
Abstract
Introduction
Scientific evidence of the effectiveness of the tumor necrosis factor inhibitor adalimumab (ADA) in pediatric patients with non-infectious non-anterior uveitis is still limited. The aim of this study is to investigate the therapeutic role of ADA in a cohort of pediatric patients with non-anterior uveitis.
Methods
This is an international multicenter study analyzing real-life data referred to pediatric patients treated with ADA for intermediate uveitis/pars planitis, posterior uveitis and panuveitis. Data were drawn from the AutoInflammatory Disease Alliance (AIDA) registry for patients with uveitis.
Results
Twenty-one patients (36 affected eyes) were enrolled, and all patients benefited from ADA administration. In detail, 11 patients (19 affected eyes) did not experience further ocular inflammation after ADA introduction; 10 cases (17 affected eyes) showed a significant clinical improvement consisting of a decrease in severity and/or frequency of ocular relapses. The number of ocular flares dropped from 3.91 to 1.1 events/patient/year after ADA introduction (p = 0.0009); macular edema and retinal vasculitis were respectively observed in 18 eyes and 20 eyes at the start of ADA and in 4 eyes and 2 eyes at the last assessment. The mean daily glucocorticoid dosage significantly decreased from 26.8 ± 16.8 mg/day at the start of ADA to 6.25 ± 6.35 mg/day at the last assessment (p = 0.002). Intermediate uveitis/pars planitis (p = 0.01) and posterior uveitis (p = 0.03) were more frequently observed in patients with full response to ADA; panuveitis (p = 0.001) was significantly more frequent among patients continuing to experience uveitic flares. This could be related to a higher use of systemic glucocorticoids (p = 0.002) and conventional immunosuppressants (p = 0.007) at the start of ADA when treating intermediate uveitis/pars planitis. Regarding the safety profile, only one adverse event was reported during ADA treatment, consisting of the development of generalized adenopathy.
Conclusions
ADA proved to have an effective therapeutic role in all pediatric patients with non-anterior uveitis enrolled in the study. An overall glucocorticoid-sparing effect was observed despite the severity of cases enrolled. A more aggressive treatment of panuveitis and posterior uveitis at start of ADA could increase the likelihood of full response to therapy.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
To further enlarge evidence on the effectiveness of adalimumab in a multi-center cohort of pediatric patients with non-infectious non-anterior uveitis. |
What was learned from the study? |
Adalimumab is highly effective in completely controlling or reducing the frequency and/or severity of ocular flares in pediatric patients with non-infectious non-anterior uveitis. |
A glucocorticoid-sparing effect may be observed even when resistant and/or severe cases are treated with adalimumab. |
A more aggressive treatment at the start of adalimumab could increase the likelihood of obtaining a full response when dealing with posterior uveitis and panuveitis. |
Introduction
Non-infectious uveitis represents a group of immune-mediated diseases characterized by intraocular inflammation primarily involving the uveal tract [1]. Uveitis is rare in children, accounting for 5–10% of all patients with this condition; however, it has a significant impact on patients’ quality of life, as visual function may be severely affected by intraocular inflammation and uveitis-related complications including cataract, glaucoma, amblyopia and cystoid macular edema. Retinal vasculitis may also represent a serious inflammatory manifestation of patients with uveitis [2, 3].
Uveitis in children often involves the anterior segment of the eye, as generally observed in the context of juvenile idiopathic arthritis (JIA) [4, 5]. Nevertheless, intermediate uveitis/pars planitis, posterior uveitis and panuveitis may also occur as either an idiopathic entity or a manifestation of an early-onset Behçet’s disease, or in connective tissue diseases, inflammatory bowel diseases, Vogt-Koyanagi-Harada disease, sarcoidosis and Blau syndrome [5,6,7]. These conditions are rarer than JIA in children, with a subsequent lower amount of scientific evidence about the most proper therapeutic management.
Generally, the goal of treatment of non-anterior uveitis is to suppress active intraocular inflammation and decrease the risk of eye complications. Local and/or systemic glucocorticoids often represent the first line of treatment. However, glucocorticoids may favor the development of ocular complications and even induce retarded growth and development in children in addition to other metabolic or endocrine side effects. To allow a rapid decrease in the corticosteroid dosage and avoid steroid-associated side effects, conventional disease-modifying anti-rheumatic drugs (cDMARDs) including methotrexate, mycophenolate mofetil, azathioprine and cyclosporine are used in chronic or recurrent cases [5].
During the last decade, biotechnologic agents, especially tumor necrosis factor (TNF) inhibitors, have increasingly emerged as useful therapeutic approaches in pediatric patients with resistant or severe uveitis [5, 8, 9]. The fully humanized monoclonal antibody adalimumab (ADA) is the only biotechnologic agent currently approved by the US Food and Drug Administration and the European Medicines Agency for the treatment of adult non-infectious non-anterior uveitis [10, 11].
Despite recent and ongoing clinical trials, non-infectious uveitis treatment is already based on experts’ opinion and algorithms proposed by multidisciplinary panels. In this context, on-line international registries dedicated to patients with uveitis may facilitate the acquisition of new strong evidence from real-life experience and may also facilitate enrolling in randomized clinical trials [12, 13]. In this perspective, the AutoInflammatory Disease Alliance (AIDA) has supported the development of international registries dedicated to uveitis, scleritis and Behçet’s disease [12, 14, 15]. Based on data currently accrued in the AIDA registry dedicated to uveitis, we have conducted a study aimed at further enlarging the current evidence about effectiveness of ADA in pediatric patients with intermediate uveitis/pars planitis, posterior uveitis and panuveitis.
Methods
Study Design
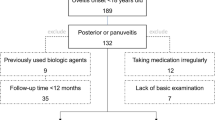
This is a multicenter international study enrolling pediatric patients with non-infectious non-anterior uveitis treated with ADA. Data of patients were drawn from the AIDA international registry specifically dedicated to uveitis [14]. All patients with pediatric-onset, non-infectious, non-anterior uveitis were included in the study when ADA was started before the age of 16 years and after having obtained the consent from patients and the informed consent from parents (or the legal guardians). Patients with anterior uveitis were excluded, along with those with a treatment duration < 3 months. Figure 1 summarizes the flow leading to the selection of patients on October 1, 2022.

Study flow diagram illustrating the process leading to the selection of patients included in the present study among all patients recruited in the International AutoInflammatory Disease Alliance (AIDA) Registry dedicated to uveitis until October 1, 2022. Abbreviations: ADA adalimumab, AIDA AutoInflammatory Disease Alliance, NIU non-infectious uveitis
Medical records of 21 pediatric patients from six centers involved in the AIDA Network were reviewed from the retrospective phase of the AIDA project. The following demographic, clinical and therapeutic data were collected at the start of ADA and at each subsequent follow-up assessment (every 3 months or more frequently if clinically needed): age, gender, age at disease onset, disease duration, concomitant systemic diseases, features of uveitis, concomitant and previous treatments, dosages of glucocorticoids and cDMARDs used, ADA treatment duration, use of originator or biosimilar ADA, the specific ADA biosimilar, eventual ADA discontinuation, ocular relapses before and after ADA treatment and best corrected visual acuity (BCVA) assessed in decimal fractions.
Uveitis had been diagnosed by uveitis-trained ophthalmologists, who have also filled in the instruments of the AIDA registry dedicated to uveitis. Ophthalmologists were required to provide details about the anatomical and pathogenic classification of uveitis, the type of presentation and ocular disease course according to the standardized uveitis nomenclature (SUN) working group. Ophthalmologists were also required to provide BCVA values and details about the local treatment at the start of treatment and at the subsequent assessments [16]. Ophthalmologists had to specify the presence of macular edema, retinal vasculitis and any ocular complication induced by inflammation and/or glucocorticoid treatment.
The choice of ADA was at the discretion of the patient’s physician. All patients had undergone a full hematologic and urinary screening, chest x-ray and abdominal ultrasound to rule out infectious or neoplastic diseases; all specific tests required to identify systemic diseases associated with uveitis were also performed according to the clinical picture. Patients with pars planitis and/or retinal vasculitis underwent brain magnetic resonance imaging to exclude multiple sclerosis.
The response to ADA was interpreted and judged by uveitis-trained ophthalmologists according to complete ophthalmologic examination including ocular fundus evaluation, optical coherence tomography (OCT) evaluation, autofluorescence and retinal angiography.
An ocular flare was defined as a worsening of inflammatory activity after a period of inactive disease, as classified in the SUN working group nomenclature and according with the standardization of vitreal inflammatory activity proposed by Nussenblatt et al. [16, 17]. In case of ocular relapse, data recruited referred to findings observed before starting corticosteroid treatment adjustments. Patients were considered “fully responsive” when no ocular relapses occurred after ADA introduction; patients were considered “less responsive” when inflammation occurred in at least one eye, despite the decrease in the number of flares and/or of inflammation. Patients showing no decrease in the frequency and/or severity of ocular inflammation were considered “unresponsive.”
Aims and Endpoints
The primary aim of the study was to investigate the efficacy and safety of ADA in a pediatric cohort of patients affected by non-infectious non-anterior uveitis. Secondary aims were: (1) to quantify the frequency of ocular flare-ups during the first 12 months of ADA and to compare it with the frequency observed during the previous 12 months; (2) to evaluate how BCVA changed after ADA treatment; (3) to assess any glucocorticoid sparing effect; (4) to assess the frequency of macular edema and retinal vasculitis at the start of ADA and thereafter; (5) to find differences in the clinical presentation of patients fully responsive to ADA and those with a partial response.
The primary endpoint was represented by the frequency of patients with a complete and persistent control of ocular inflammation, those with a decrease in severity and/or frequency of ocular flares and those with no improvement after ADA introduction. The secondary endpoints were: (1) a statistically significant difference in the frequency of ocular flares (standardized as number of events/patient/year) during the 12 months preceding and following the start of ADA; (2) to assess any significant change in the BCVA value from the start of ADA to the last assessment while on ADA treatment; (3) to identify any decrease in the number of patients administered with systemic glucocorticoids and in the daily dosage (prednisone or equivalent); (4) to describe the frequency of macular edema (according to clinical and OCT assessments) and retinal vasculitis (identified as a vascular leakage at fluorescein angiography) at the start of ADA and at the last assessment while on ADA treatment; (5) to identify statistically significant differences in the demographic features and in the baseline features of uveitis between patients with full and persistent control of ocular inflammation after ADA introduction and those with a partial or a failing response.
Ethics
As previously described for the AIDA registry dedicated to uveitis [12], this study has been approved by the Ethics Committee of Azienda Ospedaliera Universitaria Senese, Siena, Italy (ref. no. 14,951) and was conducted in accordance with the recommendations by the Declaration of Helsinki and subsequent updates. All patients provided their assent; the informed consent was obtained from the parents (or by the legal guardian).
Statistical Analysis
Descriptive statistics with computation of samples sizes, percentages, means, interquartile ranges (IQR) and standard deviations were carried out. Qualitative data were analyzed with Fisher exact test and 2 × 2 and 2 × 3 contingency tables to search for differences between groups. Pairwise comparisons of quantitative data were performed by using Student’s two-tailed t-test or Mann-Whitney two-tailed U-test or Wilcoxon test, as appropriate, after having assessed normality distribution with the Shapiro-Wilk test. The threshold for statistical significance was set at 95% (p < 0.05). Bonferroni correction was used for post hoc analysis; in this case, statistical significance required p < 0.016. Statistical computations were performed with STATA 17/MP2 (StataCorp. 2021. Stata Statistical Software: Release 17. College Station, TX: StataCorp LLC).
Results
Twenty-one patients (36 affected eyes) were recruited in the study as suffering from non-anterior uveitis treated with the anti-TNF agent ADA. Figure 1 summarizes the flow diagram leading to the selection of patients included in this study; Table 1 provides information about demographics and clinical features of patients enrolled; Table 2 describes features of the 36 eyes involved with uveitis.
The mean duration of the ADA treatment was 30.47 ± 26.01 (range 3–125) months. ADA was the first biologic agent employed in all patients enrolled. A biosimilar molecule was used in ten cases since the start of treatment, while ADA originator was used in all the other cases with no switches.
In 19 patients (34 affected eyes), biologic therapy with ADA was started for the active ocular disease, while a systemic active disease also occurred in the other 2 cases (2 eyes). In all cases, clinical efficacy was observed as soon as the 3-months follow-up assessment.
As a whole, 11 subjects (19 affected eyes) did not experience further ocular inflammation after the start of ADA treatment and were considered fully responsive patients. Ten cases (17 affected eyes) showed a significant clinical improvement as follows: 4 patients experienced a reduced frequency and severity of relapses; 3 patients showed reduced frequency of relapses and 3 patients a reduced severity of relapses. These subjects were included in the group of less responsive patients, while no patients were unresponsive to treatment. Less responsive patients with bilateral involvement variably experienced inflammation in both eyes during the study period. Table 3 highlights differences of ocular features between patients with no further flares after ADA introduction (group 1) and patients with no complete control of relapses (group 2). No significant differences were observed in the use of biosimilars between the two patient groups (more precisely: biosimilars were used in 6 patients in group 1 and 4 patients in group 2, p = 0.52).
In total, 94 relapses had been recorded during the 12 months preceding the start of ADA, corresponding to 3.91 ocular flares/patient/year (range 1–10 flares/patient/year); the total number of relapses occurring during ADA treatment was 33, corresponding to 1.1 flares/patient/year (p = 0.0009).
In two patients (4 affected eyes), ADA treatment was discontinued: the first patient experienced a loss of efficacy on ocular manifestations after 44 months of treatment; the second patient discontinued ADA for non-medical reasons (not specified). One further patient discontinued ADA because of long-term remission after 24 months of treatment; nevertheless, he experienced ocular disease reactivation after nearly 12 months, with the subsequent need to resume ADA treatment, which proved to be effective again.
The BCVA was available for 12 patients (22 eyes) at the start of ADA, and its mean value was 0.625 ± 0.35. The mean BCVA value was 0.84 ± 0.23 at the last assessment (p = 0.21). The improvement of BCVA was observed in 8 eyes, while a decrease was recorded in 2 eyes.
At the start of treatment macular edema and retinal vasculitis were observed in 18 eyes and 20 eyes, respectively. At the last assessment macular edema was found in 4 eyes and retinal vasculitis in 2 eyes.
Table 4 summarizes data concerning other concomitant treatments (glucocorticoids and cDMARDs) used at the start of ADA and at the last follow-up assessment.
At the start of ADA, 14 patients were treated with glucocorticoids and 16 with concomitant cDMARDs; at the last follow-up, 8 patients were administered concomitant glucocorticoids and 15 patients concomitant cDMARDs. Among partially responsive patients, cDMARDs were used in 9/11 (81.8%) cases at the start of ADA and 8.0 (IQR: 9) mg/day at the last assessment.
Non-statistically significant decrease was observed in the number of patients requiring glucocorticoids at the last assessment compared with what was observed at the start of ADA (p = 0.12). Conversely, the mean daily glucocorticoid dosage significantly decreased among patients already treated with steroids during the study period (from 26.8 ± 16.8 mg/day at the start of ADA to 6.25 ± 6.35 mg/day at the last assessment, p = 0.002). Among partially responsive patients, the mean daily glucocorticoids passed from 21.7 (IQR: 35) mg/day at the start of ADA to in 7/11 (63.6%) cases at the last assessment.
Regarding cDMARD therapy, the results did not show a significant change in the frequency of patients requiring a concomitant cDMARD at the start of ADA compared with what observed at the last assessment.
At the start of treatment, ADA was administered subcutaneously at a dose of 24 mg/m2 up to the maximum dose of 40 mg every 2 weeks in all cases. Posology changes were performed in 2 patients during the study period. The first patient decreased the frequency of administration from 40 mg every other week to 40 mg every 21 days because of long-term clinical efficacy (after 99 months of ADA treatment). The clinical efficacy was maintained after posology adjustment. The second patient decreased the frequency of administration from 20 mg every other week to 20 mg every 21 days because of long-term clinical efficacy (after 11 months of ADA treatment). The clinical benefit persisted, and a second decrease from 20 mg every 21 days to 20 mg every 28 days was performed after a further 6 months. An ocular exacerbation occurred within 3 months, and an increase of ADA administrations every other week was required to resume disease control.
In our cohort, during the study period 2 patients (3 eyes with panuveitis) were treated with peribulbar/intravitreal injections of glucocorticoids, and 1 patient (1 eye with panuveitis) was treated with intravitreal injection of ranibizumab; in 3 patients phacoemulsification surgery was also performed. These therapeutic practices were performed as soon as the start of ADA. Table 5 provides information about systemic treatments performed on eyes involved with uveitis at the start and at the last assessment while on ADA treatment.
Regarding safety, an adverse event was reported in one patient during the study period, consisting of a generalized adenopathy developed during the course of ADA treatment.
Discussion
At current, ADA is the only biotechnologic agent approved by the US Food and Drug Administration (FDA) for the treatment of non-infectious uveitis in adults [10, 11, 18, 19]. The use of this biologic agent is also licensed for other chronic diseases of childhood, including JIA. In this context, ADA has shown benefit in JIA-associated anterior uveitis [8, 20,21,22], while very few data are available regarding the role of ADA in management of non-anterior non-infectious uveitis. Therefore, although based on a limited number of patients, the present study widens the current evidence about the effectiveness of ADA in controlling uveitis in the pediatric scenario.
In particular, our results confirm that ADA treatment is highly effective in either avoiding or reducing ocular inflammatory activity in patients with refractory and severe uveitis. Indeed, the majority of patients did not experience any ocular flare after the start of ADA during a median follow-up of 17.5 months; the other patients experienced a decrease in the frequency and/or in severity of ocular inflammatory episodes. As a whole, none of the patients enrolled experienced a lack of response to ADA. This is even more remarkable when considering the severity of patients included in the study. In fact, many patients showed ocular inflammatory complications as soon as the start of ADA, and the mean BCVA value was quite low when this biologic agent was introduced. In the same way, many eyes were burdened by macular edema and retinal vasculitis at the beginning of treatment.
Most patients included in the present study were diagnosed with idiopathic uveitis, while seven patients had an associated systemic immune-mediated systemic disorder. On this basis, our data support the use of ADA in idiopathic cases of non-anterior uveitis and further enlarge the current literature about the effective role of ADA in controlling uveitis in the context of pediatric Behçet’s disease, Vogt-Koyanagi-Harada disease, inflammatory bowel diseases and scleroderma [23,24,25]. Of note, a systemic disease related to non-anterior uveitis occurred in a similar proportion in both groups. Therefore, the existence of a systemic disease did not seem to represent a predictor of poor response in terms of occurrence of non-anterior uveitis relapses when patients were treated with ADA.
Despite the poor visual acuity at the start of ADA, BCVA improved in many eyes during follow-up, and only two eyes underwent a further sight worsening. Definitely, the BCVA value was maintained during ADA treatment. This result conflicts with that of Kouwenberg et al. [26]. These authors assessed the role of ADA in 28 patients with non-anterior uveitis observing a statistically significant improvement at 9 and 24 months from the start of treatment. No statistically significant improvement was observed in the change of BCVA in our cohort of patients, and this could be related to the severity of subjects enrolled, the high number of ocular complications already developed at the start of ADA and the inclusion of patients with severe ocular comorbidities, as for one child with X-linked retinoschisis (mutated CRB1 gene).
About one half of patients were treated with a biosimilar ADA agent. The good response to treatment with lack of difference in the frequency of full and partial ocular disease control further supports the effectiveness of biosimilar agents also in the context of pediatric non-anterior uveitis, as previously described for pediatric rheumatic diseases and for adult patients with uveitis [27, 28].
Of note, ADA proved to be effective also in controlling retinal vasculitis and macular edema in non-anterior pediatric uveitis. Based on a relatively wide number of patients, these findings resemble similar results obtained with ADA in five children with Behçet’s disease associated retinal vasculitis and point out the effective role of ADA in pediatric patients with macular edema associated with non-infectious uveitis [24].
As also observed in adult patients and in JIA patients in the era of biotechnologic treatment [22, 29], significant glucocorticoid sparing has been observed during follow-up despite the severity of cases. In particular, the mean daily glucocorticoids dosage was reduced to about one third of the starting dosage. Of note, the number of patients requiring glucocorticoids did not decrease in a statistically significant fashion, but their mean dosage was significantly spared among patients already requiring this concomitant treatment. In the same way, the number of patients concomitantly treated with glucocorticoid eye drops significantly decreased, and only one patient underwent glucocorticoid eye drops at the last assessment. Sparing glucocorticoids is even more essential in pediatric patients to avoid iatrogenic ocular and systemic side effects. Indeed, systemic and local corticosteroids may lead to eye structural complications as severe as those induced by the inflammation itself. In this regard, ocular hypertension, cataract and consequent amblyopia frequently occur when glucocorticoids cannot be spared [5]. Regarding the cDMARD-sparing effect, patients included in the present study did not discontinue concomitant immunosuppressants. This is probably related to severity of the uveitic disease and the need for maintaining a high immunosuppressive load to avoid ocular flares in patients already burdened by multiple ocular complications at the start of ADA.
Looking at the safety profile, ADA proved to be a safe treatment, with generalized lymphadenopathy being the only adverse event retrospectively reported. Of note, a possible reactivation of uveitis after discontinuation of anti-TNF therapy occurred in one patient discontinuing ADA because of long-term remission. This confirms the possible reactivation of non-anterior uveitis even in patients with a prompt and complete response, as previously reported in the literature for patients with non-infectious uveitis undergoing withdrawal of anti-TNF agents, especially ADA [30]. As a whole, only one patient discontinued ADA because of effectiveness after a long period of optimal response. This is different from the report by Kouwenberg et al. [26], who highlighted the suspension of ADA because of ineffectiveness in about one third of patients with non-JIA uveitis during a similar period of observation. While the reason for this difference is not clear, our results further support the therapeutic role of ADA even in the most severe cases of non-anterior uveitis.
Notably, Table 3 points out the differences between patients experiencing a full ocular disease control and those continuing to experience less severe and/or less frequent ocular flares. In particular, intermediate uveitis/pars planitis and posterior uveitis proved to be more frequent among patients with a full response to ADA, while panuveitis was more frequent among patients continuing to experience flares despite the improvement in the frequency and/or severity of inflammatory episodes. However, this could be related to a more aggressive treatment at the start of ADA introduction, as observed in Table 5. Indeed, both cDMARDs and systemic corticosteroids were more frequently employed on eyes with pars planitis/intermediate uveitis than on eyes with panuveitis or posterior uveitis. These differences lost statistical significance during follow-up. These findings seem to suggest that a more aggressive treatment of panuveitis and posterior uveitis at start of ADA could increase the likelihood of fully respond to therapy.
Subjects with a recurrent course and non-granulomatous uveitis seemed to be less prone to full control of ocular flares. However, the presence of unknown data, especially interesting the pathogenic classification, prompts us not to consider these statistically significant differences, inviting the scientific community to further investigate these issues in future studies. In the same way, patients with earlier disease onset seemed to belong more frequently to the group of patients with incomplete control. This last observation was not supported by statistical significance and deserves to be studied on a much larger number of patients.
The main limitation of the study is represented by the relatively limited number of patients. Other flaws consist of the etiologic variety of the cases and the frequent use of combination therapy between ADA and cDMARDs. However, uveitis is a quite rare condition, especially in the pediatric context, and collecting wide numbers of patients is not trivial even in the context of an international registry. Regarding the frequent use of combination therapy, patients included in this study are particularly severe, and treatment with both adalimumab and cDMARDs was required to control the ocular disease. However, the combination therapy was able to control disease manifestations and obtain good ophthalmologic results even in an unfavorable condition. In addition, as combination therapy did not change during follow-up, this did not account for a confounding factor in assessing ADA's therapeutic role. In a few cases, retrospectively collected data were lacking, especially in relation to the description of the uveitis features, as for some variables in Table 3. However, these lacking data tend to be missing in a random manner, so they did not influence statistical analysis in advance. Despite limitations, this study substantially enlarges the current literature on the role of the anti-TNF agent ADA in pediatric patients with severe and/or resistant non-anterior uveitis.
Conclusion
In conclusion, ADA was often used in combination with cDMARDs because of the severe clinical picture; however, the treatment proved to be an effective agent in all pediatric patients with non-anterior uveitis consecutively enrolled in this study. In particular, ADA introduction led to a full control of ocular inflammation in about half of the patients, with the other patients experiencing a reduction in the severity and/or frequency of ocular flares. ADA allowed a significant overall glucocorticoid-sparing effect despite the severity of most cases enrolled. A more aggressive treatment at the start of ADA could enhance the likelihood to reach full response when dealing with posterior uveitis and panuveitis.
References
Hoy SM. Adalimumab: a review in non-infectious non-anterior uveitis. BioDrugs. 2017;31(2):135–42. https://doi.org/10.1007/s40259-017-0213-x.
Edelsten C, Lee V, Bentley CR, Kanski JJ, Graham EM. An evaluation of baseline risk factors predicting severity in juvenile idiopathic arthritis associated uveitis and other chronic anterior uveitis in early childhood. Br J Ophthalmol. 2002;86(1):51–6. https://doi.org/10.1136/bjo.86.1.51.
Fabiani C, Vitale A, Orlando I, et al. Impact of uveitis on quality of life: a prospective study from a tertiary referral rheumatology-ophthalmology collaborative uveitis center in Italy. Isr Med Assoc J. 2017;19(8):478–83.
Abdwani R. Challenges of childhood uveitis. Sultan Qaboos Univ Med J. 2009;9(3):247–56.
Maleki A, Anesi SD, Look-Why S, Manhapra A, Foster CS. Pediatric uveitis: a comprehensive review. Surv Ophthalmol. 2022;67(2):510–29. https://doi.org/10.1016/j.survophthal.2021.06.006.
Sardar E, Dusser P, Rousseau A, Bodaghi B, Labetoulle M, Koné-Paut I. Retrospective study evaluating treatment decisions and outcomes of childhood uveitis not associated with juvenile idiopathic arthritis. J Pediatr. 2017;186:131–7. https://doi.org/10.1016/j.jpeds.2017.03.052. (e1).
Kahwage PP, Ferriani MP, Furtado JM, et al. Uveitis in childhood-onset systemic lupus erythematosus patients: a multicenter survey. Clin Rheumatol. 2017;36(3):547–53. https://doi.org/10.1007/s10067-016-3534-0.
Lerman MA, Burnham JM, Chang PY, et al. Response of pediatric uveitis to tumor necrosis factor-α inhibitors. J Rheumatol. 2013;40(8):1394–403. https://doi.org/10.3899/jrheum.121180.
Sood AB, Angeles-Han ST. An update on treatment of pediatric chronic non-infectious uveitis. Curr Treatm Opt Rheumatol. 2017;3(1):1–16. https://doi.org/10.1007/s40674-017-0057-z.
Nguyen QD, Merrill PT, Jaffe GJ, et al. Adalimumab for prevention of uveitic flare in patients with inactive non-infectious uveitis controlled by corticosteroids (VISUAL II): a multicentre, double-masked, randomised, placebo-controlled phase 3 trial. Lancet. 2016;388(10050):1183–92. https://doi.org/10.1016/S0140-6736(16)31339-3.
Suhler EB, Adán A, Brézin AP, et al. Safety and efficacy of adalimumab in patients with noninfectious uveitis in an ongoing open-label study: VISUAL III. Ophthalmology. 2018;125(7):1075–87. https://doi.org/10.1016/j.ophtha.2017.12.039.
Della Casa F, Vitale A, Guerriero S, et al. Autoinflammatory Diseases Alliance (AIDA) Network. Development and implementation of the AIDA international registry for patients with non-infectious uveitis. Ophthalmol Ther. 2022;11(2):899–911. https://doi.org/10.1007/s40123-022-00459-1.
Zhang Z, Ng Ming Sheng S, Kempen JH, et al. Uveitis registries—a digital tool for patient care, education, research, and collaboration. Ocul Immunol Inflamm. 2022;3:1–11. https://doi.org/10.1080/09273948.2022.2140062.
Della Casa F, Vitale A, Pereira RM, et al. Development and implementation of the AIDA international registry for patients with non-infectious scleritis. Ophthalmol Ther. 2022;11(2):887–97. https://doi.org/10.1007/s40123-022-00466-2.
Vitale A, Della-Casa F, Ragab G, et al. Autoinflammatory Diseases Alliance (AIDA) Network. Development and implementation of the AIDA international registry for patients with Behçet’s disease. Intern Emerg Med. 2022;17(7):1977–86. https://doi.org/10.1007/s11739-022-03038-1.
Jabs DA, Nussenblatt RB, Rosenbaum JT, Standardization of Uveitis Nomenclature (SUN) Working Group. Standardization of uveitis nomenclature for reporting clinical data. Results of the First International Workshop. Am J Ophthalmol. 2005;140(3):509–16. https://doi.org/10.1016/j.ajo.2005.03.057.
Nussenblatt RB, Palestine AG, Chan CC, Roberge F. Standardization of vitreal inflammatory activity in intermediate and posterior uveitis. Ophthalmology. 1985;92(4):467–71. https://doi.org/10.1016/s0161-6420(85)34001-0.
Díaz-Llopis M, Salom D, Garcia-de-Vicuña C, et al. Treatment of refractory uveitis with adalimumab: a prospective multicenter study of 131 patients. Ophthalmology. 2012;119(8):1575–81. https://doi.org/10.1016/j.ophtha.2012.02.018.
Jari M, Shiari R, Salehpour O, Rahmani K. Epidemiological and advanced therapeutic approaches to treatment of uveitis in pediatric rheumatic diseases: a systematic review and meta-analysis. Orphanet J Rare Dis. 2020;15(1):41. https://doi.org/10.1186/s13023-020-1324-x.
Ramanan AV, Dick AD, Jones AP, et al. SYCAMORE Study Group. Adalimumab plus methotrexate for uveitis in juvenile idiopathic arthritis. N Engl J Med. 2017;376(17):1637–46. https://doi.org/10.1056/NEJMoa1614160.
Cecchin V, Zannin ME, Ferrari D, et al. Longterm safety and efficacy of adalimumab and infliximab for uveitis associated with juvenile idiopathic arthritis. J Rheumatol. 2018;45(8):1167–72. https://doi.org/10.3899/jrheum.171006.
Del Giudice E, Simio C, Scala A, et al. Juvenile idiopathic arthritis-associated uveitis in the era of biological therapy: how the disease changed in more than 20 years of observation in a tertiary referral center in Rome (Italy). Int Ophthalmol. 2022;42(3):775–84. https://doi.org/10.1007/s10792-021-02043-1.
Jeroudi A, Angeles-Han ST, Yeh S. Efficacy of adalimumab for pediatric Vogt-Koyanagi-Harada syndrome. Ophthalmic Surg Lasers Imaging Retina. 2014;45(4):332–4. https://doi.org/10.3928/23258160-20140709-09.
Ho M, Chen LJ, Sin HPY, et al. Experience of using adalimumab in treating sight-threatening paediatric or adolescent Behcet’s disease-related uveitis. J Ophthalmic Inflamm Infect. 2019;9(1):14. https://doi.org/10.1186/s12348-019-0181-z.
Hu YC, Yang YH, Lin YT, et al. Clinical manifestations and anti-TNF alpha therapy of juvenile Behçet’s disease in Taiwan. BMC Pediatr. 2019;19(1):232. https://doi.org/10.1186/s12887-019-1613-5.
Kouwenberg CV, Koopman-KalininaAyuso V, de Boer JH. Clinical benefits and potential risks of adalimumab in non-JIA chronic paediatric uveitis. Acta Ophthalmol. 2022;100(4):e994–1001. https://doi.org/10.1111/aos.15012.
Fabiani C, Vitale A, Emmi G, et al. The role of biosimilars in uveitis: long-term real-world outcomes of the switch from original to biosimilar TNF-alpha inhibitors. Front Pharmacol. 2019;10:1468. https://doi.org/10.3389/fphar.2019.01468.
Ulu K, Çakan M, Çağlayan Ş, et al. Real-life data on efficacy and safety of original Adalimumab and biosimilar Adalimumab (ABP 501) in pediatric rheumatic diseases. Expert Opin Biol Ther. 2022. https://doi.org/10.1080/14712598.2022.2123703.
Sota J, Gentileschi S, Perfetti MO, et al. Role of adalimumab biosimilar in the treatment of non-anterior uveitis associated with Behçet’s syndrome. Ophthalmol Ther. 2021;10(4):1129–35. https://doi.org/10.1007/s40123-021-00387-6.
Lerman MA, Lewen MD, Kempen JH, Mills MD. Uveitis reactivation in children treated with tumor necrosis factor alpha inhibitors. Am J Ophthalmol. 2015;160(1):193–200. https://doi.org/10.1016/j.ajo.2015.04.016. (e1).
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Author Contributions
All the authors substantially contributed to the conception or design of the work and critically revised the paper. All the authors approved the final version and agreed to be responsible for all the aspects of the work. In addition, Antonio Vitale and Francesca Della Casa wrote the first draft of the manuscript and performed the preliminary data analysis and interpretation; Claudia Fabiani, Antonio Vitale, Silvana Guerriero, Gaafar Ragab, Angela Mauro, Valeria Caggiano, Giuseppe Lopalco, Irene Bellicini, Rossella Favala, Mohamed Tharwat Hegazy, Rana Hussein Amin, Marco Cattalini, Emanuela Del Giudice, Carla Gaggiano, Maria Pia Paroli, Jurgen Sota, Francesco La Torre, Maria Tarsia, Bruno Frediani, Gian Marco Tosi, HA and Alex Fonollosa were involved in the study according to their active role in enrolling pediatric patients with uveitis treated with adalimumab for nonanterior uveitis in the AIDA Registries by September 1, 2022; Abdurrahman Tufan, Ibrahim Almaghlouth, Francesco La Torre, Ewa Więsik-Szewczyk, HA, Gian Marco Tosi, JAR, Antonio Vitale, Jurgen Sota, Maria Tarsia, Valeria Caggiano, Carla Gaggiano, Claudia Fabiani, Gaafar Ragab were involved in the study as investigators from the top contributor centers for any of the other AIDA Registries; Alberto Balistreri is also the bioengineer involved in the technical management of the platform and registries; Donato Rigante edited and supervised the first version of the manuscript; Claudia Fabiani and Luca Cantarini took care of the final revision of the manuscript and accounted for AIDA Registries Coordinator.
Disclosures
All named authors confirm that they have no conflicts on interest to declare.
Compliance with Ethics Guidelines
This study is part of the AIDA project, approved by the Ethics Committee of Azienda Ospedaliera Universitaria Senese, Siena, Italy (ref. no. 14,951). The study has been conducted in accordance with the recommendations by the Declaration of Helsinki and subsequent updates. All patients provided their assent; the informed consent was obtained from the parents (or by the legal guardian).
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Vitale, A., Casa, F.D., Guerriero, S. et al. Efficacy and Safety of Adalimumab in Pediatric Non-infectious Non-anterior Uveitis: Real-life Experience From the International AIDA Network Uveitis Registry. Ophthalmol Ther 12, 1957–1971 (2023). https://doi.org/10.1007/s40123-023-00712-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-023-00712-1