
Abstract
Introduction
Ranibizumab is an inhibitor of vascular endothelial growth factor-A (anti-VEGF) approved for the treatment of neovascular age-related macular degeneration (nAMD). The treat and extend (T&E) regimen can potentially reduce the burden of clinic visits compared with a pro re nata (PRN) regimen. Retrospective, interim analyses of clinical effectiveness, treatment and resource use patterns were conducted using real-world data in England and Wales from the TERRA study.
Methods
Two cohorts, those switching from a PRN to a T&E regimen (‘prior PRN’) and those initiating ranibizumab on the T&E regimen as their first anti-VEGF therapy (‘anti-VEGF-naïve’) were enrolled in TERRA. Retrospective clinical assessments were gathered from medical records, while resource use patterns were collected via an operating cost questionnaire completed by each study site.
Results
At the interim analysis cut-off date (15 November 2016), 11 sites had enrolled 145 patients (prior PRN: n = 110; anti-VEGF-naïve: n = 35). Mean change from baseline (date of first injection) in visual acuity and central subfield retinal thickness to 12 months was +7.6 Early Treatment Diabetic Retinopathy Study letters [95% confidence interval (CI) 2.8, 12.4; p = 0.003; n = 27] and −67.7 μm (95% CI −106.5, −28.9; p = 0.001, n = 29), respectively, in the anti-VEGF-naïve cohort. Most T&E clinics were run as one-stop services (same-day monitoring and injection), whereas 4/10 PRN clinics were run as two-stop services (monitoring and injection on different days). In general, one-stop clinics used less staff resources and were likely to be shorter in duration for healthcare providers than the cumulative time spent for two-stop clinics.
Conclusion
This is the first real-world observational study conducted in England and Wales demonstrating the effectiveness of the ranibizumab T&E regimen in anti-VEGF-naïve patients. T&E is compatible with one-stop clinic services, which these real-world data suggest to be less resource intensive than two-stop clinic services, possibly providing a dosing regimen beneficial to both patients and resource burden in UK clinical practice.
Funding: Novartis Pharmaceuticals UK Limited.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Age-related macular degeneration (AMD) is the leading cause of irreversible vision loss in people aged 50 years or older [1]. In the UK, the wet or neovascular (nAMD) form of the disease has an estimated prevalence of 1.2% in patients aged over 50 years, rising to 6.3% in patients aged over 80 years, corresponding to a total number of 263,000 cases [1]. Progressive loss of visual acuity (VA) during nAMD progression can have a significant impact on patient quality of life and may result in blindness [2]. Due to the long-term nature of the disease, it is now recognised that the current status of nAMD care in the UK places significant burden on both healthcare services and patients alike; optimising service models whilst avoiding compromises to patient care is therefore desirable [3].
Intravitreal vascular endothelial growth factor-A (VEGF-A) inhibitors (anti-VEGF), have proven effective in the treatment of nAMD by improving VA [4], and are now standard first line therapy for nAMD in the UK [5, 6]. In Europe, the anti-VEGF ranibizumab is licensed to be administered according to three regimens: ‘fixed dosing’, ‘pro re nata’ (PRN; meaning treat as needed) and ‘treat and extend’ (T&E) [5]. The PRN regimen requires regular monitoring visits in order to assess disease activity and determine whether an injection should be given. In contrast, the T&E regimen involves the gradual extension of time between treatment visits for patients who have achieved maximum VA and/or there are no signs of disease activity without the need for additional monitoring [5, 7], and has also been shown in both randomised controlled trials [8,9,10] and recent real-world observational studies [11,12,13] to be effective at improving VA. The T&E regimen has been shown to be either non-inferior or more effective in terms of VA gains in comparison with the PRN regimen, in addition to resulting in fewer clinic visits [13,14,15]. The ranibizumab T&E regimen is also most commonly associated with a one-stop clinic service, in which both monitoring and treatment can occur on the same day. As such, the T&E regimen may be a more viable strategy in the real world for the long-term management of nAMD, possessing the potential to increase ophthalmology clinic capacity, reduce resource use [14], provide flexibility with regard to scheduling subsequent visits, and allow for virtual clinic monitoring [16]. Furthermore, the ranibizumab T&E regimen has been shown to be cost-effective compared to the licensed dosing for aflibercept (fixed monthly dosing for three consecutive doses, followed by one injection every two months; the treatment interval may then be gradually increased to maintain stable visual and/or anatomic outcomes after 12 months) from a UK health service perspective [6, 17].
Since the update to the European licence of ranibizumab in September 2014 to include the T&E posology, there has been widespread adoption of the T&E regimen within the National Health Service (NHS) in England and Wales. This change has resulted in three distinct groups of patients within nAMD services in the real world: (1) those that have remained on the PRN regimen, usually with regular visits for monitoring; (2) those that have been switched to a T&E regimen in an attempt to reduce the frequency of clinic visits; and (3) those that have started on a T&E regimen from the outset, with three loading doses and further injections according to the T&E posology.
At this early stage of experience with the T&E regimen in the UK, it was desirable to evaluate the impact of the licence update on the treatment patterns and performance of ranibizumab T&E in terms of visual outcomes, and to investigate whether the majority of patients could truly be extended (i.e., have increased intervals between injection visits without the need for additional monitoring) regardless of whether they were switched from a PRN to a T&E regimen or were initiated on T&E from the outset.
The objective of the observational TERRA (Treat and extend ranibizumab regimen for neovascular age-related macular degeneration) study was to describe the real-world clinical effectiveness and resource utilisation patterns of the ranibizumab T&E regimen both in patients who have switched from a PRN to a T&E regimen as well as in anti-VEGF-naïve patients initiated on ranibizumab T&E from the outset. This paper provides an overview of treatment and resource use patterns across large teaching centres and smaller district hospitals in England and Wales, and presents the interim results of visual outcomes from the retrospective data collection component of the TERRA study.
Methods
TERRA is a phase 4, multicentre, observational, non-interventional study conducted at 13 sites in England and Wales, with both retrospective and prospective components. The study protocol (MREC: 16/YH/0336; IRAS ID: 205633) was approved by the Multicentre Research Ethics Committee and written consent was obtained from all patients prior to enrolment and data collection.
Patient Eligibility
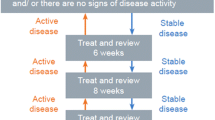
The study enrolled two cohorts of patients with nAMD that were being treated with ranibizumab: a cohort that switched from a PRN to a T&E regimen (‘prior PRN cohort’) and a cohort that received no prior anti-VEGF therapy before initiating ranibizumab according to the T&E regimen (‘anti-VEGF-naïve cohort’) (Fig. 1).

TERRA Study Design. Enrolled set. aIn the prior PRN cohort, baseline visit was defined as the date of switch from the PRN to the T&E regimen (i.e., the date of the first ranibizumab injection under the T&E regimen) after at least 12 months of treatment on the PRN regimen; bIn the anti-VEGF-naïve cohort, the baseline visit for the anti-VEGF-naïve cohort was defined as the initiation date (and first ranibizumab injection) on the T&E regimen; at least 12 months of treatment on the T&E regimen was required for the anti-VEGF-naïve cohort. PRN pro re nata, T&E treat and extend, VEGF vascular endothelial growth factor
Eligibility criteria included: (1) adult patients ≥50 years of age with active choroidal neovascularisation secondary to AMD in one or both eyes at the time of diagnosis; (2) medical records providing at least 12 months of retrospective data on either the ranibizumab PRN regimen prior to switching to T&E (prior PRN cohort), or the T&E regimen (anti-VEGF-naïve cohort) immediately prior to enrolment; and (3) no prior treatment with an anti-VEGF agent (i.e., bevacizumab or aflibercept) other than ranibizumab in the study eye (defined as the eye first treated with a T&E regimen). Patients were able to withdraw consent and discontinue from the study at any time without affecting their medical care.
Study Design
In the prior PRN cohort, the primary endpoint of the study was change in VA [in Early Treatment Diabetic Retinopathy Study (ETDRS) letters] from regimen switch to 12 months of treatment on the T&E regimen. In the anti-VEGF-naïve cohort, the primary endpoint of the study was change in VA from initiation of ranibizumab treatment on the T&E regimen to 12 and 24 months of treatment. The endpoints will be evaluated separately in their respective cohorts. The period required to achieve 24 months’ follow-up data from all patients was estimated to be from 31 August 2016 to 28 February 2018. It should be noted that the definition of baseline was different for the two cohorts; for the prior PRN cohort, baseline was defined as the date of switch from a PRN to a T&E regimen (i.e., the date of the first ranibizumab injection under the T&E schedule), whereas baseline for the anti-VEGF-naïve cohort was defined as the initiation date (and first ranibizumab injection) on the T&E regimen. Thus, these two baseline definitions represent different time points in the patient cohorts’ respective treatment pathways.
The results of both the pre-specified and non-pre-specified interim analyses (with a data cut-off date of 15 November 2016) from the retrospective component of the study are presented here. Full efficacy and safety results from both cohorts will be presented in a future publication following completion of the prospective observational component of the study. The results of the interim analyses reported here will not impact the conduct of the ongoing study. Final study conclusions will be drawn from the primary analyses performed on the full data set following completion of the prospective phase of the study.
For each patient in both cohorts, the date of study enrolment was defined as the time point at which the consent form was signed. In the anti-VEGF-naïve cohort, data [e.g., patient demographics, medical history, VA, central subfield retinal thickness (CSRT)] were collected from patients’ case notes retrospectively for all visits that occurred, beginning from the first ranibizumab injection administered. For the prior PRN cohort, these data (e.g., patient demographics, medical history, VA, CSRT) were collected retrospectively for 12 months of PRN treatment prior to study enrolment. For visits after the date of study enrolment, i.e., during the prospective part of the study, data for both cohorts were collected from patients’ case notes after each completed visit in an ongoing, observational manner.
The study protocol did not influence the way that patients were treated in terms of any re-treatment criteria. Therefore, the frequencies of clinic visits, ranibizumab injections, and clinical assessments (i.e., VA and CSRT) were also not pre-specified by the study protocol. It was assumed that the patients were treated according to routine medical practice and local prescribing information based on the licensed ranibizumab posology, which for the T&E regimen is a monthly injection until maximum VA is achieved and/or there are no signs of disease activity (i.e., no change in VA, optical coherence tomography, and other signs and symptoms of the disease) [5]. Therefore, patients on the T&E regimen had their treatment intervals and monitoring determined by their physician accordingly. To accurately capture real-world evidence, only data that were available in the case notes documented as part of patients’ routine standard care were collected to avoid any influence from data collection on clinical decision-making.
Handling of Missing Data
If the data from the baseline visit were incomplete or there was no visit on the recorded baseline date, data from no more than 30 days earlier were considered valid. In the prior PRN cohort, the date of the 12-month pre-baseline time point was defined as baseline (switch date) minus 365 days. If there was no visit exactly on this date, the nearest data from a visit plus or minus 30 days were considered valid. In the event of two equidistant visits from minus 365 days, data from the earlier visit were prioritised. In the anti-VEGF-naïve cohort, the date of the 12-month post-baseline time point was defined as baseline plus 365 days. If there was no visit exactly on this date, the nearest data from a visit plus or minus 90 days were considered valid. In the event of two equidistant visits from plus 365 days, data from the later visit were prioritised. Missing data outside of these criteria were not imputed.
Assessment of Healthcare Resource Use
Resource use was assessed through an operating cost questionnaire (Supplementary Table 1) [18], which was completed by the principal investigator or a member of their study team once at each study site on initiation of the study (enrolment of the first patient). The questionnaire collected information on healthcare staff resource use required to administer ranibizumab and monitor outcomes, such as clinic type (e.g., one-stop and separate monitoring and injection clinics, defined as a two-stop clinic service), the length of time to deliver each clinic, the estimated time patients spent in clinic at each visit, and the staff resource use [including type (e.g., clinicians, administrative coordinators, healthcare assistants, etc.), post, NHS grade/band, roles in clinic, and time spent by staff in a clinic session] required to run the clinics. For the purposes of the questionnaire, a one-stop clinic service was defined as a service in which monitoring and investigations were conducted on the same day as ranibizumab was administered. A two-stop clinic service was defined as a visit where the monitoring and investigations occurred on separate days to the administration of ranibizumab.
Assessment of Clinical Outcomes
Clinical outcomes were measured using VA and CSRT. VA was assessed and manually converted on-site to ETDRS letters via a standard conversion chart [19], where applicable (VA data not provided in ETDRS letters were excluded from analyses). CSRT was measured by spectral domain optical coherence tomography.
Statistical Analysis
The overall target sample size for this study was set at 300 patients. The cut-off for the interim analyses was defined as 15 November 2016 or when 150 patients were enrolled, whichever occurred first. Assuming a standard deviation (SD) of 12.9 letters for the change in VA from baseline to 24 months [11], and conservatively assuming the same SD for the change in VA from baseline to 12 months, the expected width of a two-sided 95% confidence interval (CI) for 100 prior PRN and 50 anti-VEGF-naïve patients was calculated to be 5.0 and 7.2 letters, respectively.
There were no adjustments made for multiple analyses, due to the descriptive nature of this study. All statistical analyses were performed in SAS software, Version 9.4 (SAS Institute, Cary, NC, USA). For patients with missing values or who withdrew from the study, no imputation was used. The p values and 95% CIs were calculated using a paired t test.
The full analysis set for the interim analysis consisted of all patients who were enrolled in the study and received at least one dose of ranibizumab treatment as part of a PRN or T&E regimen.
Results
At the pre-specified interim analysis data cut-off point of 15 November 2016, 11 out of 13 sites across England and Wales had enrolled 145 patients, of whom 35 patients (24.1%) were in the anti-VEGF-naïve cohort, and 110 patients (75.9%) were in the prior PRN cohort. The full analysis set of the anti-VEGF-naïve cohort consisted of 34 patients; one patient was excluded from the originally-enrolled 35 patients due to missing clinical data, whereas all 110 patients enrolled in the prior PRN cohort were included in the full analysis set. The mean age of patients in the anti-VEGF-naïve cohort was 77.5 years, and 52.9% (n = 18/34) of patients were female. In the prior PRN cohort, the mean age was 82.2 years, and 65.5% (n = 72/110) of patients were female.
Here, the results of the analyses on the retrospectively-collected data from the operating cost questionnaire and on the visual outcomes of both cohorts are presented.
Resource Use
Across the 11 study sites analysed, only one site ran a two-stop T&E clinic; the other ten sites ran one-stop T&E clinics, suggesting that this format is preferred for the T&E regimen. Ten of the 11 sites also ran PRN clinics and, in contrast to the T&E clinics, six sites ran one-stop and four sites ran two-stop PRN clinics.
The mean duration of a one-stop clinic was 4.56 h, while the mean duration for an injection clinic was 3.75 h, and for a monitoring clinic was 3.89 h. From this, a total mean duration of 7.64 h for a two-stop clinic was derived, indicating a longer clinic time for cases when both visits are required.
In terms of the time that patients spent in the different clinic types, patients spent on average 2.6 h (range 2–4, median 2.0, SD 0.9) per visit in one-stop clinics (n = 9 clinics), whereas patients spent on average 1.5 h in the monitoring clinic (range 1–4, median 1.0, SD 1.1, n = 8 clinics) and 1.3 h in the injection clinic (range 1–2, median 1.0, SD 0.5, n = 6 clinics). Although not all patients will require both a monitoring and injection visit each time they attend, these mean values indicate that when patients do require both visits, they are likely to spend a similar, albeit slightly longer, amount of time attending clinics under a two-stop clinic service system (2.8 h).
The total and mean changes in resource use for one-stop clinics and for separate injection and monitoring clinics are shown in Table 1. The total resource use required to run a two-stop clinic service was defined as the sum of the total resource for running the separate injection and monitoring clinics. It should be noted that one other type of clinic was also included under monitoring clinic. The distribution of staff resource types appears to differ between one stop and two-stop clinic services and, curiously, the two-stop clinic services used 2 less clinicians than the one-stop clinic services. Total resource use over almost all staff resource types was greater in the two-stop clinic services versus the one-stop clinic services, particularly amongst administrative coordinators (32 versus 19, respectively) and healthcare assistants (35 versus 26, respectively).
Visual Outcomes After 12 Months of Ranibizumab Treatment on the T&E Regimen in the Anti-VEGF-Naïve Cohort
In the anti-VEGF-naïve cohort, there was a mean (SD) of 12.1 (3.9) clinic visits and 11.4 (4.2) injections in the period before study enrolment (which was longer than 12 months for some patients), supporting the assumption that, in general, these patients received one injection per clinic visit on the T&E regimen. Following study enrolment, patients received a mean (SD) 8.9 (1.4) injections (n = 30) in the period from baseline to 12 months of T&E treatment in this cohort. It should be noted that due to the definition of baseline in this cohort as the start date of ranibizumab injection, these numbers include the monthly ranibizumab loading phase, and thus may be higher than expected for a T&E regimen.
Mean (SD) VA at baseline was 58.5 (16.9) ETDRS letters (n = 27) and mean (SD) CSRT was 364.9 (105.4) μm (n = 29) for patients treated according to the T&E regimen who had valid baseline and 12-month post-baseline assessments (Table 2). During this retrospective period, the mean change from baseline in VA was a gain of 7.6 ETDRS letters (95% CI 2.8, 12.4; p = 0.003; n = 27), and the mean change from baseline in CSRT was −67.7 μm (95% CI −106.5, −28.9; p = 0.001, n = 29), representing statistically significant improvements in both VA and CSRT over the initial 12-month treatment period for patients in the anti-VEGF-naïve cohort.
Additional, non-pre-specified analyses revealed that a large proportion (n = 26/27, 96.3%) of anti-VEGF-naïve patients treated for 12 months on the T&E regimen did not lose 15 ETDRS letters or more from baseline. Furthermore, 59.3% (n = 16/27), 48.1% (n = 13/27), and 25.9% (n = 7/27) of these patients gained five, ten, and 15 ETDRS letters or more after 12 months of treatment, respectively (Fig. 2).

Patients in the anti-VEGF-naïve cohort (n = 27) achieving VA gains (in ETDRS letters) after 12 months of treatment with ranibizumab on the T&E regimen. ETDRS Early Treatment Diabetic Retinopathy Study, T&E treat and extend, VEGF vascular endothelial growth factor
Visual Outcomes from 12 Months Pre-Baseline to Baseline in Patients Treated with Ranibizumab on the PRN Regimen (Prior PRN Cohort)
In the prior PRN cohort, there was a mean (SD) of 7.2 (4.0) clinic visits and 4.7 (1.9) injections in the 12 months prior to baseline (switch from PRN to T&E regimen). It should be noted that these patients were more advanced in their treatment pathway than the anti-VEGF-naïve cohort and had a variable length of treatment history, providing an explanation for why visit and treatment frequencies were lower than in the anti-VEGF-naïve cohort.
At 12 months prior to baseline (date of switch from PRN to T&E regimen), mean (SD) VA in the prior PRN cohort was 57.9 (20.0) ETDRS letters (n = 38) and mean (SD) CSRT was 246.2 (50.8) μm (n = 30); only patients with both valid 12-month pre-baseline and baseline data are included here. At baseline, these patients had a VA of 57.7 (20.6) ETDRS letters (n = 38) with a CSRT of 252.8 (59.4) μm (n = 30) (Table 2). The mean change from pre-baseline to baseline, therefore, was a VA loss of 0.2 ETDRS letters (95% CI −1.9, 1.6; p = 0.855; n = 38), whereas the mean change in CSRT was a gain of 6.6 μm (95% CI −7.5, 20.6; p = 0.348, n = 30), suggesting that the visual outcomes for these patients were stable prior to switching regimens. Results of 12 months treatment on the T&E regimen for this cohort will be presented in the future full publication.
Discussion
The interim analyses presented here represent the first peer-reviewed and published real-world data on the use of the ranibizumab T&E regimen for the treatment of nAMD in the NHS in England and Wales. These data show that the T&E regimen was organised predominantly as a one-stop clinic service, in contrast to the PRN regimen, which was delivered as a two-stop clinic service in over one-third of the study sites surveyed. These data suggest that the T&E regimen is more commonly associated with a one-stop clinic service model. The operating cost questionnaire also evaluated the time spent by patients attending each type of clinic; patients spent a similar, albeit slightly shorter, period of time in a one-stop clinic compared with a two-stop clinic (2.6 versus 2.8 h). Though the one-stop clinic took longer to deliver than each monitoring or injection only clinic, it was shorter than the two-stop combination of a monitoring and injection clinic delivered on separate days.
The questionnaire also evaluated the difference between one-stop and two-stop clinic services in terms of the number and type of personnel required. Interestingly, the data demonstrate that both service models were very similar in terms of the number of clinicians, nurses and optometrists required per individual clinic. However, the two-stop clinic service model needed considerably more administrative staff responsible for patients’ appointments and telephone queries, and more healthcare assistants to perform VA assessments and ensure patient movement between clinical stations, by 13 and 9 staff members, respectively. The findings from this study, therefore, provide a quantitative aspect to the evidence and experience that could be used to support the implementation of a one-stop clinic T&E regimen model to provide a sustainable nAMD service, in preference to the PRN regimen, which was found to be delivered as a two-stop clinic model in several centres.
An important limitation of this study was that healthcare resource use was solely analysed with the data collected from the operating cost questionnaire, which was modified from the questionnaire developed for a different clinical study (IVAN), a large, randomised clinical trial [18]. For the purposes of this study, the questionnaire was modified and deployed to capture resource use between different ranibizumab service models. The use of this non-standard questionnaire with the small sample of study sites could have led to reporting bias, as the subjective interpretation of service pressures may have varied across sites. Nevertheless, in terms of the relative comparison between the one-stop and two-stop clinic service models, it appears the results reported reflect the popular impression amongst healthcare providers that the one-stop clinic service model requires less resources.
As the data have shown that the majority of sites delivered the T&E regimen using a one-stop clinic service model, it could be inferred with some level of confidence that one-stop clinic services and a T&E regimen would be the optimal model for both the provider and the patient. In real-world clinical practice, however, there will be some patients more suited to a PRN regimen due to the low grade nature of their macular disease. Many of the sites participating in this study appeared to be successful in delivering both one-stop and two-stop clinic services; it may be inferred, therefore, that a distinct, dedicated one-stop clinic T&E regimen model can be coordinated alongside an existing PRN two-stop clinic model service with some gains in resource utilisation.
Another limitation of this study was that amongst the 11 sites contributing to this interim analysis, there were no NHS sites in Scotland and Northern Ireland (the sample included sites from England and Wales only); nevertheless, the sample may be considered representative of the UK as a whole, as both large teaching centres and smaller district hospitals were included. In addition, where study sites did not have VA data recorded in ETDRS letters in the case notes, manual conversion to ETDRS letters was required to complete the electronic case report form. Finally, it is acknowledged that there is a scarcity of comparative quantitative studies on resource use in the real world; this is a difficult area to evaluate and further learning points may be discussed in subsequent future publications resulting from the TERRA study.
Focus group discussions have shown that many healthcare professionals believe the current model of nAMD care in the UK is burdensome and therefore not appropriate for the elderly patient population [20]. The time commitment for clinic visits has been shown to place a significant burden on patients and their caregivers; a survey conducted in the United States reported that 72% of nAMD patients were driven to their appointment by a caregiver, and that the average time commitment was 11.7 h, including 9 h of post-appointment recovery time [21]. Studies have shown that one-stop clinic services and the frequency of clinic visits factor in both patient-reported treatment preferences [22, 23] and physician treatment decision-making [21].
A T&E regimen administered as a one-stop clinic service may also reduce patients’ anxiety regarding receiving injections [24, 25], as the likelihood of an injection is already known and understood, which is in contrast to the PRN regimen; however, this may vary from patient to patient and, conversely, those on a PRN regimen may have a sense of hope that they will not receive an injection, compared with a T&E regimen in which they know they will always be treated. Nevertheless, the T&E regimen represents an individualised treatment approach, ensuring that patients receive injections at the frequency most appropriate to their disease status [12]. In addition, the prospective nature of the T&E regimen allows the service to predict the number of patients requiring injections within the next 4 weeks. Thus, the T&E regimen enables personalised treatment while also assisting with planning by enabling the balance of capacity versus demand and ensuring more timely treatment delivery than can be achieved within a reactive service model, such as with the PRN regimen.
In terms of visual outcomes, the interim analyses of the retrospectively-collected data in the anti-VEGF-naïve cohort found a VA gain of 7.6 ETDRS letters (n = 27) and CSRT reduction of 67.7 µm (n = 29) following 12 months of ranibizumab treatment on the T&E regimen. Although this sample size was relatively small, these outcomes are comparable to the early outcomes achieved by treatment-naïve patients on a T&E regimen in other randomised, controlled and real-world studies [9, 10, 13, 26]. It is anticipated that the sample size of this cohort will increase following the end of data collection for the full retrospective and prospective observational components of the TERRA study. For the prior PRN cohort in the 12 months leading up to the switch from a PRN to a T&E regimen, a VA loss of 0.2 ETDRS letters (n = 38) and an increase in CSRT of 6.6 µm (n = 30) were found, indicating that VA and CSRT remained relatively stable for this period.
The results of this interim analysis are potentially of great interest to the NHS in the UK, particularly because the long-term nature of nAMD is projected to result in increased pressure on NHS capacity, and the UK has the fewest consultant ophthalmologists per 100,000 population in the European Union [3]. Studies of retinal specialist centres in the United States have shown that nAMD poses a significant burden on resources, with the management of care accounting for around one-fifth of the work week, and many physicians feeling that resource use in terms of materials and office space was disproportionate compared with other non-nAMD ophthalmology services [21]. This feeling is consistent with the consensus in the UK that innovations in models of care and treatment are needed to address the burgeoning shortcomings of the healthcare system and the increased cost burden of nAMD to the NHS [3].
Conclusion
This is the first real-world observational study conducted in the English and Welsh setting to quantify both healthcare resource use and early clinical outcomes of ranibizumab administered according to the T&E regimen in patients who are anti-VEGF-naïve. The ranibizumab T&E regimen, when delivered as a one-stop clinic service, could provide a practical, effective and patient-friendly dosing schedule that may be beneficial to healthcare resource for nAMD services.
References
Owen CG, Jarrar Z, Wormald R, Cook DG, Fletcher AE, Rudnicka AR. The estimated prevalence and incidence of late stage age related macular degeneration in the UK. Br J Ophthalmol. 2012;96:752–6.
Bunce C, Xing W, Wormald R. Causes of blind and partial sight certifications in England and Wales: April 2007–March 2008. Eye (Lond). 2010;24(11):1692–9.
Amoaku W, Blakeney S, Freeman M, Gale R, Johnston R, Kelly SP, et al. Action on AMD. Optimising patient management: act now to ensure current and continual delivery of best possible patient care. Eye (Lond). 2012;26(Suppl 1):S2–21.
Solomon SD, Lindsley K, Vedula SS, Krzystolik MG, Hawkins BS. Anti-vascular endothelial growth factor for neovascular age-related macular degeneration. Cochrane Database Syst Rev. 2014;(8):CD005139.
European Medicines Agency (EMA). Lucentis 10 mg/ml solution for injection. Summary of Product Characteristics. 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000715/WC500043546.pdf. Accessed Feb 2016.
European Medicines Agency (EMA). Eylea 40 mg/ml solution for injection in pre-filled syringe. Summary of product characteristics. 2012. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002392/WC500135815.pdf. Accessed Feb 2016.
Spaide R. Ranibizumab according to need: a treatment for age-related macular degeneration. Am J Ophthalmol. 2007;143(4):679–80.
Berg K, Hadzalic E, Gjertsen I, Forsaa V, Berger LH, Kinge B, et al. Ranibizumab or bevacizumab for neovascular age-related macular degeneration according to the lucentis compared to avastin study treat-and-extend protocol: two-year results. Ophthalmology. 2016;123(1):51–9.
Berg K, Pedersen TR, Sandvik L, Bragadottir R. Comparison of ranibizumab and bevacizumab for neovascular age-related macular degeneration according to LUCAS treat-and-extend protocol. Ophthalmology. 2015;122(1):146–52.
Wykoff CC, Croft DE, Brown DM, Wang R, Payne JF, Clark L, et al. Prospective trial of treat-and-extend versus monthly dosing for neovascular age-related macular degeneration: TREX AMD 1-year results. Ophthalmology. 2015;122(12):2514–22.
Abedi F, Wickremasinghe S, Islam AF, Inglis KM, Guymer RH. Anti-VEGF treatment in neovascular age-related macular degeneration: a treat-and-extend protocol over 2 years. Retina. 2014;34(8):1531–8.
Arnold JJ, Campain A, Barthelmes D, Simpson JM, Guymer RH, Hunyor AP, et al. Two-year outcomes of “treat and extend” intravitreal therapy for neovascular age-related macular degeneration. Ophthalmology. 2015;122(6):1212–9.
Johnston RL, Carius HJ, Skelly A, Ferreira A, Milnes F, Mitchell P. A retrospective study of ranibizumab treatment regimens for neovascular age-related macular degeneration (nAMD) in Australia and the United Kingdom. Adv Ther. 2017;34(3):703–12.
Hatz K, Prunte C. Changing from a pro re nata treatment regimen to a treat and extend regimen with ranibizumab in neovascular age-related macular degeneration. Br J Ophthalmol. 2016;100(10):1341–5.
Hatz K, Prunte C. Treat and extend versus Pro Re Nata regimens of ranibizumab in neovascular age-related macular degeneration: a comparative 12 month study. Acta Ophthalmol. 2017;95(1):e67–72.
Tsaousis KT, Empeslidis T, Konidaris VE, Kapoor B, Deane J. The concept of virtual clinics in monitoring patients with age-related macular degeneration. Acta Ophthalmol. 2016;94(5):e353–5.
Ghosh W, Wickstead R, Claxton L, Kusel J, Taylor M, Fleetwood K, et al. The cost-effectiveness of ranibizumab treat and extend regimen versus aflibercept in the UK. Adv Ther. 2016;33(9):1660–76.
Chakravarthy U, Harding SP, Rogers CA, Downes S, Lotery AJ, Dakin HA, et al. A randomised controlled trial to assess the clinical effectiveness and cost-effectiveness of alternative treatments to inhibit VEGF in age-related choroidal neovascularisation (IVAN). Health Technol Assess. 2015;19(78):1–298.
Gregori NZ, Feuer W, Rosenfeld PJ. Novel method for analyzing snellen visual acuity measurements. Retina. 2010;30(7):1046–50.
Townsend D, Reeves BC, Taylor J, Chakravarthy U, O’Reilly D, Hogg RE, et al. Health professionals’ and service users’ perspectives of shared care for monitoring wet age-related macular degeneration: a qualitative study alongside the ECHoES trial. BMJ Open. 2015;5(4):e007400.
Prenner JL, Halperin LS, Rycroft C, Hogue S, Williams Liu Z, Seibert R. Disease burden in the treatment of age-related macular degeneration: findings from a time-and-motion study. Am J Ophthalmol. 2015;160(4):725–31.
Baxter JM, Fotheringham AJ, Foss AJ. Determining patient preferences in the management of neovascular age-related macular degeneration: a conjoint analysis. Eye (Lond). 2016;30(5):698–704.
Vennedey V, Danner M, Evers SM, Fauser S, Stock S, Dirksen CD, et al. Using qualitative research to facilitate the interpretation of quantitative results from a discrete choice experiment: insights from a survey in elderly ophthalmologic patients. Patient Prefer Adherence. 2016;10:993–1002.
Droege KM, Muether PS, Hermann MM, Caramoy A, Viebahn U, Kirchhof B, et al. Adherence to ranibizumab treatment for neovascular age-related macular degeneration in real life. Graefes Arch Clin Exp Ophthalmol. 2013;251(5):1281–4.
Thetford C, Hodge S, Harding S, Taylor S, Knox PC. Living with age-related macular degeneration treatment: patient experiences of being treated with ranibizumab (Lucentis)(R) intravitreal injections. Br J Vis Impair. 2013;31(2):89–101.
Kim LN, Mehta H, Barthelmes D, Nguyen V, Gillies MC. Metaanalysis of real-world outcomes of intravitreal ranibizumab for the treatment of neovascular age-related macular degeneration. Retina. 2016;36(8):1418–31.
Acknowledgements
This study and all costs associated with the development of this manuscript, including the article processing charges, were funded by Novartis Pharmaceuticals UK Limited. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval to the version to be published. The authors acknowledge the principal investigators and co-investigators at all of the TERRA study sites for their ongoing contributions and assistance with this study. The authors also acknowledge Rose Wickstead, MPharm, and Lisa Wulund, PhD, from Costello Medical Consulting Ltd (Cambridge, UK), for medical writing and editorial assistance in preparing this manuscript for publication, based on the authors’ input and direction, with funding from Novartis Pharmaceuticals UK Limited. Dr. Praveen J. Patel is supported by the National Institute for Health Research (NIHR) Biomedical Research Centre based at Moorfields Eye Hospital NHS Foundation Trust and the UCL Institute of Ophthalmology. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health.
Compliance with Ethics Guidelines
This study was conducted in accordance with the current version of the applicable regulatory and International Conference on Harmonisation (ICH)-Good Clinical Practice (GCP) requirements, the ethical principles that have their origin in the principles of the Declaration of Helsinki, and the local laws of the countries involved. Written consent was obtained from all patients prior to enrolment and data collection.
Disclosures
Yit Yang: Research grants from Alcon, Allergan, Alimera Sciences, Bayer, Thrombogenics; Consulting fees from Allergan, Alimera Sciences, Bayer, Pfizer; Honoraria from Alcon, Allergan, Alimera Sciences, Bayer, Novartis, Pfizer, Thrombogenics; Advisory board for Novartis. Louise Downey: Travel support from Novartis, Bayer, Allergan; Advisory board and honoraria from Novartis, Bayer, Allergan, Alimera, Alcon, Oraya, Thrombogenics; Speaker’s fees from Bayer, Novartis, Alimera. Hemal Mehta: Research grants, honoraria and travel support from Allergan, Bayer, Novartis. Bushra Mushtaq: Consulting fees from and advisory board for Novartis, Bayer, Alimera Sciences, ORAYA Therapeutics. Niro Narendran: Advisory board and travel support from Novartis, Bayer; Speaker’s fees from Novartis, Heidelberg. Nishal Patel: Consulting and speaker’s fees from Novartis, Bayer, Roche, Allergan. Praveen J. Patel: Consulting fees and travel support from Bayer; Honoraria from Bayer, Novartis. Filis Ayan: Employee of Novartis Pharmaceuticals UK Limited. Kara Gibson: Employee of Novartis Pharmaceuticals UK Limited. Franklin Igwe: Employee of Novartis Pharmaceuticals UK Limited. Pete Jeffery: Consultant/Contractor at Novartis, with services also currently provided to GlaxoSmithKline.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/8C18F0603703D331.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Yang, Y., Downey, L., Mehta, H. et al. Resource Use and Real-World Outcomes for Ranibizumab Treat and Extend for Neovascular Age-Related Macular Degeneration in the UK: Interim Results from TERRA. Ophthalmol Ther 6, 175–186 (2017). https://doi.org/10.1007/s40123-017-0091-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-017-0091-9