
Abstract
Dyskeratosis congenita is a syndrome of bone marrow failure secondary to unstable telomeres. It is characterized by a range of mucocutaneous diseases. Due to premature telomere shortening, these patients have limbal stem cell deficiency leading to poor regeneration and maintenance of the cornea. Many of these patients will require hematopoietic stem cell transplant in their lifetime, which poses a significant risk for acute and chronic graft-versus-host disease with and without ocular manifestations. We advise against elective corneal refractive surgery in patients with dyskeratosis congenita due to the compounded and long-term risks of delayed healing secondary to limbal stem cell deficiency and ocular complications of graft-versus-host disease post-allogeneic hematopoietic stem cell transplant.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Dyskeratosis congenita (DC) is a complex syndrome of bone marrow failure (BMF) [1]. Disease prevalence at birth is approximately one in 1 million with mean age at diagnosis of 15 years [2, 3]. However, it is not uncommon for clinicians to treat DC patients in their career. Patients with DC commonly present with features secondary to mucocutaneous involvement. These mucocutaneous features can include the classic triad of dystrophic nails with longitudinal ridges, lacy reticular pigmentation of the skin (especially of the neck and chest), and oral leukoplakia (premalignant) (Fig. 1) [1, 4,5,6].

Image adapted from Savage, 2018 under the license of Creative Commons
Mucocutaneous triad of DC. a Dystrophic nails. b Lacy reticular pigmentation of the skin. c Oral leukoplakia.
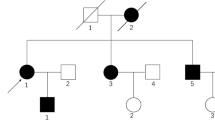
A defect in telomere stability causes the systemic nature of DC [1, 7, 8]. Telomeres are the redundant strands of DNA at the ends of chromosomes that preserve genetic material cycle after cycle of cellular proliferation [8]. The enzyme telomerase is a DNA polymerase that maintains the integrity of telomeres [8]. The telomerase complex consists of proteins that stabilize the DNA [9]. Therefore, in DC, telomere instability leads to accelerated shortening of DNA, which mainly impacts tissue with high cell proliferation rates such as bone marrow and mucocutaneous tissues [1]. While telomere shortening is a natural process with age, the rate of shortening in DC is higher than normal [8]. The most common inheritance pattern of DC is X-linked recessive with a defect in the DKC1 gene (chromosome q28) that encodes for dyskerin, a component of the telomerase complex [1]. There also exist autosomal dominant and recessive inheritance patterns with other genes affected [7, 10]. Twelve known genes cause DC, and this list continues to grow (Fig. 2) [11].

DC is phenotypically heterogeneous, and patients also present with features such as developmental delay, pulmonary fibrosis, and diseases affecting multiple organs like the eyes, liver, esophagus, and periodontal areas [1, 5]. Ocular findings in DC vary according to patient phenotype, with common ocular findings of obstruction of the lacrimal system with epiphora (31%), conjunctivitis, entropion and ectropion, trichiasis, blepharitis, cataract, glaucoma, and exudative retinopathy [1, 3, 12]. Specifically, bilateral exudative retinopathy and intracranial calcifications are associated with a subtype of DC named Revesz syndrome [4, 5]. While some of these findings are contraindications themselves to corneal refractive surgery, we will also address additional complications of DC that we believe to be significant contraindications to elective corneal refractive surgery. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Limbal epithelial stem cells (LESCs), located at the junction of the cornea and conjunctiva, maintain and regenerate the corneal epithelium in response to corneal injury or normal wear over time [13]. The stem cells exist in a niche referred to as the limbal palisades of Vogt that provides a protective microenvironment for these cells [13]. LESCs are undifferentiated and have an unlimited proliferative potential owed to the stability of their telomeres. This process is abnormal in patients with DC, a heritable limbal stem cell deficiency (LSCD) disease [13, 14]. Complications of LSCD, both in DC patients and LSCD secondary to other conditions, consist of corneal epithelial defects, corneal pannus, breakdown, neovascularization, and delayed healing capacity (Fig. 3) [14, 15]. Epithelial trauma secondary to corneal refractive surgery in a DC patient, especially in the setting of subclinical LSCD, will likely not heal as well as a patient without DC.

Photo from the author, Majid Moshirfar, MD
Corneal pannus and ulceration secondary to limbal stem cell deficiency.
Secondary to the underlying genetic cause of DC, patients are highly susceptible to BMF (up to 80% of patients) with hematopoietic stem cell transplantation (HSCT) as definitive therapy [16, 17]. Due to disease-associated eye disorders in addition to the considerable likelihood of BMF with complications of LSCD and graft-versus-host disease (GVHD) post-bone marrow transplant, we must analyze if a patient with DC is an appropriate candidate for corneal refractive surgery [12,13,14]. We conclude that DC patients are not suitable candidates, and we do not recommend any elective corneal refractive surgery such as laser in situ keratomileusis (LASIK), photorefractive keratectomy (PRK), or small-incision lenticule extraction (SMILE) for DC patients.
At least 80% of patients with DC will experience BMF. Allogeneic HSCT is done to treat this, which puts patients at risk for both acute (< 100 days since transplant) and chronic (> 100 days) GVHD [18, 19]. GVHD is the clinical manifestation of donor T-cells attacking recipient tissue, and ocular manifestations are common [19]. Approximately 40% of individuals who undergo HSCT will experience acute GVHD, and 30% will experience chronic GVHD [20, 21]. Of patients who undergo allogeneic HSCT, 40–60% will have ocular involvement (without GVHD), and 60–90% with acute or chronic GVHD will have ocular manifestations [22]. Ocular GVHD can present as dry eye (most common), keratoconjunctivitis, corneal ulceration, peripheral corneal melting, corneal vascularization, and acquisition of ocular allergy from the donor [19, 22, 23]. Using the mentioned percentages, the lifetime probability of a patient with DC having acute GVHD is 19–29%, and the probability of chronic GVHD with ocular manifestations is 14–22%.
Conclusion
In summary, DC represents a range of mucocutaneous manifestations of BMF secondary to premature telomere shortening. Figure 4 outlines the main clinical manifestations and clinical course in patients with DC.

Potential clinical course and outcomes of DC
While some patients with DC may not have prominent ocular findings, others will present primarily with eye complaints. Regardless of patient phenotype, the significant likelihood of a DC patient having bone marrow failure and needing hematopoietic stem cell transplant puts these patients at substantial risk for acute and chronic GVHD with or without ocular complications. When considering the impact of limbal stem cell deficiency on healing, in conjunction with allogeneic transplant complications, we do not recommend elective corneal refractive surgery such as LASIK, PRK, or SMILE in patients with DC.
References
Savage SA, Alter BP. Dyskeratosis congenita. Hematol Oncol Clin N Am. 2009. https://doi.org/10.1016/j.hoc.2009.01.003.
Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: dyskeratosis congenita. Eur J Hum Genet. 2011. https://doi.org/10.1038/ejhg.2011.90.
Mialou V, Leblanc T, de Latour RP, Dalle J-H, Socié G. Dyskeratosis congenita: an update. Arch Pédiatrie. 2013;20(3):299–306. https://doi.org/10.1016/j.arcped.2012.12.003.
Savage SA. Dyskeratosis congenita. University of Washington, Seattle; 1993. http://www.ncbi.nlm.nih.gov/pubmed/20301779. Accessed May 17, 2019.
García MSF, Teruya-Feldstein J. The diagnosis and treatment of dyskeratosis congenita: a review. J Blood Med. 2014;5:157. https://doi.org/10.2147/JBM.S47437.
Savage SA. Beginning at the ends: telomeres and human disease. F1000Research. 2018. https://doi.org/10.12688/f1000research.14068.1.
Vulliamy TJ, Knight SW, Mason PJ, Dokal I. Very short telomeres in the peripheral blood of patients with X-linked and autosomal dyskeratosis congenita. Blood Cells Mol Dis. 2001. https://doi.org/10.1006/bcmd.2001.0389.
Hornsby PJ. Telomerase and the aging process. Exp Gerontol. 2007;42(7):575–81. https://doi.org/10.1016/j.exger.2007.03.007.
Kirwan M, Dokal I. Dyskeratosis congenita, stem cells and telomeres. Biochim Biophys Acta. 2009;1792(4):371–9. https://doi.org/10.1016/j.bbadis.2009.01.010.
Dyskeratosis Congenita—NORD (National Organization for Rare Disorders). https://rarediseases.org/rare-diseases/dyskeratosis-congenita/. Accessed 17 May 2019.
Ratnasamy V, Navaneethakrishnan S, Dushyanthi Sirisena N, et al. Dyskeratosis congenita with a novel genetic variant in the DKC1 gene: a case report. BMC Med Genet. 2018. https://doi.org/10.1186/s12881-018-0584-y.
Tsilou ET, Giri N, Weinstein S, Mueller C, Savage SA, Alter BP. Ocular and orbital manifestations of the inherited bone marrow failure syndromes: Fanconi anemia and dyskeratosis congenita. Ophthalmology. 2010;117(3):615–22. https://doi.org/10.1016/j.ophtha.2009.08.023.
Daniels JT, Harris AR, Mason C. Corneal epithelial stem cells in health and disease. 2006. https://link.springer.com/content/pdf/10.1007%2Fs12015-006-0053-4.pdf. Accessed 20 May 2019.
Aslan D, Akata RF, Holme H, Vulliamy T, Dokal I. Limbal stem cell deficiency in patients with inherited stem cell disorder of dyskeratosis congenita. Int Ophthalmol. 2012;32(6):615–22. https://doi.org/10.1007/s10792-012-9611-8.
Aslan D, Akata RF. Dyskeratosis congenita and limbal stem cell deficiency. Exp Eye Res. 2010;90(3):472–3. https://doi.org/10.1016/J.EXER.2009.12.008.
Alter BP, Giri N, Savage SA, Rosenberg PS. Cancer in dyskeratosis congenita. Blood. 2009. https://doi.org/10.1182/blood-2008-12-192880.
Dokal I. Dyskeratosis congenita. ASH Educ Progr B. 2011;2011(1):480–6. https://doi.org/10.1182/ASHEDUCATION-2011.1.480.
Ayas M. Hematopoietic cell transplantation in Fanconi anemia and dyskeratosis congenita: a minireview. Hematol Oncol Stem Cell Ther. 2017. https://doi.org/10.1016/j.hemonc.2017.05.014.
Nassar A, Tabbara KF, Aljurf M. Ocular manifestations of graft-versus-host disease. Saudi J Ophthalmol Off J Saudi Ophthalmol Soc. 2013;27(3):215–22. https://doi.org/10.1016/j.sjopt.2013.06.007.
Jacobsohn DA, Vogelsang GB. Acute graft versus host disease. Orphanet J Rare Dis. 2007;2:35. https://doi.org/10.1186/1750-1172-2-35.
Arai S, Arora M, Wang T, et al. Increasing incidence of chronic graft-versus-host disease in allogeneic transplantation: a report from the Center for International Blood and Marrow Transplant Research. Biol Blood Marrow Transplant. 2015;21(2):266–74. https://doi.org/10.1016/j.bbmt.2014.10.021.
Nassiri N, Eslani M, Panahi N, Mehravaran S, Ziaei A, Djalilian AR. Ocular graft versus host disease following allogeneic stem cell transplantation: a review of current knowledge and recommendations. J Ophthalmic Vis Res. 2013;8(4):351–358. http://www.ncbi.nlm.nih.gov/pubmed/24653823. Accessed 20 May 2019.
Tabbara KF, Al-Ghamdi A, Al-Mohareb F, et al. Ocular findings after allogeneic hematopoietic stem cell transplantation. Ophthalmology. 2009;116(9):1624–9. https://doi.org/10.1016/J.OPHTHA.2009.04.054.
Acknowledgements
Funding
Research to Prevent Blindness NY, NY funded the Rapid Service Fees.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosure
Madeline B. Heiland, Majid Moshirfar, David B. Rosen, Yasmyne C. Ronquillo, and Phillip C. Hoopes have nothing to declare.
Compliance with Ethical Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article, go to https://doi.org/10.6084/m9.figshare.8668274.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License ( http://creativecommons.org/licenses/by-nc/4.0/ ), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Heiland, M.B., Moshirfar, M., Rosen, D.B. et al. Dyskeratosis Congenita and Corneal Refractive Surgery. Ophthalmol Ther 8, 361–365 (2019). https://doi.org/10.1007/s40123-019-0200-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-019-0200-z