
Abstract
A 10-year-old male was referred for evaluation of a right orbital mass present for 3 weeks with associated tenderness to palpation. Magnetic resonance imaging (MRI) and computed tomography imaging (CT) revealed a solid mass centered in the frontal bone with extension into the orbit. Surgical excision and histologic analysis of the lesion was consistent with a diagnosis of a Giant Cell Tumor (GCT) of the frontal bone. The patient tolerated the procedure without complication and is doing well upon follow-up.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary orbital bone tumors compose less than 2% of all orbital tumors [1]. Of these, GCT is an extremely rare osseous neoplasm that can cause significant bone destruction and has a high propensity for recurrence if not treated appropriately. While GCTs are commonly believed to be benign, there are rare case reports of possible malignant transformation [2–4]. Due to its rarity, much uncertainty and debate exist as to the proper management of these and other primary orbital bone tumors. We describe our experience treating a 10-year-old male who presented with a rare GCT of the frontal bone that was treated with surgical excision. This report is in full compliance with the Declaration of Helsinki and the current Health Insurance Portability and Accountability Act regulations. Informed consent was obtained from the patient for being included in the study.
Case Report
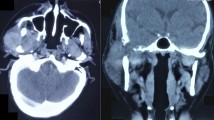
A 10-year-old male was referred for evaluation of a 3-week history of an enlarging right orbital mass (Fig. 1). Past ocular history was notable for a long-standing exotropia (XT). The patient endorsed mild pain to palpation of the lesion but denied vision changes. There was no recent or remote history of periorbital trauma, and he was otherwise healthy. His visual acuity was measured to be 20/25 without correction in each eye. Intraocular pressures were 19 and 20 mmHg in the right (OD) and left (OS) eyes, respectively. Extraocular movements were full, and alternate cover testing revealed a 15-prism diopter XT. Hertel exophthalmometry was symmetric without relative proptosis. He exhibited 2–3 mm of hypoglobus. Cranial nerves V1, V2, V3, and VII were found to be normal. The mass was firm and fixed to the superolateral orbital rim, and was tender to palpation. The remainder of the anterior and posterior segment examinations of both eyes was unremarkable.

External photographs show right brow lesion from before (top), 1-month (middle) and 4-months (bottom) after surgical excision
The patient underwent magnetic resonance imaging (MRI) with and without contrast as well as computed tomography (CT) without contrast for further evaluation and surgical planning. On MRI, there was a 2.7 cm mass centered in the bone of the superolateral right orbit with associated bone destruction and extension into the superolateral right extraconal orbit and abnormal signal in the right frontal bone and sphenotemporal buttress (Fig. 2). CT revealed bone destruction with only thin bone remaining between the tumor bed and the intracranial space. The patient underwent an uncomplicated right orbitotomy with excision of the mass. Given his lack of an eyelid crease, a sub-brow incision was used to access the mass, which was found to be firmly adherent to the bone. The surrounding periosteum was incised around the mass circumferentially with cutting cautery, and the mass was then dissected free using a Freer periosteal elevator. There was a residual defect in the superolateral orbital rim. The incision was closed in layers. Histopathology revealed the lesion to be composed of osteoclast-type giant cells evenly distributed among mononuclear cells with ill-defined cell borders and associated fragments of reactive woven bone rimmed by osteoblasts (Fig. 3). Both the mononuclear and giant cells stained positively for CD68 on immunohistochemistry. Only rare scattered cells stained with CD1a. Ki-67 demonstrated a high proliferation index with 70% of the mononuclear cells being positive. Further analysis of the lesion revealed a loss of one normal copy of chromosomes 5 and 12 with additional material of unknown origin added to the long arm of chromosome 16, and gain of a chromosome 21 and of a marker chromosome of unknown origin. The chromosomal findings indicate a neoplastic process which, in the setting of the imaging and histopathology features, was consistent with giant cell tumor of the bone. Given that the mass was grossly resected, and further curettage would necessitate a craniotomy with reconstruction, observation was elected.

Post-contrast T1 sequence with fat suppression MR images before (top), 1 month (middle) and 4 months (bottom) after surgical resection

Intraoperative photographs (a) highlight appearance of lesion (top) and the extent of surrounding bone destruction after excision (middle), and gross pathology of excised lesion is shown (bottom). Histologic analysis (b) at low magnification (top) reveals areas of reactive woven bone (star), and high magnification (bottom) shows multinucleated giant cells (arrows) scattered among mononuclear cells (H&E staining; scale bars are 200 μm top and 50 μm for bottom)
At the 1-month follow-up, MRI with and without contrast revealed residual enhancement that reduced dramatically by his 4-month follow-up scan. Examination at 4 months revealed a palpable defect in the superolateral bony rim; otherwise, he is asymptomatic and continues to have his longstanding well-controlled XT (Figs. 1, 2). He is healing appropriately and denies pain or other symptoms of concern.
Discussion
As the incidence of GCT of the frontal bone is extremely rare, the majority of studies in the literature have been case reports [5–7]. The World Health Organization recognizes three distinct types of GCTs arising from the bone, tendon sheath, or soft tissue [8]. GCT most frequently occurs in the epiphyses of long bones and rarely occur in the orbit [9]. Radiographic features of GCT include an osteolytic lesion that is radiolucent and results in bony erosions with sharp margins on CT scan. MRI shows a well-circumscribed lesion that exhibits isointensity on T1-weighted images and hypointensity on T2-weighted images. Typically, the lesion enhances with contrast on both imaging modalities [10, 11]. Histologically, GCT arising from bone are composed of stromal mononuclear cells and giant cells whose histogenesis is controversial. The mononuclear cell represents the true neoplastic component, while the multinucleated giant cells have an osteoclast-like phenotype and express histocytic lineage markers. The histologic differential diagnosis of GCT include other processes in which multinucleated giant cells can be found, such as giant cell granuloma, “brown tumor” of hyperparathyroidism, non-ossifying fibroma, osteoblastoma, aneurysmal bone cyst, and Langerhans cell histiocytosis [12, 13]. While GCT is generally considered a benign tumor, it has the potential for malignant transformation [3, 4, 11]; therefore, careful observation is essential. The lesion often grows slowly and can cause significant bone destruction. The current treatment goal is for total surgical resection of the tumor without adjuvant radiation therapy, which has been shown to have the lowest recurrence rate. Indeed, Kamoshima et al. described a 2-year-old female patient with recurrent GCT of the frontal bone that underwent two partial surgical removals of the tumor with recurrence before total resection of the lesion, surrounding bone, and frontal base dura mater was curative [14]. The reported incidence in the literature of non-recurrence after total resection of a frontal bone lesion has been up to 30 months [15]. If total resection cannot be achieved, the combination of subtotal resection and radiation therapy shows a similar low recurrence. Other therapeutic strategies may lead to increased recurrence rates [16]. Continued long-term follow-up will be important in the ongoing management of our patient.
Conclusion
In summary, we present a rare case of a rapidly enlarging superotemporal orbital mass in a ten-year-old boy, which proved to be GCT of the frontal bone. While total surgical resection could not be achieved without a risk of added morbidity, gross resection, followed by close surveillance has resulted in no recurrence with 4 months of follow-up. Although rare, the ophthalmologist should be aware that GCT might present with orbital involvement.
References
Selva D, White VA, O’Connell JX, Rootman J. Primary bone tumors of the orbit. Surv Ophthalmol. 2004;49:328–42.
Herize M, Fein JM. Malignant giant cell tumor of the frontal bone. NY State J Med. 1984;84:201–2.
Brien EW, Mirra JM, Kessler S, Suen M, Ho JK, Yang WT. Benign giant cell tumor of bone with osteosarcomatous transformation (“dedifferentiated” primary malignant GCT): report of two cases. Skeletal Radiol. 1997;26:246–55.
Dahlin DC, Cupps RE, Johnson-Jr EW. Giant-cell tumor: a study of 195 cases. Cancer. 1970;25:1061–70.
Ueno I, Toyoshima S, Fuchinoue T, Yahagi Y, Nakamura T, Kubo O. Primary giant cell tumor of the frontal bone with adjacent intracranial involvement. No Shinkei Geka. 1979;7:1101–8.
Miyazaki Y, Takada I. Surgically treated case of a very rare giant cell tumor of the frontal bone. Geka Chiryo. 1970;22:112–6.
Ferrington E, Mufson JA. Relationship of the so-called giant-cell tumor to the frontal bone; report of a case. Am J Clin Pathol. 1945;15:529–33.
Vinogradova TP. The international classification of bone tumors and some remarks concerning it. Arkh Patol. 1975;37:34–7.
Campanacci M, Baldini N, Boriani S, Sudanese A. Giant-cell tumor of bone. J Bone Joint Surg Am. 1987;69:106–14.
Kashiwagi N, Hirabuki N, Andou K, Yoshifumi N, Tanaka H, Morino H, et al. MRI and CT findings of the giant cell tumors of the skull; five cases and a review of the literature. Eur J Radiol. 2006;58:435–43.
Tang JY, Wang CK, Su YC, Yang SF, Huang MY, Huang CJ. MRI appearance of giant cell tumor of the lateral skull base: a case report. Clin Imaging. 2003;27:27–30.
Chawla B, Khurana S, Kashyap S. Giant cell reparative granuloma of the orbit. Ophthal Plast Reconstr Surg. 2013;29:e94–5.
Wen Y, Yan J. Primary tumors of the orbital bone. J Craniofac Surg. 2016;27:e344–8.
Kamoshima Y, Sawamura Y, Imai T, Furukawa H, Kubota K, Houkin K. Giant cell tumor of the frontal bone in a girl: case report. Neurol Med Chir (Tokyo). 2011;51:798–800.
Ulu MO, Biceroglu H, Ozlen F, Oz B, Gazioglu N. Giant cell tumor of the frontal bone in an 18-month-old girl: a case report. Cent Eur Neurosurg. 2010;71:104–7.
Pai SB, Lalitha RM, Prasad K, Rao SG, Harish K. Giant cell tumor of the temporal bone—a case report. BMC Ear Nose Throat Disord. 2005;5:8.
Acknowledgements
No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Disclosures
Peter H. Tang, Pradeep Mettu, Amanda C. Maltry, Andrew R. Harrison and Ali Mokhtarzadeh have nothing to disclose.
Compliance with Ethics Guidelines
This report is in full compliance with the Declaration of Helsinki and current Health Insurance Portability and Accountability Act regulations. Informed consent was obtained from the patient for being included in the study.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced content
To view enhanced content for this article go to http://www.medengine.com/Redeem/EA87F060643674FD.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Tang, P.H., Mettu, P., Maltry, A.C. et al. Giant Cell Tumor of the Frontal Bone Presenting as an Orbital Mass. Ophthalmol Ther 6, 215–220 (2017). https://doi.org/10.1007/s40123-017-0081-y
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40123-017-0081-y