mengli Zhu, Zenghua Wang, and Yang Yan are employees of Qilu Pharmaceutical Co., Ltd. Bing Li, Ke Fan, Tonghe Zhang, Zhifeng Wu, Siming Zeng, Mingwei Zhao, Qian Ren, Dongping Zheng, Lifei Wang, Xiaoling Liu, Mei Han, Yanping Song, Jian Ye, Cheng Pei, Jinglin Yi, Xian Wang, Hui Peng, Hong Zhang, Zhanyu Zhou, Xiaoling Liang, Fangliang Yu, Miaoqin Wu, Chaopeng Li, Chunling Lei, Jilong Hao, Luosheng Tang, Huiping Yuan, Shanjun Cai, Qiuming Li, Jingxiang Zhong, Suyan Li, Lin Liu, Min Ke, Feng Wang, and Youxin Chen declare no competing interests.
Ethical Approval
The study protocol was approved by the ethics committee in each center (the approval number of the leading center Peking Union Medical College Hospital: KS2021031). The study was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice. All patients provided written informed consent before participation.
Additional information
Prior Presentation: Previously presented as an oral presentation at the World Ophthalmology Congress 2022 (Virtual, September 9–12).
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Li, B., Fan, K., Zhang, T. et al. Efficacy and Safety of Biosimilar QL1207 vs. the Reference Aflibercept for Patients with Neovascular Age-Related Macular Degeneration: A Randomized Phase 3 Trial. Ophthalmol Ther 13, 353–366 (2024). https://doi.org/10.1007/s40123-023-00836-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-023-00836-4