
Abstract
Introduction
The use of oral nonsteroidal anti-inflammatory drugs (NSAIDs) for acute musculoskeletal pain should be at the lowest effective dosage and for the shortest duration to minimize potential adverse effects. This study evaluated treatment satisfaction, effectiveness, and tolerability of a low-dose diclofenac epolamine 12.5-mg soft capsule formulation (DHEP 12.5-mg capsules) using patient-reported outcome measures in a real-life setting over a short period (3 days) in subjects with mild-to-moderate acute musculoskeletal pain.
Methods
A prospective, open-label, phase IV clinical study in adult outpatients at hospital clinic departments/general practitioner’s clinics at eight sites in Italy. The primary efficacy variable was the degree of satisfaction with treatment at 72 ± 7 h after initiation of treatment, assessed using the Overall Satisfaction Question of the Pain Treatment Satisfaction Scale (PTSS) and described by classic descriptive statistics. Secondary objectives were to evaluate the analgesic effect after the first administration and over time; the time to and satisfaction with the onset of pain relief, amount of and duration of pain relief; pain intensity differences over time; and safety and tolerability. The investigator’s satisfaction with the treatment was also assessed. Subjects initially took 1–2 capsules of the study treatment and then one or two soft capsules every 4–6 h according to their needs. Not more than six soft capsules were to be taken in any 24-h period.
Results
A total of 182 subjects (mean age, 56.2 years; 54.4% female) took ≥ 1 dose of DHEP capsule and were included in the full analysis set. The most common musculoskeletal conditions were arthralgia (39.0%) and low back pain (23.1%). All subjects completed the study, and 165/182 (90.7%, 95% CI 0.86, 0.95) were satisfied or very satisfied with the treatment at 72 ± 7 h after the first dose (primary efficacy variable). Similar percentages were recorded for treatment satisfaction concerning other efficacy parameters. The onset of the analgesic effect was rapid, with complete pain relief reached after a mean of 49.45 min. Investigators rated their overall treatment satisfaction as 92.9%. Treatment was well tolerated.
Conclusions
The low-dose (12.5 or 25 mg) oral diclofenac epolamine soft capsules formulation exerted rapid, effective, and safe analgesic activity in patients with mild-to-moderate musculoskeletal pain, with subjects’ overall satisfaction with treatment more than 90%.
Trial registration
EudraCT Number: 2018-004886-15 (Study 18I-Fsg08). Registered 04/09/2018.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Worldwide health authorities have advised that nonsteroidal anti-inflammatory drugs (NSAIDs) should be prescribed at the lowest effective dosage and for the shortest duration to minimize the risk of unwanted side effects, which may include serious gastrointestinal, cardiovascular, and cerebrovascular events. |
Several low-dose NSAID formulations, including low-dose diclofenac preparations, have been developed and are available to relieve mild-to-moderate pain. |
This study was conducted to determine how effective subjects rated a new low-dose diclofenac epolamine soft capsule formulation for treating their mild-to-moderate acute musculoskeletal pain. |
What was learned from the study? |
Although the safety and pain-relieving effectiveness of some other low-dose diclofenac formulations have been well studied, there was little information on the use of the low-dose diclofenac epolamine soft capsule formulation. |
This study showed that the low-dose (12.5 or 25 mg) oral diclofenac epolamine soft capsules formulation provided fast, effective, and safe pain relief in subjects with mild-to-moderate musculoskeletal pain, with subjects’ rating their overall satisfaction with treatment at over 90%. |
These results confirm and reinforce the use of a low-dose NSAID formulation in a real-life setting of different musculoskeletal conditions. |
Introduction
Acute pain following a musculoskeletal injury or mechanical/postural issues is very common, affecting large numbers of people from the teenage years or younger through to old age [1, 2]. Musculoskeletal pain is responsible for substantial direct and indirect economic and societal costs for individuals, their employers, the healthcare system, and the broader economy related to medical treatment, impacts on function, and impaired productivity [2]. The consequences of patients ignoring or denying symptoms of acute musculoskeletal pain should not be underestimated, and appropriate pain management is a vital component of patient care, particularly in an emergency setting. It is important to patients that acute pain is recognized, acknowledged, and adequately assessed by clinicians and that the most appropriate medication with the correct dosage is available for the individual patient [3]. Acute pain demands quick relief that should be tailored based on severity.
The many treatment options for acute musculoskeletal pain include oral, topical, or intramuscular pain medications and various nonpharmacological strategies, including physical therapies and cognitive behavioral interventions. Numerous guidelines, algorithms, flow charts, and position papers recommending pharmacological and nonpharmacological therapeutic approaches for managing musculoskeletal pain have been developed to guide the management of acute pain [4,5,6,7,8]. Oral nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used for pain management, although there are concerns about potential side effects, including increased risk of gastrointestinal, cardiovascular, and renal adverse events, particularly at higher doses or with prolonged use [8,9,10]. However, NSAIDs can be an effective treatment option for acute musculoskeletal pain when used appropriately.
Diclofenac is an NSAID with pronounced analgesic, anti-inflammatory, and antipyretic activity that has been shown in wide-ranging clinical trials to be an effective treatment for various acute and chronic inflammatory and painful conditions [11]. As is common to the NSAID class, the use of diclofenac at higher doses is limited by the risk of adverse events, mainly to the gastrointestinal tract and the hepatic, renal, and cardiovascular systems [8, 10]. Due to the linear dose–toxicity relationship between diclofenac dose and the risk of serious gastrointestinal, cardiovascular, and cerebrovascular events, these adverse events diminish with a dose reduction [12, 13].
Worldwide health authorities have advised that NSAIDs, including diclofenac, should be used at the lowest effective dosage and for the shortest duration [5, 14, 15]. Therefore, the recommended dose should be individually adjusted, and the lowest effective dose should be given for the shortest possible duration.
To improve analgesic efficacy and patient convenience and address these safety concerns and warnings, a number of diclofenac products have been developed over the years using pharmaceutical technology, including the development of low-dose diclofenac preparations. Immediate-release preparations (tablets and capsules) containing 12.5 and 25 mg of diclofenac (sodium or potassium salt) are currently available for the relief of mild-to-moderate pain, such as headache, dental pain, period pain, rheumatic pain, and muscular pain. Available data on the analgesic efficacy and safety of diclofenac potassium 12.5-mg tablets and soft capsules containing diclofenac potassium 25 mg or diclofenac sodium 25 mg demonstrate that low-dose diclofenac is safe and effective in various pain models [16,17,18,19,20]. In particular, data from thirteen randomized, double-blind, placebo-controlled studies with active comparators (such as ibuprofen, paracetamol, and aspirin) and/or placebo, reviewed in Moore N, Clinical Drug Investigation 2007 [17], have confirmed that ‘low-dose’ oral diclofenac preparations administered as a single dose (12.5 and 25 mg), or in multiple doses of 25 mg up to 75 mg per day short term, are safe and effective in established acute mild-to-moderate pain and fever models, without appreciably increasing the risk of upper gastrointestinal bleeding [21].
The epolamine salt of diclofenac has higher solubility and aqueous stability than the parent compound [22,23,24]. Diclofenac epolamine (diclofenac hydroxy-ethyl pyrrolidine, DHEP) has been used for around 30 years in medicinal products, including topical plasters and granules for oral solution. A liquid capsule formulation containing 15.38 and 30.76 mg diclofenac epolamine (DHEP), equivalent to 12.5 and 25 mg diclofenac potassium, respectively, has been developed by IBSA, Switzerland, to provide patients with a fast-acting low-dose NSAID formulation in an easier to swallow form than tablets. The soft capsules were developed using PEARLTec® technology (Precision Encapsulation technology for the Application and Release of a Liquid Formulation), a process for obtaining softgel capsules (soft capsules) in which a liquid matrix in a suspension or a gel is encapsulated into a continuous soft gelatin shell providing high dosing precision along with better oral bioavailability than other oral solid dosage forms. The ease of oral administration and rapid dissolution of the soft capsule diclofenac formulation is desirable in an analgesic for acute pain relief and contributes to the product’s acceptance and subject compliance. Soft capsule formulations have been shown to deliver rapid absorption of medications with a shorter time to peak plasma concentration, along with other benefits such as ease of swallowing, neutral or flavorless taste, and convenience in use compared with other solid dosage forms [25,26,27,28].
Diclofenac epolamine soft capsule formulations at dosages equivalent to 12.5 and 25 mg diclofenac potassium have been authorized in the European Union (EU) by the European Medicines Agency (https://www.ema.europa.eu/en/human-regulatory/post-authorisation/data-medicines-iso-idmp-standards/public-data-article-57-database) based on bioequivalence established versus an already marketed, similar diclofenac-containing immediate-release oral formulation (diclofenac potassium capsules, Voltaren® Dolo Liquid currently marketed in Germany by GlaxoSmithKline). In Italy, where the drug was launched first, diclofenac epolamine soft capsules are marketed as an over-the-counter (OTC) drug with the brand name FLECTORGO®Footnote 1 and are indicated for the short-term symptomatic treatment of mild-to-moderate pain, such as headache, dental pain, period pain, rheumatic pain, and muscular pain.
The approved maximum treatment duration of the diclofenac epolamine soft capsules is 3 days, taken on demand at intervals of 4 to 6 h, up to a maximum daily dose equivalent to 75-mg diclofenac potassium (i.e., no more than six 12.5 mg or three 25-mg capsules during 24 h).
Available data on the analgesic efficacy and safety of diclofenac potassium 12.5-mg tablets and soft capsules containing diclofenac potassium 25 mg or diclofenac sodium 25 mg demonstrate that low-dose diclofenac is safe and effective in various pain models [16,17,18,19,20]. Although the analgesic efficacy and safety of low-dose formulations of other diclofenac salts have been well documented, it was the intention of the manufacturer to support the diclofenac epolamine soft capsule formulation with new clinical data specifically related to the product.
Therefore, we evaluated the IBSA 12.5 mg soft capsule formulation (DHEP 12.5 mg capsules) in a real-life setting in subjects with mild-to-moderate acute musculoskeletal pain to gain greater insight into the effectiveness and tolerability of the formulation in daily practice and to measure the subject’s satisfaction with treatment using patient-reported outcome measures (PROMs).
Methods
Study Design and Objectives
This was a prospective, open-label, single-arm, phase IV interventional study in adult outpatients attending hospital clinic departments or general practitioner’s clinics at eight sites in Italy for mild-to-moderate acute musculoskeletal pain. The study was designed to evaluate the subject’s treatment satisfaction, effectiveness, and tolerability of DHEP soft gel capsules given at a low dose (12.5 mg) in a real-life setting over a short period (3 days). Subjects were recommended by the investigators to take the lowest effective dose for the shortest possible duration. Although FLECTORGO® 12.5 mg is approved for various painful conditions, only subjects with mild or moderate acute musculoskeletal pain were included in order to ensure a homogeneous population. Patient-reported outcome measures (PROMs) were used to assess the subject’s experience with this new product (i.e., the subject’s satisfaction and assessment of effectiveness), and product utilization.
The primary objective was to assess subjects’ overall satisfaction with 12.5-mg DHEP soft gel capsules when used as needed according to the approved dose regimen and administration conditions.
The study did not include a control group because the aim was to investigate the subject’s overall satisfaction with the treatment 3 days after the start of treatment, compared to baseline conditions.
Ethical Considerations
All study-specific documents, including the protocol, proposed patient information and consent form, were submitted for review and approval to the Competent Authority, the Agenzia Italiana del Farmaco (AIFA), the relevant Competent Ethics Committee (CEC) of the coordinating center, Bergamo, and all other participating centers, in agreement with local legal requirements. The CEC Bergamo formally approved the study, confirming the required ethics approval for all participating centers in a single covering decision (opinion no. 12019FA00169 of the 14th of March, 2019). The trial was carried out following the ethical principles of the International Conference on Harmonisation (ICH) Harmonised Tripartite Guideline on the Structure and Content of Clinical Trial Reports on Good Clinical Practice, the current version of the Declaration of Helsinki and following all other requirements of local laws of Italy. All subjects were informed about the study’s purpose and procedures and provided written informed consent before participating.
Project Management, Protocol and Report Medical Writing, Statistics, Regulatory and Monitoring activities of the study were carried out by the Contract Research Organisation (CRO), Medical Trials Analysis Swiss SA, Lugano, Switzerland. Advice Pharma Group S.r.l, Milan, Italy, carried out data management.
The study, Study 18I-Fsg08, was registered with the European Union Clinical Trials Register (EudraCT Number: 2018-004886-15). No amendments to the study protocol were implemented during the study.
Trial Population
Subjects who provided informed consent and fulfilled all the specified inclusion and exclusion criteria were enrolled in the study. Adult male and female outpatients aged ≥ 18 years with acute mild or moderate musculoskeletal pain such as back pain, neck pain, shoulder pain, tendon or ligament pain, and myalgia) that started ≤ 72 h prior to inclusion in the trial. Pain could be either traumatic (e.g., due to an injury) or postural/mechanical pain (e.g., due to postural strain, changes in posture or poor body mechanics, repetitive movements, prolonged immobilization). At baseline, mild or moderate acute musculoskeletal pain was defined as ≥ 20 mm and ≤ 60 mm 0 to 100 mm on a patient-assessed visual analog scale (VAS), where 0 represented no pain and 100 the worst pain possible. The use of reliable contraception was required for women of childbearing age.
The key exclusion criteria were the following: musculoskeletal pain of neuropathic or post-surgical origin or non-musculoskeletal pain; musculoskeletal pain requiring treatment with NSAIDs for more than 3 days; pain associated with chills or fever, or dysmenorrhea or endometriosis; known or suspected hypersensitivity or intolerance to diclofenac and/or any other ingredient in the formulation to be tested; prior use of OTC or prescription NSAIDs within 36 h of baseline assessment; the use of prohibited treatments, including any systemic or topical drugs with muscle relaxant properties; active, suspected or history of peptic ulcer disease, bleeding or perforation related to previous NSAID therapy; history of recurrent peptic ulcer/hemorrhage; established cardiac and/or cerebrovascular disease; severe hepatic, renal or cardiac failure; known allergy or hypersensitivity to aspirin or NSAIDs; any clinical condition that might interfere with the study drug; participation in any other protocol involving the administration of an investigational agent within 3 months before Visit 1; pregnancy or breastfeeding.; unable to comply with the study procedures. Prior use of acetaminophen (paracetamol) ≤ 1000 mg, ibuprofen ≤ 400 mg, and aspirin ≤ 600 mg were not grounds for exclusion if the last dose was taken > 6 h before the VAS assessment.
To allow the analysis of all endpoints and to take into account possible dropouts, the study population consisted of a balanced number of male and female subjects.
Study Procedures
The study treatment (FLECTORGO® 12.5 mg, IBSA; batch no. 190112/001D19-043) was a clear, yellowish-colored capsule with a size of about 0.8 cm containing a slightly viscous solution with 15.38 mg diclofenac epolamine, equivalent to 12.5 mg diclofenac potassium. Non-active constituents included macrogol 600, glycerol anhydride, purified water (fill solution), gelatin, anhydrous glycerol, liquid sorbitol, hydroxypropyl betadex, and sodium hydroxide (shell components).
The study schedule comprised two visits, a screening/enrolment visit (Visit 1/D0) and an end-of-study/evaluation visit (Visit 2/72 ± 7 h). At both visits, the same investigator conducted procedures for each subject.
During the screening visit, the subjects were informed about the aims, procedures, and possible risks of the study and instructed in the patient-reported outcome measures to be used. Written informed consent was required for inclusion in the trial. All inclusion/exclusion criteria were checked, and the subject’s demographic characteristics were collected, including relevant medical history and ongoing concomitant disease, physical examination and check of vital signs, and concomitant treatments. Female subjects were required to have a negative urine pregnancy test. Once all inclusion/exclusion criteria were assessed and the investigator had ensured that the assessment measures had been clearly explained and understood, each subject completed an assessment of pain severity using the 0 to 100 mm VAS-pain scale.
The study Investigators then established the initial dose (one or two capsules) to be swallowed with liquid during Visit 1 based on their assessment of pain intensity and origin. They recommended that subjects adjusted the dose in subsequent administrations using one or two capsules at intervals of 4 to 6 h according to their needs up to a maximum daily dose of six capsules per day, ensuring that the lowest effective dose was taken and participants were not exposed to unnecessary higher diclofenac doses.
Using the lowest possible dose of DHEP, corresponding to one or two capsules of 12.5 mg diclofenac potassium during a short period (3 days) was intended to obtain the maximum benefit for the subjects, lowering the possible side effects and thus the risks for the study participation.
Subjects were instructed to record the number of diclofenac 12.5-mg capsules taken and the date/time of intake, the VAS and the time to onset of the analgesic effect at determined time points after capsule administration, and any adverse events (AEs) in a “Subject’s Treatment and Pain Assessment” logbook (patient diary).
An appointment for the next visit was fixed for 3 days after (72 ± 7 h).
At Visit 2 (the day 3 evaluation point), the logbook and medication boxes (used and unused) were collected. The subject was asked to measure their pain intensity utilizing VAS-pain, and to evaluate the overall subject’s satisfaction, satisfaction with efficacy in terms of time to pain relief, amount of pain relief, duration of pain relief, and satisfaction with the form (capsule) of the treatment. The investigator then assessed their overall satisfaction with the treatment.
Subjects were asked to describe any problem or change in health status since informed consent was recorded, regardless of the relationship to the treatment, as well as any changes in the musculoskeletal pain condition, use of concomitant treatments, and occurrence of any AE. A complete physical examination, including vital signs, was performed during the visit to evaluate any significant changes. Compliance was assessed by the investigator asking the subject and confirmed by checking the subject logbook, and any medication returned.
Subjects were treated with the most appropriate treatment based on their status and symptoms, according to the standard of care and as determined by the Investigator. Any subjects taking the investigational medical product on the day of Visit 2 were followed up by phone call to ensure the collection of AEs until the day after the last intake of diclofenac capsules.
Outcome Measures
The primary efficacy variable of the study was the degree of satisfaction with the treatment of participants at 72 ± 7 h after initiation of treatment, assessed using the Overall Satisfaction Question of the Pain Treatment Satisfaction Scale (PTSS) [29], a reliable and validated patient-reported outcome measure of patient satisfaction for patients receiving treatment for either acute or chronic pain. Responses were rated utilizing a five-point Likert-like scale where 1) represented “very satisfied” and 5) represented “very dissatisfied.” A positive outcome of this variable consisted of an overall satisfaction answer, defined as 1) very satisfied or 2) satisfied.
Secondary objectives were to (1) evaluate the analgesic effect after the first diclofenac soft capsule administration (between 0 and 3 h) at each scheduled assessment time point after the first dose; (2) evaluate the time to onset of the analgesic effect experienced at each drug intake; (3) assess the subject’s satisfaction with efficacy in terms of time to pain relief, amount of, and duration of pain relief, using the Efficacy subscale of the PTSS; and (4) to evaluate safety and tolerability. In addition, the subject’s assessment of pain level by VAS over the 3 days study period was compared to the baseline pain level of the same subject to detect any statistically significant difference.
Specifically, assessments consisted of the VAS pain intensity difference (PID), based on the subject’s self-assessment of pain intensity by means of VAS at each scheduled assessment time point after the first dose; the sum of the pain intensity difference (VAS-SPID), calculated as the area under the concentration–time curve (AUC) over 0–3 h (SPID 0–3 h), over 0–24 h (SPID 0–24 h), over 0–48 h (SPID 0–48 h), and over 0–72 h (SPID 0–72 h) or until the last VAS time point after the last intake if treatment was not taken over 3 days; the time to onset of the analgesic effect as expressed by asking the subject 1 h after each diclofenac intake the time in minutes he/she experienced an initial slight pain relief, a significant pain relief, and a complete pain relief; the proportion of participants satisfied (very satisfied or satisfied) with the Time to Pain Relief, Amount of Pain Relief and Duration of Pain Relief 72 ± 7 h after initiation of treatment with DHEP soft gel capsules as assessed using the Satisfaction question of the PTSS; the number of doses and timing of DHEP soft gel capsules 12.5 mg taken per day and within 72 ± 7 h; the subject’s satisfaction with the form (capsule) of the treatment assessed 72 ± 7 h after initiation of treatment with DHEP soft gel capsules using one of the Medication characteristics satisfaction questions of the PTSS; and the Investigator’s assessment of satisfaction with the treatment, as expressed at the end of the study using a five-point Likert-like scale.
Safety Variables
The safety variables assessed were: discontinuation of study treatment because of AEs; incidence, nature, and severity of treatment-emergent AEs (TEAEs); incidence, nature, and severity of serious AEs (SAEs); incidence, nature, and severity of adverse drug reactions (ADRs); change from baseline in vital signs. Adverse events were defined according to the ICH Topic E2A Definitions and Standards for Expedited Reporting of Clinical Safety Data of the European Medicines Agency (EMA) website: http://www.ema.europa.eu and categorized according to the Medical Dictionary for Regulatory Activities (MedDRA) classification system. All AEs had to be collected, thoroughly investigated, and documented in the study’s source documents and case report forms.
Sample Size
It was anticipated that powering the study to detect 70% of a heterogeneous group of subjects with mild-to-moderate acute pain of musculoskeletal origin, with or without comorbidities, who were overall satisfied with the treatment (very satisfied or satisfied), would require enrolment of 177 subjects. The estimates were obtained by setting the probability of type I error α = 0.05 (two-tailed) and a confidence interval (CI) width of 0.14. Assuming a dropout rate of 12%, a total of 200 subjects enrolled was judged adequate to provide the required power.
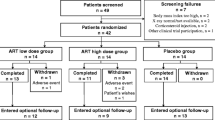
Since there were no dropouts, it was decided to terminate the study after the required sample size for statistical calculation was reached (n = 177). Therefore, recruitment was stopped when 182 subjects were enrolled.
Statistical Methods
Analysis was planned for three populations. All subjects eligible for the study made up the Inclusion set. The full analysis set (FAS) included all subjects of the Inclusion set having taken at least one dose of study medication and who had a reliable baseline value and at least one post-baseline reliable value of at least one parameter over the 72-h study period. The per-protocol set (PPS) included all FAS subjects with reliable pain values at baseline and at 72 h and not associated with a major protocol deviation.
Descriptive statistics and graphical analyses were used to summarize data and results. Analysis of the primary endpoint and all secondary parameters were presented with their 95% CI for both the FAS and PP populations. The Wald Χ2 test was used for comparison between qualitative variables. All statistical analyses and data processing were performed using SAS® Software (SAS Institute Inc., Cary, NC, USA).
A last observation carried forward (LOCF) approach was employed for subjects not completing the study. For the secondary parameter (VAS-pain), intermittent missing data were imputed via linear interpolation. Other missing data remained missing.
Results
Demographics
The study was conducted at eight investigational study sites in the Lombardia and Liguria regions of Italy. Two other sites were initially planned but withdrew due to internal organizational issues related to the COVID-19 pandemic. The subjects were outpatients of hospital clinic departments or selected general practitioner’s clinics.
The first subject was enrolled on the 5th of September 2019, and the last subject on the 27th of February 2021. A total of 182 subjects were enrolled in the study, took at least one dose of DHEP capsule, and were included in the Inclusion set and FAS analyses. Nineteen subjects had major protocol deviations related to inclusion criteria (n = 4), schedule of assessment (4), and treatment compliance (12). Thus, the PP population consisted of 163 subjects.
All the subjects completed the study, no withdrawal occurred during the whole study duration, and no prohibited treatment was taken during the study. Therefore, the Inclusion and FAS populations were the same, and analyses were conducted on only the FAS and PP populations.
Subject demographics at baseline are presented in Table 1. The mean age was 56.2 (range 20–92) years, and 54.4% of enrolled subjects were female. The majority of subjects (91.8%) had pain conditions classified as musculoskeletal and connective tissue disorders according to the MedDRA classification system, in particular, arthralgia (39.0%) and low back pain (23.1%).
Pain was reported to have started between 1 and 79 h prior to inclusion in the trial. In 69.8% of subjects, the pain started within 48 h before inclusion. There was a prior therapeutic intervention in only five subjects, and only 8.2% of subjects had experienced a relevant pain event in the last year. However, no relevant previous musculoskeletal pain events were ongoing at baseline.
Vital signs were normal or without clinically relevant abnormality in 181 (99.5%) subjects (one subject had missing data). The most frequent comorbidities included: cardiovascular disorders (n = 46, 25.3%), metabolic disorders (n = 14, 7.7%), and reproductive disorders (n = 9, 4.9%). The most common concomitant treatments were for hypertension, peptic syndrome, diabetes, and dyslipidemia.
Efficacy
One hundred sixty-five of the 182 subjects (90.7%, 95% CI 0.86, 0.95) in the FAS were satisfied or very satisfied with DHEP soft capsule treatment at 72 ± 7 h after initiation of treatment (primary efficacy variable) (Table 2). Overall treatment satisfaction in the PP population was 92.0% Table 2. The Investigators rated overall treatment satisfaction as 92.9% (n = 169, 95% CI 0.89, 0.97) (Table 2).
Similar percentages were also registered for treatment satisfaction concerning other efficacy parameters such as time to, amount of, and duration of pain relief (respectively 86.8, 87.9, and 82.4%) and for subject satisfaction with the form (capsule) of the treatment (Medication Characteristics) (98.4%) (Table 2).
At baseline, the mean VAS pain in the FAS population was 46.45 ± 11.04 (range 20–65). This was a similar distribution of VAS pain to that in the PP population (mean 45.34 ± 10.91, range 20–60).
Within 3 h from the first intake, there was a 46.5% mean reduction in pain intensity, with a pain intensity difference (PID) from 0 to 3 h of 21.61 (95% CI 18.79, 24.43) (Table 3). Due to missing data in subject logbooks, the VAS sum of pain intensity difference (SPID) was calculated only for the first intake of DHEP capsules (i.e., from 0 to 3 h from baseline) to avoid potential bias. The SPID 0–3 h was 2970 (95% CI 2547, 3393). Overall pain intensity reduced progressively over 3 days, with a reduction in VAS pain score of 75% (PID 0–72 h, 34.36).
The onset of the analgesic effect was rapid. One hour after the first intake, 143 (78.6%) subjects reported relief from the initial pain, while five (2.7%) subjects reported no relief. In the remaining 34 subjects, no answer was given, or the answer was not applicable. The mean time to reach a slight relief was 29.4 min, and the mean time to reach a moderate pain relief was 42.32 min. Complete pain relief was reached after a mean of 49.45 min.
At the first intake, 29.1% of subjects took one capsule (12.5 mg), and 70.9% took two capsules (25 mg). In the 24 h after the first intake, 152 subjects had further intakes, 133 subjects continued the treatment up to 48 h, and 89 subjects until 72 h. In addition, eight subjects took the treatment over the allowed time per protocol. Of those continuing treatment, 63.8% used two capsules in the following 24 h, 56.4% between 24 and 48 h, 52.8% between 48 and 72 h, and two of the four subjects that continued to take the drug after 72 h used 25 mg.
The mean number of doses was 9.16 ± 5.07, and the mean time between intakes was 11.55 h between 0 and 24 h, 15.53 h between 24 and 48 h, 14.01 h between 48 and 72 h, and 11.63 h after 72 h.
Subgroup analyses using generalized additive models (GAM) to verify the impact of center, baseline pain intensity, etiology of pain, and age on subjects’ overall satisfaction with treatment found no significant impacts of any of these factors. Furthermore, no correlations were detected between subjects’ overall satisfaction, time to pain relief, PID, and number of doses used.
Safety
Oral treatment with DHEP 12.5-mg soft capsules was well tolerated, with only two AEs reported, only one of which was potentially correlated with treatment (nausea of mild intensity, self-resolved within 20 min). The other AE was an asymptomatic COVID-19 infection, considered unrelated to treatment. There were no significant differences in vital signs and clinical evaluation of subjects between Screening/enrolment and at the End of Study Visit (data not shown).
Discussion
The results from this study show that oral treatment with diclofenac epolamine soft capsules given at a low dose (DHEP soft gel capsule 12.5 mg) and taking one (12.5 mg) or two (25 mg) capsules according to the subject’s need for a maximum of 3 days, was assessed by the subjects and investigator to be satisfactory, effective, and safe in patients with mild or moderate musculoskeletal pain. Ninety-point-seven percent of patients expressed overall satisfaction with the treatment, and the Investigators were satisfied with the treatment for 92.9% of subjects. Similar satisfaction rates were registered for time to the onset of pain relief, amount and duration of pain relief, and satisfaction with the soft capsule medication form.
The onset of the analgesic effect following treatment with DHEP soft gel capsules was rapid; slight pain relief was reported within 30 min from the first intake and complete relief within 50 min on average. Within 3 h after the first intake, pain intensity was almost halved, from a mean baseline VAS of 46.45 to 24.24 after 3 h, a pain intensity difference (PID) of 21.6, corresponding to a mean reduction of 46.5%. Analgesic activity continued to reduce overall pain intensity over time, with a 75% decrease in VAS pain score after 3 days.
The treatment was also well-tolerated and safe. Only two AEs were registered during the study; both were mild and self-resolving. Only one AE was potentially correlated with treatment (nausea of mild intensity, resolving within 20 min). Such a safety profile for an oral NSAID is important, particularly for a medicine that may be approved for OTC use, as is the case in Italy for DHEP soft capsules.
Using the lowest dose for the shortest period to achieve analgesia is vital to an NSAID’s safe and effective use in treating acute pain [5]. The findings of our study that show the safety and efficacy of low-dose, short-duration treatment with diclofenac epolamine soft gel capsules is supported by the published evidence of the analgesic efficacy and safety of other oral low-dose NSAID preparations [16,17,18,19,20].
Appropriate and validated assessment tools were used to evaluate pain in the study, including the visual analog scale (VAS), considered the gold standard in everyday practice and all pain settings [3, 30]. Moreover, patient-reported outcome measures (PROMs) such as VAS and the PTSS are considered to provide the most accurate, reliable, and relevant evidence of pain and its intensity, and patient-centered care and patient participation are playing an increasing role in achieving healthcare goals [31,32,33,34,35].
Therefore, the results of this study provided evidence that the majority of subjects were satisfied or very satisfied with this analgesic treatment and that the intake of one or two capsules of DHEP soft gel capsules 12.5 mg is sufficient to exert a fast, effective, and safe analgesic activity.
Limitations and Strengths
As the medicinal product under investigation was a well-known molecule with proven efficacy in different pain conditions, a control group was not considered necessary in a clinical study designed to confirm and reinforce the use of a low-dose NSAID formulation in a real-world context of different musculoskeletal conditions. Nevertheless, as a single-arm study that did not include a control group, it shares the potential limitations of open-label studies, and no definitive conclusions on the efficacy of DHEP soft gel capsule 12.5 mg can be made. It is also acknowledged that acute, mild, or moderate musculoskeletal pain may be self-limiting, regardless of medication. However, the times of assessment (0–3 h, 0–24 h, 0–48 h, and 0–72 h) were designed to capture medication-related analgesic effects, and the methodology was prospectively designed with inclusion and exclusion criteria intended to provide a homogeneous population in terms of mild-to-moderate acute musculoskeletal pain but across a broad range of musculoskeletal pain conditions involving several body areas. The use of patient-reported outcome measures to assess the effectiveness of the intervention reflects the increasing use of this method in health care studies [31,32,33,34,35], and the patient-reported outcomes in this study were strengthened by including an Investigator-rated assessment of how satisfactory the treatment was considered to be. Together with a high adherence level, there was broad agreement between subjects and Investigators on treatment satisfaction.
The Investigator’s selection of the subject population could be considered a possible source of selection bias, although stringent inclusion and exclusion criteria were applied, and the duration of treatment exposure for potential AEs to become apparent was limited. Notwithstanding, low-dose, short-duration treatment was shown to be effective without safety concerns.
Despite the study’s limitations, the study provides support for the real-life effectiveness and safety of low-dose diclofenac salts. Specifically, the results support the efficacy and safety of the low-dose soft capsule formulation of DHEP in a broadly representative subject population consistent with patients presenting with acute musculoskeletal pain at emergency departments or general practice.
Conclusions
Low-dose (12.5 or 25 mg) oral diclofenac epolamine soft capsules taken on demand up to every 4 to 6 h provide rapid and effective analgesic activity in patients with mild or moderate musculoskeletal pain and are well tolerated. Subjects report a high level of satisfaction with treatment, including time to onset of analgesia, amount and duration of pain relief, and characteristics of the medication form. Similarly, the study Investigators reported high levels of satisfaction with treatment effectiveness. Diclofenac epolamine soft capsules are a practical treatment option for patients with mild-to-moderate acute musculoskeletal pain, and future controlled studies against an active comparator would allow further evaluation of their benefits in this patient population.
Notes
Other available tradenames: Flector® Dolo Rapid, Flector® 12.5–25 mg, Flector® Rapidcaps.
References
GBD 2019 Diseases and Injuries Collaborators. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet. 2020;396(10258):1204–22. https://doi.org/10.1016/S0140-6736(20)30925-9.
McMahon SB, Dargan P, Lanas A, Wiffen P. The burden of musculoskeletal pain and the role of topical non-steroidal anti-inflammatory drugs (NSAIDs) in its treatment. Ten underpinning statements from a global pain faculty. Curr Med Res Opin. 2021;37(2):287–92. https://doi.org/10.1080/03007995.2020.1847718.
Hachimi-Idrissi S, Coffey F, Hautz WE, Leach R, Sauter TC, Sforzi I, et al. Approaching acute pain in emergency settings: European Society for Emergency Medicine (EUSEM) guidelines-part 1: assessment. Intern Emerg Med. 2020;15(7):1125–39. https://doi.org/10.1007/s11739-020-02477-y.
Amaechi O, Huffman MM, Featherstone K. Pharmacologic therapy for acute pain. Am Fam Physician. 2021;104(1):63–72.
Hachimi-Idrissi S, Dobias V, Hautz WE, Leach R, Sauter TC, Sforzi I, et al. Approaching acute pain in emergency settings; European Society for Emergency Medicine (EUSEM) guidelines-part 2: management and recommendations. Intern Emerg Med. 2020;15(7):1141–55. https://doi.org/10.1007/s11739-020-02411-2.
Hsu JR, Mir H, Wally MK, Seymour RB, Orthopaedic Trauma Association Musculoskeletal Pain Task F. Clinical practice guidelines for pain management in acute musculoskeletal injury. J Orthop Trauma. 2019;33(5):e158–82. https://doi.org/10.1097/BOT.0000000000001430.
Machado GC, Abdel-Shaheed C, Underwood M, Day RO. Non-steroidal anti-inflammatory drugs (NSAIDs) for musculoskeletal pain. BMJ. 2021;372:n104. https://doi.org/10.1136/bmj.n104.
Qaseem A, McLean RM, O’Gurek D, Batur P, Lin K, Kansagara DL, et al. Nonpharmacologic and pharmacologic management of acute pain from non-low back, musculoskeletal injuries in adults: a clinical guideline from the American College of Physicians and American Academy of Family Physicians. Ann Intern Med. 2020;173(9):739–48. https://doi.org/10.7326/M19-3602.
Hatt KM, Vijapura A, Maitin IB, Cruz E. Safety considerations in prescription of NSAIDs for musculoskeletal pain: a narrative review. Pm r. 2018;10(12):1404–11. https://doi.org/10.1016/j.pmrj.2018.06.011.
Gan TJ. Diclofenac: an update on its mechanism of action and safety profile. Curr Med Res Opin. 2010;26(7):1715–31. https://doi.org/10.1185/03007995.2010.486301.
Rainsford KD. Chapter 5: mode of action, uses, and side effects of anti-inflammatory drugs. In: Rainsford KD, editor. Advances in anti-rheumatic therapy. Boca Raton: CRC Press; 1996. p. 59–111.
Fanelli A, Ghisi D, Aprile PL, Lapi F. Cardiovascular and cerebrovascular risk with nonsteroidal anti-inflammatory drugs and cyclooxygenase 2 inhibitors: latest evidence and clinical implications. Ther Adv Drug Saf. 2017;8(6):173–82. https://doi.org/10.1177/2042098617690485.
Odom DM, Mladsi DM, Saag KG, Sherif BN, Miles L, Ronquest N, et al. Relationship between diclofenac dose and risk of gastrointestinal and cardiovascular events: meta-regression based on two systematic literature reviews. Clin Ther. 2014;36(6):906–17. https://doi.org/10.1016/j.clinthera.2014.04.012.
European Medicines Agency (EMA) 2013. New safety advice for diclofenac. https://www.ema.europa.eu/ Accessed Jul 2022.
van der Gaag WH, Roelofs PD, Enthoven WT, van Tulder MW, Koes BW. Non-steroidal anti-inflammatory drugs for acute low back pain. Cochrane Database Syst Rev. 2020;4(4): Cd013581. https://doi.org/10.1002/14651858.Cd013581.
Altman R, Bosch B, Brune K, Patrignani P, Young C. Advances in NSAID development: evolution of diclofenac products using pharmaceutical technology. Drugs. 2015;75(8):859–77. https://doi.org/10.1007/s40265-015-0392-z.
Moore N. Diclofenac potassium 12.5mg tablets for mild to moderate pain and fever: a review of its pharmacology, clinical efficacy and safety. Clin Drug Investig. 2007;27(3):163–95. https://doi.org/10.2165/00044011-200727030-00002.
Zuniga JR, Malmström H, Noveck RJ, Campbell JH, Christensen S, Glickman RS, et al. Controlled phase III clinical trial of diclofenac potassium liquid-filled soft gelatin capsule for treatment of postoperative dental pain. J Oral Maxillofac Surg. 2010;68(11):2735–42. https://doi.org/10.1016/j.joms.2010.05.075.
Zuniga JR, Noveck RJ, Schmidt WK, Boesing SE, Hersh EV. Onset of action of diclofenac potassium liquid-filled capsules in dental surgery patients. Curr Med Res Opin. 2011;27(9):1733–9. https://doi.org/10.1185/03007995.2011.600300.
Dreiser RL, Marty M, Ionescu E, Gold M, Liu JH. Relief of acute low back pain with diclofenac-K 12.5 mg tablets: a flexible dose, ibuprofen 200 mg and placebo-controlled clinical trial. Int J Clin Pharmacol Ther. 2003;41(9):375–85. https://doi.org/10.5414/cpp41375.
Lewis SC, Langman MJ, Laporte JR, Matthews JN, Rawlins MD, Wiholm BE. Dose-response relationships between individual nonaspirin nonsteroidal anti-inflammatory drugs (NANSAIDs) and serious upper gastrointestinal bleeding: a meta-analysis based on individual patient data. Br J Clin Pharmacol. 2002;54(3):320–6. https://doi.org/10.1046/j.1365-2125.2002.01636.x.
Fini A, Fazio G, Orienti I, Zecchi V, Rapaport I. Chemical properties-dissolution relationship. IV. Behaviour in solution of the diclofenac/N-(2-hydroxyethyl) pyrrolidine salt (DHEP). Pharm Acta Helv. 1991;66(7):201–3.
Fini A, Fazio G, Rapaport I. Diclofenac/N-(2-hydroxyethyl)pyrrolidine: a new salt for an old drug. Drugs Exp Clin Res. 1993;19(3):81–8.
Maggi CA, Lualdi P, Mautone G. Comparative bioavailability of diclofenac hydroxyethylpyrrolidine vs diclofenac sodium in man. Eur J Clin Pharmacol. 1990;38(2):207–8. https://doi.org/10.1007/BF00265987.
Kienzler JL, Gold M. Diclofenac potassium 12.5 mg liquid capsules: earlier and higher exposure to diclofenac. A fasted, single-dose, comparative, bioavailability study versus diclofenac potassium 12.5 mg tablets. Int J Clin Pharmacol Ther. 2012;50(6):438–44. https://doi.org/10.5414/cp201641.
Lissy M, Scallion R, Stiff DD, Moore K. Pharmacokinetic comparison of an oral diclofenac potassium liquid-filled soft gelatin capsule with a diclofenac potassium tablet. Expert Opin Pharmacother. 2010;11(5):701–8. https://doi.org/10.1517/14656561003614773.
Bende G, Biswal S, Bhad P, Chen Y, Salunke A, Winter S, et al. Relative bioavailability of diclofenac potassium from softgel capsule versus powder for oral solution and immediate-release tablet formulation. Clin Pharmacol Drug Dev. 2016;5(1):76–82. https://doi.org/10.1002/cpdd.215.
Gullapalli RP. Soft gelatin capsules (softgels). J Pharm Sci. 2010;99(10):4107–48. https://doi.org/10.1002/jps.22151.
Evans CJ, Trudeau E, Mertzanis P, Marquis P, Pena BM, Wong J, et al. Development and validation of the Pain Treatment Satisfaction Scale (PTSS): a patient satisfaction questionnaire for use in patients with chronic or acute pain. Pain. 2004;112(3):254–66. https://doi.org/10.1016/j.pain.2004.09.005.
Hjermstad MJ, Fayers PM, Haugen DF, Caraceni A, Hanks GW, Loge JH, et al. Studies comparing Numerical Rating Scales, Verbal Rating Scales, and Visual Analogue Scales for assessment of pain intensity in adults: a systematic literature review. J Pain Symptom Manage. 2011;41(6):1073–93. https://doi.org/10.1016/j.jpainsymman.2010.08.016.
Emshoff R, Bertram S, Emshoff I. Clinically important difference thresholds of the visual analog scale: a conceptual model for identifying meaningful intraindividual changes for pain intensity. Pain. 2011;152(10):2277–82. https://doi.org/10.1016/j.pain.2011.06.003.
Haywood KL. Patient-reported outcome I: measuring what matters in musculoskeletal care. Musculoskeletal Care. 2006;4(4):187–203. https://doi.org/10.1002/msc.94.
Haywood KL. Patient-reported outcome II: selecting appropriate measures for musculoskeletal care. Musculoskeletal Care. 2007;5(2):72–90. https://doi.org/10.1002/msc.101.
Patrick DL, Burke LB, Powers JH, Scott JA, Rock EP, Dawisha S, et al. Patient-reported outcomes to support medical product labeling claims: FDA perspective. Value Health. 2007;10(Suppl 2):S125–37. https://doi.org/10.1111/j.1524-4733.2007.00275.x.
Wiering B, de Boer D, Delnoij D. Patient involvement in the development of patient-reported outcome measures: a scoping review. Health Expect. 2017;20(1):11–23. https://doi.org/10.1111/hex.12442.
Acknowledgements
We thank the participants of the study. We also acknowledge and thank the Study 18I-Fsg08 Investigators: Gemma Baldari, Azienda Sociosanitaria Ligure n. 3 (ASL3), Genoa, Italy; Pierclaudio Brasesco, ASL3, Genoa, Italy; Andrea Carraro, Azienda Sociosanitaria Ligure n. 3 (ASL3), Genoa, Italy; Giuseppina Caruso, ASST Bergamo Est, Italy; Leo Fisichella, Ospedale SS. Capitanio e Gerosa, Lovere, Italy; Francesco Gatani, ASL1, Imperia, Italy; Vittorio Guida, ASL3, Genoa, Italy; Ettore Perreca, Azienda Sociosanitaria Ligure n. 1 (ASL1), Imperia, Italy; Carlo Trevisan, Ospedale Bolognini Seriate ASST Bergamo Est, Seriate, Italy.
The Study 18I-Fsg08 Investigators: Carlo Trevisan, Andrea Carraro, Gemma Baldari, Leo Fisichella, Ettore Perreca, Francesco Gatani, Pierclaudio Brasesco, Vittorio Guida, and Giuseppina Caruso
Funding
The study and the Rapid Service Fee were funded by IBSA Institut Biochimique S.A. (Lugano, Switzerland).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Medical Writing and Editorial Support
The authors thank Ray Hill, an independent medical writer, who provided technical writing support funded by IBSA Institut Biochimique S.A. (Lugano, Switzerland).
Author Contributions
Carlo Trevisan, Andrea Carraro, and Gemma Baldari all contributed to the design of the study, interpreted the data, and discussed, reviewed and agreed to the content of the final manuscript before submission.
Disclosures
Carlo Trevisan, Andrea Carraro and Gemma Baldari declare that they have no competing interests.
Compliance with Ethics Guidelines
The study was approved by the Agenzia Italiana del Farmaco (AIFA), the concerned Competent Ethics Committee, and other participating centers. The trial was conducted in accordance with the ethical principles of the International Conference on Harmonisation (ICH) Harmonised Tripartite Guideline on the Structure and Content of Clinical Trial Reports on Good Clinical Practice, the current version of the Declaration of Helsinki and following all other requirements of local laws of Italy. All subjects were informed about the purpose and procedures of the study and provided written informed consent before participation.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The members of the Study 18I-Fsg08 Investigators are listed in “Acknowledgements”.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Trevisan, C.L.M., Carraro, A., Baldari, G.L.A. et al. Treatment Satisfaction, Efficacy, and Tolerability of Low-Dose Diclofenac Epolamine Soft Capsules in Acute, Mild, or Moderate Musculoskeletal Pain: A Prospective Open-Label, Single-Arm Interventional Study. Pain Ther 12, 1149–1163 (2023). https://doi.org/10.1007/s40122-023-00531-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-023-00531-z