
Abstract
Introduction
To evaluate real-life efficacy, safety, and treatment patterns with the dexamethasone intravitreal implant (DEX) in posterior segment inflammation due to non-infectious uveitis (treatment-naïve or not) in French clinics.
Methods
In this prospective, multicenter, observational, non-comparative, post-reimbursement study, consecutive patients with posterior segment inflammation due to non-infectious uveitis were enrolled and evaluated at baseline (day 0). Those who received DEX on day 0 were re-evaluated at months 2, 6, and 18. Retreatment with DEX and/or alternative therapies was allowed during follow-up. Primary outcome: patients (%) with at least a 15-letter gain in best corrected visual acuity (BCVA) at 2 months. Secondary outcomes included patients (%) with at least 15-letter BCVA gains at 6 and 18 months; mean BCVA change from baseline at 2, 6, and 18 months; and patients (%) retreated, mean central retinal thickness (CRT), and adverse events (AEs) at all post-baseline visits.
Results
Ninety-seven of 245 enrolled patients with posterior segment inflammation due to non-infectious uveitis (80% previously treated) and disease duration of 5 years (average) received DEX on day 0 and were included in efficacy analyses. At month 2 (n = 91), 20.5% of patients (95% CI 12.0–28.9) gained at least 15 letters from a baseline mean of 60.9 letters; the mean gain was 6.2 letters (95% CI 3.5–8.9). At month 6, 50.0% (n = 38/76) of patients did not receive alternative treatment or DEX retreatment, mostly because inflammation had sufficiently subsided (n = 27/38, 71.1%). Although early study termination prevented efficacy analysis at 18 months (n = 12), CRT reductions persisted throughout follow-up. From baseline to month 18, 21/245 (8.6%) patients had DEX-related AEs; 17/245 (6.9%) had ocular hypertension (most common AE).
Conclusion
LOUVRE 2 confirms DEX efficacy on visual acuity and CRT in predominantly DEX-pretreated patients with relatively old/stabilized uveitis. DEX tolerability was consistent with known/published data, confirming treatment benefits in posterior segment inflammation due to non-infectious uveitis.
ClinicalTrials.gov Identifier
NCT02951975.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
First-line treatment of inflammation of the posterior segment due to non-infectious uveitis and associated macular edema mainly relies on local administration of corticosteroids. |
The dexamethasone intravitreal implant (DEX) is the first intravitreal treatment approved for inflammation of the posterior segment due to non-infectious uveitis in Europe. |
The LOUVRE 2 study evaluated real-world efficacy, safety, and treatment patterns with DEX in inflammation of the posterior segment due to non-infectious uveitis in France. |
What was learned from the study? |
LOUVRE 2 confirmed that DEX improves visual acuity and central retinal thickness in individuals with inflammation of the posterior segment due to non-infectious uveitis, even in a study population consisting mostly of previously treated patients. |
Although follow-up was shorter than anticipated, the study findings show that positive outcomes are achievable with DEX in this patient population. |
Digital Features
This article is published with a digital feature, i.e., an infographic to facilitate understanding of the article. To view the digital feature for this article, go to https://doi.org/10.6084/m9.figshare.20078987
Introduction
Uveitis is an internal inflammation of the eye that can lead to severe and sudden vision loss if left untreated [1]. It can be infectious or non-infectious in origin and is categorized on the basis of the site of primary ocular inflammation, i.e., anterior uveitis (affecting the anterior segment), intermediate uveitis (involving the vitreous, peripheral retina, and pars plana of the ciliary body), posterior uveitis (involving the choroid and/or retina), and panuveitis [1]. Despite being the least common form, posterior uveitis is the most vision-threatening and challenging form to treat, due in part to the location of the target tissues in the back of the eye and the lack of effective delivery with topical treatments [1,2,3].
Uveitis-related loss of vision is most commonly due to cystoid macular edema, inflammatory vitreous haze and associated debris, and cataract [4, 5]. Corticosteroids have been shown to control both inflammation and macular edema (ME) [6], but the low bioavailability [7] and side effects [8, 9] of topical and oral formulations, respectively, have led to development of alternative therapies. In Europe, the immunosuppressant adalimumab (Humira®, AbbVie, North Chicago, IL, USA) has been approved for subcutaneous injection in non-infectious intermediate, posterior, and panuveitis since 2017, but only in adult patients who have insufficient response or intolerance to corticosteroid therapy, and those for whom corticosteroid therapy is contraindicated. First-line treatment of inflammation of the posterior segment and ME associated with non-infectious uveitis still mainly relies on local administration of corticosteroids such as the dexamethasone intravitreal implant 0.7 mg (DEX; Ozurdex®, Allergan, an AbbVie company) [10, 11], which improves bioavailability and tolerability, compared with the aforementioned topical and oral formulations.
Following extension (in 2011) of the indication of DEX as the first local/intravitreal treatment for inflammation of the posterior segment due to non-infectious uveitis in Europe, the French Haute Autorité de Santé requested that an observational study (LOUVRE 2) be conducted to provide information on DEX treatment patterns in this disease in French clinical settings, as well as characteristics/profile of patients treated with DEX (compared with patients not treated with DEX) during the study, changes in best corrected visual acuity (BCVA) and anatomic outcome (central retinal thickness [CRT]) from baseline in those patients, and adverse events (AEs). Although the study was terminated early, because of a product recall [12] that impacted the number of patients with data available at 18 months (end of study prespecified in the protocol), efficacy and safety findings at months 2 (prespecified primary time point) and 6 are presented herein.
Methods
Statement of Ethics Compliance
Before the study start, the protocol was approved centrally by the Comité Consultatif sur le Traitement de l’Information en Matière de Recherche dans le Domaine de la Santé (CCTIRS), Commission Nationale de l’Informatique et Libertés (CNIL), and Conseil National de l’Ordre des Médecins (CNOM). The study was conducted in accordance with the ethical principles of the Helsinki Declaration of 1964, and its later amendments [13], French Public Health Code and French Act on Data Processing, Data Files, and Individual Liberties [14], Good Epidemiological Practices [15], and guidelines from the Haute Autorité de Santé on post-registration studies [16]. Each patient provided written informed consent to participate in the study before study initiation, and all authors consented to publication of the manuscript.
Study Design
This prospective, multicenter, observational, longitudinal, post-reimbursement study (ClinicalTrials.gov Identifier NCT02951975) was conducted in metropolitan areas of France between 25 January 2017 and 19 December 2018.
Overall, 43 ophthalmologists from 20 representative injection centers (stratified as public vs. private status) participated. Randomly selected from a comprehensive list, the centers included were 70% public and 30% private, consistent with the prespecified ratio (75% public; 25% private).
Study Population and Treatment
Consecutively presenting adults (at least 18 years of age) with inflammation of the posterior segment due to non-infectious uveitis (treatment-naïve or previously treated with DEX or other agents) were recruited at each center. Excluded were patients who did not reside in metropolitan France and those who were concurrently participating in a non-observational study.
Upon enrollment in the study, patients completed a questionnaire about their disease history to help physicians determine whether treatment of inflammation of the posterior segment due to non-infectious uveitis with DEX or an alternative therapy was required/appropriate. Due to the observational nature of the study, all decisions related to treatment, i.e., whether treatment was indicated or not, as well as selection of therapy (including type and frequency) were made at the investigators’ discretion. All patients not treated with DEX on day 0 were included as a control group to collect information on patient characteristics leading to treatment with DEX or not. DEX was supplied as usual by the clinic or practitioner.
Visits and Assessments
Study visits were scheduled at baseline (day 0) for all patients enrolled, and at months 2, 6, and 18 (per typical follow-up in French clinical practice) for the subgroup that received DEX treatment on day 0. Additional visits, including those after retreatment with DEX and/or an alternative therapy, were scheduled as needed (per investigator judgement) and documented. Patients not treated with DEX on day 0 (per investigator’s decision) were only evaluated on day 0.
Collected at baseline were patient demographics and characteristics, including reasons for not treating with DEX. Assessed bilaterally at each visit were BCVA (per the Early Treatment Diabetic Retinopathy Study [preferably] or Monoyer scale), intraocular pressure (IOP; per standard practice), CRT (by optical coherence tomography), and vitreous haze score (per a modified version of the photographic scale published by Nussenblatt et al. [17] in which 0 = no inflammation; + 0.5 = trace inflammation; + 1 = mild disorder of the retinal vessels and optic nerve; + 1.5 = disorder of the optic nerve head and the posterior retina > + 1 but < + 2; + 2 = moderate disorder of the optic nerve head; + 3 = marked disorder of the optic nerve head; and + 4 = optic nerve head not visible). Information on retreatment (if performed) was also recorded for each study eye at each visit, while quality of life was evaluated at day 0, month 2, and month 18 (using the National Eye Institute-Visual Function Questionnaire-25 [VFQ-25]).
AEs, including study discontinuations, were recorded on day 0 (baseline) in all enrolled patients, and at all post-baseline visits in the subgroup of patients who received DEX on day 0 and were followed prospectively.
Outcome Measures
The primary outcome measure was the proportion of patients with at least a 15-letter gain in BCVA from baseline at 2 months. Secondary outcome measures included the baseline characteristics of enrolled patients who received the DEX implant on day 0 (compared with those of patients who did not receive DEX on day 0); proportion of patients with at least a 15-letter gain in BCVA from baseline at 6 and 18 months; mean changes from baseline in BCVA, IOP, CRT, and vitreous haze score at months 2, 6, and 18; mean change from baseline in the VFQ-25 score at months 2 and 18; proportion of patients retreated (along with the type of and reason for retreatment); mean number of injections; and mean treatment interval. All outcomes are reported on a per-patient basis. In patients who needed bilateral treatment, the eye with the worse BCVA and/or vitreous haze score at enrollment was considered the study eye.
Statistical Analyses
Per the protocol, the primary BCVA-related outcome measure was analyzed in all patients treated with DEX on day 0 who had BCVA data available at day 0 and month 2. All secondary outcome measures were to be analyzed in all patients treated with DEX on day 0, except the proportion of patients with at least a 15-letter gain in BCVA from baseline at 6 and 18 months, which was analyzed in patients with data available at day 0 and month 6 or 18. However, as a result of the product recall and physicians being advised to consider alternative therapies for their patients (based on potential risks and benefits), switches to other therapies were expected to bias the analyses. Consequently, per decision from the study steering committee, the aforementioned analysis populations were narrowed to patients who completed the scheduled visits before 4 October 2018 (recall date). One exception was AEs, which were analyzed in all patients treated with DEX, regardless of when they completed the scheduled visits.
Statistical analyses were performed using SAS® software version 9.3 or higher (SAS Institute, Inc., Cary, NC, USA), without imputation for missing values (unless otherwise noted). Continuous variables were summarized by mean and standard deviation (SD), while categorical variables were summarized by frequency and percentage. Comparative analyses were supported by 95% confidence intervals (CIs).
The sample size was determined on the basis of information from the prospective, randomized, controlled, HURON study [18], which led to approval of DEX as the first intravitreal treatment for inflammation of the posterior segment due to non-infectious uveitis in Europe. In the HURON study, 43% of patients (95% CI 23.9–48.7) who received DEX (0.7 mg) had at least a 15-letter BCVA gain from baseline at week 9. According to the sample size equation N = 1.962 × p × (1 − p)/i2 with p = 0.43 (population proportion) and I = 0.05 (margin of error), a minimal sample size of 377 patients was required to determine the proportion of patients with at least a 15-letter gain from baseline with an accuracy of 95%. Assuming that 5% of patients would not have data available at month 2 (primary time point), enrollment of 400 patients was planned.
Results
Baseline Demographics and Characteristics in the Overall Population
Of 245 patients enrolled in the study, four were enrolled after 4 October 2018 (recall date) and excluded from all analyses, except AEs. The remaining 241 patients (overall population) had a baseline mean age of 55.9 years and mean duration of uveitis of 6.1 years, and 78.8% of patients reported having received prior treatment (Table 1). Choroidal involvement, cystoid macular edema, inflammation of the optic nerve, and retinal vasculitis were reported in 25.0% (n = 60), 58.5% (n = 141), 18.8% (n = 45), and 30.4% (n = 73) of these patients, respectively.
Of the 241 patients in the overall population, 144 (59.8%) were only evaluated on day 0 because they did not receive DEX on day 0 for the following reasons: an alternative therapy was chosen (n = 75, 52.1%); treatment was deemed unnecessary as there was no recurring edema or edema was stabilized (n = 43, 29.9%); patients were not eligible for DEX treatment (N = 14, 9.7%) because visual acuity was already too high to justify treatment (n = 9/14) or per the Summary of Product Characteristics [19]; patients refused treatment (n = 3, 2.1%); or other reasons (n = 9, 6.3%). The remaining 97 (40.2%) patients were treated with DEX on day 0 and followed prospectively (Figs. 1, 2).

Patient disposition. Recall refers to a dexamethasone intravitreal implant (DEX) recall on 4 October 2018. aReasons for ineligibility included absence of inflammation of the posterior segment due to non-infectious uveitis, age or residency criteria not met, and participation in another clinical study. bThe total adds up to more than 65 as some patients were excluded for more than one reason

Reasons for not treating with the dexamethasone intravitreal implant (DEX) on day 0. N = 144. BCVA best corrected visual acuity
Differences in Baseline Demographics and Characteristics Among Patients Treated with DEX on Day 0 and Those Who Were Not
Notably, there were statistically significantly more patients (n = 74/97; 76.3%) with cataract at baseline in the subgroup treated with DEX on day 0, compared with the subgroup not treated with DEX on day 0 (n = 81/144; 56.3%; Table 1). Patients who were treated with DEX on day 0 (N = 97) also had a statistically significantly greater age (60.6 vs. 52.7 years), CRT (424.8 vs. 333.6 µm), and mean number of prior DEX injections (7.5 vs. 2.1) than those who were not (N = 144). Moreover, the subgroup treated with DEX on day 0 included a statistically significantly greater proportion of patients with ME (70.2%) than the subgroup not treated with DEX on day 0 (44.0%; Table 1).
There were statistically significantly fewer DEX-naïve patients among patients who received DEX on day 0 (n = 24/95; 25.3%; 95% CI 16.5–34.0), compared with those who did not (n = 83/142; 58.5%; 95% CI 50.3–66.6). The subgroup treated with DEX on day 0 also presented more frequently with cystoid ME (n = 73/97; 75.3%; 95% CI 66.7–83.8) than the subgroup not treated with DEX on day 0 (n = 68/144; 47.2%; 95% CI 39.1–55.4). There was, however, no statistically significant difference in mean duration of uveitis between patients who received DEX on day 0 and those who did not.
Patient Disposition in the Subgroup Treated with DEX on Day 0 and Followed Prospectively
Of the 97 patients treated with DEX on day 0 and followed prospectively, 91 (93.8%), 76 (78.4%), and 12 (12.4%) completed the month-2, -6, and -18 visits before 4 October 2018, respectively. Sixty discontinuations were recorded through month 18, including 55/60 (91.7%) due to early termination of the study (Fig. 1). Data from 25 additional patients being followed at the time of the product recall were censored because of a follow-up visit occurring after the recall.
Treatment Patterns Among Patients Treated with DEX on Day 0
Patients treated with DEX on day 0 (N = 97) were followed for a mean (SD) of 14.9 (4.1) months and had a mean (SD) of 5.3 (3.4) visits (Table 2). During follow-up, 37 (38.1%) patients did not require retreatment, 54 (55.7%) were retreated with DEX at least once, and 6 (6.2%) were retreated with alternative therapies only. The mean (SD) number of DEX reinjections was 1.0 (1.2), with a mean (SD) injection interval of 156.3 (46.7) days. The main reason for reinjection was BCVA decrease (n = 38/93, 40.9%) despite response to treatment/reduction in CRT (Table 2). Sixty-two (63.9)% patients also received concomitant treatment(s) for uveitis or edema during follow-up.
Because DEX is typically injected at intervals of 4 months or more, there was no analysis of the reasons for not retreating at the month-2 visit. At month 6 (n = 76), 47 (62.7%) patients with available data had not received retreatment with DEX, including 38 (50.7%) who had not received alternative therapy either, mostly because—in the investigators’ judgement—additional treatment was not needed as inflammation had sufficiently subsided (n = 27/38, 71.1%). Specific reasons were available for 37 of these 38 patients (based on investigators’ judgement and an exploratory analysis) and included the following: the disease had stabilized or improved (n = 27, 73.0%), no improvement was expected at 6 months (n = 5, 13.5%), 6 months was considered too early for reinjection (n = 4, 10.8%), and other (n = 1, 2.7%). The reasons for not retreating were not analyzed at 18 months because the sample size was too small.
Efficacy and Quality of Life Among Patients Treated with DEX on Day 0
The proportion of patients treated with DEX on day 0 who gained at least 15 letters (20.5%) from baseline was statistically significant at 2 months (primary outcome measure), as was the mean (SD) gain of 6.2 (12.7) letters from baseline (Table 3). The effect of DEX on these functional outcome measures was also statistically significant at months 6 and 18 (Table 3). Although the proportion of patients who gained at least 15 letters from baseline at 2 months (primary time point) was numerically greater in patients who were DEX-naïve at baseline (n = 6/22, 27.3%), compared with those who were naïve of all treatments (n = 4/18, 22.2%) or had been previously treated with DEX (n = 8/46, 17.4%), there were no notable differences in mean BCVA gain from baseline at month 2 among these subgroups.
From an anatomic standpoint, DEX statistically significantly reduced mean CRT, with mean reductions from baseline of 27.4%, 18.5%, and 16.4% at months 2, 6, and 18, respectively. The mean vitreous haze score was also statistically significantly reduced at months 2 and 6, but not at month 18 (Table 3). An exploratory analysis indicated that, among DEX-treated patients who presented with ME (n = 63; 70.0%), 62.4% and 44.3% experienced at least a 20% decrease of ME at months 2 and 6, respectively, compared with baseline.
The mean (SD) change in the VFQ-25 quality-of-life score from baseline was statistically significant at month 2 (i.e., 4.3 [10.5]; 95% CI 1.6–7.0; n = 59). Analysis could not be performed at month 18 because of the small sample size.
Safety in All Enrolled Patients
Overall, 44 (18.0%) of all enrolled patients (N = 245) reported a total of 85 AEs, including one AE reported in a patient not treated with DEX on day 0. Of the 84 AEs reported in patients treated with DEX on day 0, 32 (38.1%) were deemed potentially DEX-related by the treating physician (Table 4). In total, 4 (4.8%) of the 84 AEs were potentially DEX-related serious AEs; those included ocular hypertension (n = 3, 3.6%) and vitreous hemorrhage (n = 1, 1.2%). No treatment-related deaths were reported during the study, and 3 (6.8%) of the 44 patients who experienced at least one AE discontinued treatment because of AEs. As shown in Table 4, the most frequently reported AE potentially related to DEX treatment was ocular hypertension (n = 20/32, 62.5%).
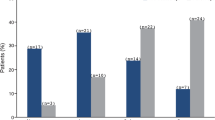
Among patients treated with DEX on day 0 with available data at month 2, the proportion of patients with an IOP increase of at least 10 mmHg or at least 25 mmHg was at most 16.3% (Fig. 3). The frequency of IOP elevation decreased to 7.0% or less at month 6 and was 18.2% or less at month 18 (Fig. 3). Notably, no laser or surgical procedures were required to control IOP.

Intraocular pressure (IOP) evolution over time in patients treated with the dexamethasone intravitreal implant (DEX) on day 0. n = number of patients with the indicated IOP increase from baseline at the indicated time point. N = total number of patients with data available at the indicated time point. CI confidence interval, M month, NA not available
Discussion
This prospective, multicenter, observational, longitudinal, post-reimbursement study (LOUVRE 2) of DEX treatment for inflammation of the posterior segment due to non-infectious uveitis found that 20.5% of patients who received DEX treatment on day 0 exhibited at least a 3-line BCVA gain from baseline at month 2. Similar results were observed at month 6, consistent with a ceiling effect on the BCVA gain due to the majority of patients treated with DEX on day 0 having received previous treatment and presented with relatively high/well-preserved BCVA and old/stabilized uveitis at baseline. Nevertheless, statistically significant mean BCVA gains from baseline were observed at months 2 and 6 in patients treated with DEX on day 0, despite prior DEX treatment in more than half of them, substantiating DEX’s efficacy in improving visual function in patients with inflammation of the posterior segment due to non-infectious uveitis in clinical settings. DEX also produced statistically significant reductions in mean vitreous haze and mean CRT, confirming its anti-inflammatory properties and efficacy in improving anatomic outcomes in patients treated with DEX on day 0. Although findings at 18 months were consistent with observations at months 2 and 6, the small sample makes it difficult to draw conclusions.
These findings were deemed clinically relevant considering that in a pivotal prospective, multicenter, masked, randomized, sham-controlled, 26-week study (HURON [18]) of DEX 0.7 mg in non-infectious intermediate and posterior uveitis, patients’ visual acuity and vitreous haze score were 58 letters and 2.1 (means) at baseline, compared with 60.9 letters and 0.6 (means), respectively, in this study. As a result, smaller improvements would be expected to be achievable in the present study. In addition, the 81% of patients with non-infectious intermediate uveitis enrolled in the HURON study may have led to greater CRT reductions at 2 months (−99.4 µm) despite the thinner baseline CRT (344.0 µm [18]), compared with our study in which no patients had non-infectious intermediate uveitis, CRT reduction was −27.4 µm, and baseline CRT was 424.8 µm. It is also noteworthy that compared with the present study, patients in HURON were younger (44 vs. 60.6 years of age herein), had a shorter duration of uveitis (50.5 vs. 60 months herein), and included a higher proportion of DEX-naïve patients (100% vs. 45.3% herein) [18].
A systematic review of the literature identified only one other prospective, observational, real-world study of DEX in non-infectious posterior segment uveitis (CONSTANCE) [20]. Although the sexes of the study populations appeared similar between the LOUVRE 2 (60.8% female) and CONSTANCE (62.9%) [20] studies, patients were noticeably older in the LOUVRE 2 study (60.6 years) than the CONSTANCE study (54.9 years [20]). In addition, the proportion of patients previously treated with DEX was remarkably larger in the LOUVRE study (54.7%) than the CONSTANCE study (25.2% [20]), which is likely because the CONSTANCE study was initiated in March 2012 [20] and thus sooner after European Union approval of DEX for inflammation of the posterior segment due to non-infectious uveitis (April 2011 [21]) than the LOUVRE 2 study. Regardless of these differences, and the fact that CONSTANCE was a safety surveillance study that did not evaluate any efficacy variables, the mean treatment interval reported herein (156.3 days) is in line with that determined in the CONSTANCE study (189.7 days) [20]. Moreover, the most common AEs of special interest reported in the CONSTANCE study (i.e., cataract formation, cataract progression, increased IOP, vitreous hemorrhage, ocular hypertension, and glaucoma [20]) are also in line with the AE profile reported herein.
The safety and efficacy findings of the LOUVRE 2 study are further reinforced by similar findings from retrospective studies of DEX in uveitis in real-word clinical practice [22,23,24,25]. Of those, the recently published RUVDEX study [24] was conducted at three centers in France and evaluated outcomes in 152 eyes with non-infectious uveitis that were treated with DEX (total of 358 implants) and followed for a mean of 19 months. In the RUVDEX study, although only 23.7% of the eyes studied had posterior uveitis, all treated eyes demonstrated substantial improvements in BCVA from a baseline mean of 60.1 letters, with mean gains of 5.3, 6.2, 4.2, 4.8, and 4.4 letters at months 2, 4, 6, 12, and 24, respectively [19]. Mean CRT also improved over time, decreasing from 422 µm at baseline to 320, 365, 381, and 351 µm at months 2, 6, 12, 24, respectively [19]. Moreover, 81.4% of patients had a vitreous haze score of 0 during follow-up, compared with a median value of 0.5 at baseline. Although cataract and ocular hypertension were reported following DEX treatment, both AEs were manageable [24].
The AEs recorded as potentially treatment related during the present study were in line with the prescribing information for DEX [10, 11] and not unexpected, except the case of macular fibrosis (ongoing at study end). However, one case of macular fibrosis in a patient with non-infectious posterior segment uveitis was reported in the CONSTANCE study as well [20]. Although cataract and conjunctival hemorrhage were also reported as common AEs following treatment with DEX 0.7 mg in the HURON study (15% and 30%, respectively), there were no cases of iridocyclitis in our study, compared with 7 (9%) in the HURON study [18]. Moreover, it is worth noting that, in this study, the definition of treatment-related AEs was broad, including those that were probably or possibly due to the injection procedure or the implant itself, as well as those for which there was uncertainty regarding causality. Although not statistically significant, the numerical reduction in the proportion of DEX-treated patients with IOP increases of at least 10 mmHg and at least 25 mmHg between months 2 and 6 suggests a transient IOP elevation at 2 months post-injection (a known side effect of intravitreal corticosteroids [26]), followed by IOP normalization. In patients treated with DEX for another indication, diabetic macular edema, IOP elevations have been shown to be transient [27,28,29,30,31,32,33], consistent with the current observations.
The greater proportion of patients with cataracts in the subgroup treated with DEX on day 0, compared with the subgroup not treated with DEX on day 0, confirms that DEX is preferentially prescribed to patients with existing cataracts, most likely because corticosteroids are often associated with cataract formation and progression [26, 34,35,36]. Otherwise, the greater proportion of patients with cystoid ME in the subgroup treated with DEX on day 0 is consistent with established anti-edema properties of DEX.
Study limitations include the sample size, which was smaller than planned at enrollment and during follow-up (especially at 18 months), due to the product recall and consequent study termination. As a result, caution is required when interpreting data at this time point. It is also worth noting that, although the study protocol originally called for recruitment of at most 20 patients per center, a few centers ultimately contributed more than 20 patients to the study, because of the low prevalence/incidence of non-infectious uveitis of the posterior segment in general and consequent difficulty in recruiting patients with the disease (245 enrolled vs. 400 planned). Nonetheless, the study was designed so that consecutive patients were recruited at each center, which prevented potential selection bias and ensured that the study population was representative of the general population of patients with non-infectious uveitis of the posterior segment. As such, the initial sample size was deemed acceptable. Although 60% of the patients enrolled did not receive DEX treatment on day 0, further contributing to the small sample size in the efficacy analyses, their inclusion as a control group was intended (per protocol) and necessary to help address the request from the French Haute Autorité de Santé for information on patient characteristics leading to treatment with DEX. Finally, considering that 63.9% of patients who received DEX treatment also received concomitant treatment for uveitis, it is possible that the observed results were due to the combination of treatments, as opposed to DEX alone, as is the case in various randomized clinical trials [18, 37,38,39].
Conclusion
Overall, our findings add to the prospective data on the effects of DEX on functional and anatomic outcomes in typical clinical settings, confirming its efficacy through month 6 and acceptable safety in patients with inflammation of the posterior segment due to non-infectious uveitis (including those previously treated with DEX) for whom treatment options remain limited.
References
National Eye Institute. Uveitis. https://www.nei.nih.gov/learn-about-eye-health/eye-conditions-and-diseases/uveitis. Accessed 7 Mar 2022.
Whitcup SM, Robinson MR. Development of a dexamethasone intravitreal implant for the treatment of noninfectious posterior segment uveitis. Ann N Y Acad Sci. 2015;1358:1–12.
Mikhail M, Sallam A. Novel intraocular therapy in non-infectious uveitis of the posterior segment of the eye. Med Hypothesis Discov Innov Ophthalmol. 2013;2:113–20.
Markomichelakis NN, Halkiadakis I, Pantelia E, et al. Patterns of macular edema in patients with uveitis: qualitative and quantitative assessment using optical coherence tomography. Ophthalmology. 2004;111:946–53.
Durrani OM, Tehrani NN, Marr JE, Moradi P, Stavrou P, Murray PI. Degree, duration, and causes of visual loss in uveitis. Br J Ophthalmol. 2004;88:1159–62.
Amoaku WM, Saker S, Stewart EA. A review of therapies for diabetic macular oedema and rationale for combination therapy. Eye (Lond). 2015;29:1115–30.
Suresh PK, Sah AK. Patent perspectives for corticosteroids based ophthalmic therapeutics. Recent Pat Drug Deliv Formul. 2014;8:206–23.
Chirikov VV, Shah R, Kwon Y, Patel D. Oral corticosteroid exposure and increased risk of related complications in patients with noninfectious intermediate, posterior, or panuveitis: real-world data analysis. Ophthalmic Epidemiol. 2019;26:27–46.
Suhler EB, Thorne JE, Mittal M, et al. Corticosteroid-related adverse events systematically increase with corticosteroid dose in noninfectious intermediate, posterior, or panuveitis: post hoc analyses from the VISUAL-1 and VISUAL-2 trials. Ophthalmology. 2017;124:1799–807.
European Medicines Agency. Ozurdex (dexamethasone) - an overview of Ozurdex and why it is authorised in the EU. https://www.ema.europa.eu/en/documents/overview/ozurdex-epar-medicine-overview_en.pdf. Accessed 7 Mar 2022.
Allergan (an AbbVie company). Highlights of prescribing information - OZURDEX® (dexamethasone intravitreal implant) for intravitreal injection. https://www.rxabbvie.com/pdf/ozurdex_pi.pdf. Accessed 19 May 2022.
Agence nationale de securite du medicament et des produits de sante. Ozurdex 700 microgrammes, implant intravitréen avec applicateur—Allergan France—Rappel de lots. https://ansm.sante.fr/informations-de-securite/ozurdex-700-microgrammes-implant-intravitreen-avec-applicateur-allergan-france. Accessed 19 May 2022.
World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. https://www.wma.net/policies-post/wma-declaration-of-helsinki-ethical-principles-for-medical-research-involving-human-subjects/. Accessed 7 Mar 2022.
Commission Nationale de l'Informatique et des Libertés. Loi informatique et libertés—Act 78-17 of 6 January 1978 on Information Technology, Data Files and Civil Liberties. http://www.audentia-gestion.fr/CNIL/Act78-17VA.pdf. Accessed 7 Mar 2022.
Association des Epidémiologistes de Langue Française (ADELF). Recommendations for professional standards and good epidemiological practices. https://www.constances.fr/base-documentaire/2015/1421662226-adelf-recommendations-english.pdf. Accessed 7 Mar 2022.
Stamenkovic S, Solesse A, Zanetti L, Zagury P, Vray M. Guide de la Haute Autorité de Santé (HAS): les études post-inscription sur les technologies de santé (médicaments, dispositifs médicaux et actes): principes et méthodes. Therapie. 2012;67:409–21.
Nussenblatt RB, Palestine AG, Chan CC, Roberge F. Standardization of vitreal inflammatory activity in intermediate and posterior uveitis. Ophthalmology. 1985;92:467–71.
Lowder C, Belfort R Jr, Lightman S, et al. Dexamethasone intravitreal implant for noninfectious intermediate or posterior uveitis. Arch Ophthalmol. 2011;129:545–53.
European Medicines Agency. Summary of Product Characteristics—OZURDEX. https://www.ema.europa.eu/en/documents/product-information/ozurdex-epar-product-information_en.pdf. Accessed 19 May 2022.
Tufail A, Lightman S, Kamal A, et al. Post-marketing surveillance study of the safety of dexamethasone intravitreal implant in patients with retinal vein occlusion or noninfectious posterior segment uveitis. Clin Ophthalmol. 2018;12:2519–34.
European Medicines Agency. Assessment report for Ozurdex. https://www.ema.europa.eu/en/documents/variation-report/ozurdex-h-c-1140-ii-0001-epar-assessment-report-variation_en.pdf. Accessed 7 Mar 2022.
Hasanreisoğlu M, Özdemir HB, Özkan K, et al. Intravitreal dexamethasone implant in the treatment of non-infectious uveitis. Turk J Ophthalmol. 2019;49:250–7.
Lam WC, Albiani DA, Yoganathan P, et al. Real-world assessment of intravitreal dexamethasone implant (0.7 mg) in patients with macular edema: the CHROME study. Clin Ophthalmol. 2015;9:1255–68.
Mathis T, Cerquaglia A, Weber M, et al. Real life study in uveitis treated with dexamethasone implant. Retina. 2020;41:620–9.
Pelegrín L, de la Maza MS, Molins B, Ríos J, Adán A. Long-term evaluation of dexamethasone intravitreal implant in vitrectomized and non-vitrectomized eyes with macular edema secondary to non-infectious uveitis. Eye (Lond). 2015;29:943–50.
Whitcup SM, Cidlowski JA, Csaky KG, Ambati J. Pharmacology of corticosteroids for diabetic macular edema. Invest Ophthalmol Vis Sci. 2018;59:1–12.
Lo Giudice G, Avarello A, Campana G, Galan A. Rapid response to dexamethasone intravitreal implant in diabetic macular edema. Eur J Ophthalmol. 2018;28:74–9.
Malclès A, Dot C, Voirin N, et al. Safety of intravitreal dexamethasone implant (Ozurdex): the Safodex study. Incidence and risk factors of ocular hypertension. Retina. 2017;37:1352–9.
Malclès A, Dot C, Voirin N, et al. Real-life study in diabetic macular edema treated with dexamethasone implant: the Reldex Study. Retina. 2017;37:753–60.
Callanan DG, Loewenstein A, Patel SS, et al. A multicenter, 12-month randomized study comparing dexamethasone intravitreal implant with ranibizumab in patients with diabetic macular edema. Graefes Arch Clin Exp Ophthalmol. 2017;255:463–73.
Bansal P, Gupta V, Gupta A, Dogra MR, Ram J. Efficacy of Ozurdex implant in recalcitrant diabetic macular edema—a single-center experience. Int Ophthalmol. 2016;36:207–16.
Scaramuzzi M, Querques G, Spina CL, Lattanzio R, Bandello F. Repeated intravitreal dexamethasone implant (Ozurdex) for diabetic macular edema. Retina. 2015;35:1216–22.
Escobar-Barranco JJ, Pina-Marín B, Fernández-Bonet M. Dexamethasone implants in patients with naive or refractory diffuse diabetic macular edema. Ophthalmologica. 2015;233:176–85.
Chawan-Saad J, Wu M, Wu A, Wu L. Corticosteroids for diabetic macular edema. Taiwan J Ophthalmol. 2019;9:233–42.
Haritoglou C, Neubauer AS, Kernt M. Fluocinolone acetonide and its potential in the treatment of chronic diabetic macular edema. Clin Ophthalmol. 2013;7:503–9.
Lattanzio R, Cicinelli MV, Bandello F. Intravitreal steroids in diabetic macular edema. Dev Ophthalmol. 2017;60:78–90.
Kuppermann BD, Blumenkranz MS, Haller JA, et al. Randomized controlled study of an intravitreous dexamethasone drug delivery system in patients with persistent macular edema. Arch Ophthalmol. 2007;125:309–17.
Lightman S, Belfort R Jr, Naik RK, et al. Vision-related functioning outcomes of dexamethasone intravitreal implant in noninfectious intermediate or posterior uveitis—randomized controlled study of an intravitreous dexamethasone drug delivery system in patients with persistent macular edema. Invest Ophthalmol Vis Sci. 2013;54:4864–70.
Thorne JE, Sugar EA, Holbrook JT, et al. Periocular triamcinolone vs. intravitreal triamcinolone vs. intravitreal dexamethasone implant for the treatment of uveitic macular edema: the PeriOcular vs. INTravitreal corticosteroids for uveitic macular edema (POINT) Trial. Ophthalmology. 2019;126:283–95.
Acknowledgements
We acknowledge the following institutions (investigators/participating physicians) for their participation in the study: Centre Hospitalier Intercommunal de Créteil (Eric Souied); Centre Hospitalier Universitaire d’Amiens (Benjamin Jany); Centre Hospitalier Universitaire de Dijon (Catherine Creuzot-Garcher); Centre Hospitalier Universitaire de Toulouse (Vincent Soler); Centre Hospitalier Universitaire Pasteur 2 (Stéphanie Baillif); Centre Pôle Vision Val d'Ouest (Pierre-Loïc Cornut); Centre Rétine Gallien à Bordeaux (Eric Fourmaux); Clinique Monticelli (Christophe Morel); Fondation Rothschild (Salah Mohamed Cherif Titah); Groupe Hospitalier Cochin St-Vincent-de-Paul (Clémence Bonnet); Groupe Hospitalier Pitié-Salpêtrière (Bahram Bodaghi); Hôpital Claude Huriez (Pierre Labalette); Hôpital de la Croix-Rousse (Laurent Kodjikian); Hôpital de la Milétrie (Naoual Tarfaoui); Hôpital de la Timone (Frédéric Matonti); Hôpital Gui de Chauliac (Christelle Schneider); Hôpital Hôtel Dieu et HME (Michel Weber); Hôpital Lariboisière (Hatem Zeghidi); Hôpital Pellegrin à Bordeaux (Marie Bénédicte Rougier); Institut Ophtalmologique de Picardie (Andrei Drimbea); SEL Ophtalliance (Jean-Michel Bosc). The authors would like to thank all the patients who participated in this study, as well as Andrew Shirlaw (AbbVie Inc.) for his support with the study oversight; Thibaut Lassalle and Karol-Anne Cage (Axonal-Biostatem) for project management; Estelle Jouanneau (Axonal-Biostatem) as clinical trial assistant; Marina Duplessis and Lilia Babouche (Axonal-Biostatem) as clinical research assistants; Marie Laborde (Axonal-Biostatem) as clinical research associate; and Gwendoline Moreau (Axonal-Biostatem) as study statistician.
Funding
The study was funded by Allergan (prior to its acquisition by AbbVie, Inc., North Chicago, IL, USA), as was funding the journal’s Rapid Service Fees. The sponsor participated in the study design, data management, analysis and interpretation, and preparation, review, and approval of the manuscript.
Medical Writing, Editorial, and Other Assistance
Writing and editorial assistance was provided to the authors by Michele Jacob, PhD, CMPP, of Evidence Scientific Solutions, Inc (Philadelphia, PA) and funded by AbbVie Inc.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) authorship criteria for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published. Neither honoraria nor payments were made for authorship.
Author Contributions
Bahram Bodaghi, Antoine P. Brézin, Michel Weber, Cécile Delcourt, Laurent Kodjikian, Marie-Ève Velard, Doris Barnier-Ripet, and Sybil Pinchinat: conception and design. Bahram Bodaghi, Antoine P. Brézin, Michel Weber, Cécile Delcourt, Laurent Kodjikian, Alexandra Provost, Marie-Ève Velard, Doris Barnier-Ripet, Sybil Pinchinat, and Laure Dupont-Benjamin: acquisition, analysis, and/or interpretation of data. Laure Dupont-Benjamin: writing (original draft). Bahram Bodaghi, Antoine P. Brézin, Michel Weber, Cécile Delcourt, Laurent Kodjikian, Alexandra Provost, Marie-Ève Velard, Doris Barnier-Ripet, Sybil Pinchinat, and Laure Dupont-Benjamin: writing (review and editing).
Disclosures
Financial arrangements of the authors with companies whose products may be related to the present report follow, as declared by the authors. Bahram Bodaghi is a consultant for Alimera, Allergan (an AbbVie company), and Santen. Antoine P. Brézin is a consultant for AbbVie, Alcon, and Laboratoires Théa. Michel Weber is a consultant for Allergan (an AbbVie company), Bayer, Horus, Laboratoires Théa, and Novartis. Cécile Delcourt is a consultant for Allergan (an AbbVie company), Bausch + Lomb, Laboratoires Théa, and Novartis. Laurent Kodjikian is a consultant for Allergan (an AbbVie company), Bayer, Laboratoires Théa, Novartis, and Roche. Alexandra Provost, Marie-Ève Velard, and Laure Dupont-Benjamin are employees of AbbVie Inc. Doris Barnier-Ripet and Sybil Pinchinat are employees of Axonal-Biostatem, the contract research organization in charge of the study.
Prior Presentation
This work was presented in part at the 20th EURETINA Congress (2–4 October 2020, virtual congress) and 126th Congress of the Société Française d’Ophtalmologie (4–7 September 2020, virtual congress).
Compliance with Ethics Guidelines
Before the study start, the protocol was approved centrally by the Comité Consultatif sur le Traitement de l’Information en Matière de Recherche dans le Domaine de la Santé (CCTIRS; reference number, 15.986), Commission Nationale de l’Informatique et Libertés (CNIL; reference number, MMS/CWR/AR162214 [Authorization number, DR-2016-138]), and Conseil National de l’Ordre des Médecins (CNOM; FR/MFG-MPC/SRMI/16-077-034). The study was conducted in accordance with the ethical principles of the Helsinki Declaration of 1964, and its later amendments [13], French Public Health Code and French Act on Data Processing, Data Files, and Individual Liberties [14], Good Epidemiological Practices [15], and guidelines from the Haute Autorité de Santé on post-registration studies [16]. Each patient provided written informed consent to participate in the study before study initiation, and all authors consented to publication of the manuscript.
Data Availability
AbbVie is committed to responsible data sharing regarding the clinical trials we sponsor. This includes access to anonymized, individual and trial-level data (analysis data sets), as well as other information (e.g., protocols and Clinical Study Reports), as long as the trials are not part of an ongoing or planned regulatory submission. This includes requests for clinical trial data for unlicensed products and indications.
This observational study data can be requested by any qualified researchers who engage in rigorous, independent scientific research, and will be provided following review and approval of a research proposal and Statistical Analysis Plan (SAP) and execution of a Data Sharing Agreement (DSA). Data requests can be submitted at any time and the data will be accessible for 12 months, with possible extensions considered. For more information on the process, or to submit a request, visit the following link: https://www.abbvie.com/our-science/clinical-trials/clinical-trials-data-and-information-sharing/data-and-information-sharing-with-qualified-researchers.html
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Bodaghi, B., Brézin, A.P., Weber, M. et al. Real-Life Efficacy, Safety, and Use of Dexamethasone Intravitreal Implant in Posterior Segment Inflammation Due to Non-infectious Uveitis (LOUVRE 2 Study). Ophthalmol Ther 11, 1775–1792 (2022). https://doi.org/10.1007/s40123-022-00525-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-022-00525-8