
Abstract
Introduction
Current external peripheral nerve stimulation devices stimulate only one nerve. This prospective, randomized, double-blind, sham-controlled trial assessed efficacy, safety, and tolerability of a novel external combined occipital and trigeminal neurostimulation (eCOT-NS) device as a self-administered home treatment for migraine (Relivion®MG, Neurolief Ltd; Netanya, Israel).
Methods
Episodic and chronic migraine subjects (N = 55) were randomized to receive active (n = 27) or sham (n = 28) treatment. Subjects received eCOT-NS devices and performed 60 ± 20-min home treatments within 45 min of migraine episode onset. The primary endpoint was relative (percent) change in mean baseline VAS pain scores 1 h after treatment initiation. Treatment outcomes assessed at 1-, 2-, and 24-h post-treatment initiation were pain reduction and proportion of pain-free subjects and treatment responders, defined as ≥ 50% pain reduction. Categorical pain ratings (none, mild, moderate, and severe pain) were also analyzed.
Results
Active stimulation was significantly more effective than sham stimulation for decreasing pain intensity at 1 h (53% vs. 10%), 2 h (52% vs. 17%), and 24 h (71% vs. 34%). Pain-free ratings were greater for the active treatment arm at 1 h (29.2% vs. 16%), 2 h (41.7% vs. 20%), and 24 h (65.2% vs. 40%). The number of subjects with baseline moderate or severe migraine pain who were pain-free at 2 h was significantly greater among active treatment subjects (43% vs. 10.5%). The responder rate was significantly higher among the active treatment group at 1 h (67% vs. 20%), 2 h (66.7% vs. 32%,), and 24 h (78.3% vs. 48%). Overall headache relief was significantly higher in the active treatment group at 1 h (67% vs. 26%) and 2 h (76% vs. 31.6%). Mild adverse events, reported by a minority of subjects, resolved spontaneously.
Conclusions
eCOT-NS provides superior clinically meaningful relief and freedom from migraine pain, offering an effective and safe therapy for acute treatment of migraine.
Trial registration
ClinicalTrials.gov Identifier NCT03398668.
Plain Language Summary
As current external nerve stimulation devices stimulate only one nerve, this study assessed the effectiveness, safety, and tolerability of a new external nerve stimulation device that stimulates two nerves (occipital and trigeminal) as a self-administered home treatment for migraine (Relivion®MG, Neurolief Ltd; Netanya, Israel). Fifty-five subjects with episodic and chronic migraine were randomly assigned to active (n = 27) or sham (dummy) treatment (n = 28). Subjects performed a 60-min home treatment within 45 min of migraine onset. The primary endpoint was the change in pain intensity 1 h after treatment initiation. Active treatment was significantly more effective than sham stimulation for decreasing pain intensity at 1 h (53% vs. 10%) and 2 h (52% vs. 17%). Pain-free ratings were also greater for the active treatment arm at 1 h (29.2% vs. 16%) and 2 h (41.7% vs. 20%). Overall headache relief was significantly higher in the active treatment group at 1 h (67% vs. 26%) and 2 h (66.7% vs. 32%). Mild, transient side effects reported by a few subjects resolved without treatment. This new external concurrent occipital and trigeminal neurostimulation (eCOT-NS) device provides superior and meaningful relief and freedom from migraine pain compared to sham treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Migraine patients often experience disabling symptoms, including moderate-to-severe headache pain that may be refractory to currently available therapies. |
Thus, a significant unmet medical need for treating migraine pain remains. |
We hypothesized that a self-administered stimulation protocol using a device delivering noninvasive, external concurrent occipital and trigeminal neurostimulation (eCOT-NS) (Relivion®MG; Neurolief Ltd, Netanya, Israel) can provide a safe, effective, and fast-acting acute reduction of migraine-related pain. |
What was learned from the study? |
The eCOT-NS device was significantly more effective than sham stimulation for decreasing pain intensity 1 h, 2 h, and 24 h after initiating treatment. Pain-free ratings were also greater for the active treatment arm at 1, 2, and 24 h. |
This novel eCOT-NS device provides non-invasive, self-administered, home-based neuromodulation of trigeminal-occipital neural networks and may represent a therapeutic option for migraine patients. |
Introduction
Migraine is an extraordinarily prevalent neurological disease, affecting 39 million men, women and children in the USA and 1 billion people worldwide [1] with a much greater prevalence among women [2]. Migraine patients experience disabling symptoms that typically consist of moderate-to-severe headache pain lasting 4 to 72 h, nausea with or without vomiting, phonophobia, and photophobia [3]. Patients with migraine are often treated with first-line therapy such as simple or combination analgesics that have variable onset and duration [4]. The triptans are effective first-line treatment for moderate to severe migraine [4]. Unfortunately, the safety and tolerability profile of these medications includes numerous potential adverse events [5], affecting approximately 50% of treated patients. Furthermore, triptans are contraindicated in patients with vascular disease [6] and have been associated with the development of headache due to medication overuse [7]. Thus, there remains a significant unmet need for treating migraine pain.
Peripheral nerve stimulation (PNS) is a clinically established medical technology for brain neuromodulation using invasively implanted electrodes or non-invasive targeted delivery of electrical current. Surgically implanted PNS electrodes for treating migraine, such as occipital nerve stimulation, significantly attenuated chronic migraine in several clinical trials [8,9,10,11,12,13] but remain a costly procedure with a high rate of complications [11, 14]. Consequently, these procedures are reserved for more severe, intractable cases of chronic migraine.
An alternative to implanted devices is the application of non-invasive, transcutaneous PNS. In sham-controlled studies, single-channel stimulation of the trigeminal nerve is similar to other drug and non-drug treatments for migraine in reducing migraine pain compared to sham [15, 16]. Among 2313 patients treated with transcutaneous stimulation of the supraorbital branch of the trigeminal nerve, there were no reports of serious adverse events [17]. A major shortcoming of these studies is that stimulation was limited to only the trigeminal nerve despite the reported efficacy of occipital nerve stimulation [18].
Concurrent invasive stimulation of both trigeminal and occipital nerves has been proposed as a more effective approach to minimize holocephalic pain due to migraine [19]. Subsequently published clinical data support the hypothesis that invasive synchronous PNS of the occipital and supraorbital nerves may provide better outcomes compared with stimulation of the occipital nerve alone [20, 21]. Using a response rate defined as ≥ 50% decrease in pain severity, subjects with intractable head-wide pain who were treated with implanted occipital and trigeminal nerve PNS achieved 70% [20] to > 90% improvement [19, 21]. By comparison, occipital nerve stimulation alone only achieved an approximately 40% response rate [9, 10, 13]. Taken together, these reports suggest the possible superiority of bi-focal PNS over uni-focal PNS [20,21,22,23,24]; however, these studies include high complication rates, which emphasizes the need for similar but noninvasive approaches.
A means for delivering noninvasive, external concurrent occipital and trigeminal neurostimulation (eCOT-NS) has been developed. The trigeminal nerve branches are relatively easy to access externally, but transferring an electric current through the hair covering the occipital area requires implanted [9,10,11,12,13] and percutaneous [25] nerve stimulators to achieve occipital nerve stimulation, which can be technologically challenging; however, topographic analysis of occipital nerve branches has challenged this assumption. As occipital nerve branches are located superficially [26, 27], accurate placement of stimulating electrodes bilaterally under the hair can effectively and noninvasively stimulate the occipital nerve. Using this method, eCOT-NS may provide the same beneficial effects of invasive procedures without the high cost and risks associated with surgically implanted stimulation [10, 11], while also avoiding the adverse events associated with medications. This is the rationale underlying the introduction of Relivion®MG (Neurolief, Ltd; Netanya, Israel), a US Food and Drug Administration-cleared eCOT-NS device for home use which includes six integrated electrodes that deliver mild electrical stimulation to the target nerves.
We hypothesized that the self-administered stimulation protocol using the Relivion®MG device can provide a safe, effective, and fast-acting acute reduction of migraine-related pain. The primary objective of this randomized, sham-controlled study was to assess changes in pain intensity following treatment with the eCOT-NS device.
Methods
This prospective randomized, double-blind, parallel-group, sham-controlled trial was conducted at a single site (Headache and Facial Pain Unit, Laniado Medical Center; Netanya, Israel). The study was conducted in accordance with principles of the Declaration of Helsinki [28] and Good Clinical Practices (ClinicalTrials.gov Identifier NCT03398668) and was approved by the Institution Ethics Committee of the Laniado Medical Center, Netanya, Israel. All subjects provided written informed consent prior to participating in any study-related activities.
Study Population
Subjects eligible for inclusion were 18 to 65 years old with a history of episodic or chronic migraine, with or without aura, with no complication (i.e., hemiplegic migraine, basilar-type migraine, ophthalmoplegic migraine, migrainous infarction), who met the International Classification of Headache Disorders (ICHD-3 beta Sect. 1, Diagnostic Criteria for Migraine) [29]. Enrolled subjects expressed their willingness to follow study requirements and were capable of following the study protocol including being able to self-operate the stimulation system at home.
Reasons for exclusion from study participation were treatment with a neurotoxin or supraorbital or occipital nerve blocks in the planned treatment area during the previous 4 months; history of medication overuse headaches; prior cerebrovascular event or neurosurgical interventions; history or current drug abuse or alcoholism; brain or facial trauma during the previous 3 months; skin lesions or inflammation in the location of the stimulating electrodes; diagnosis of epilepsy, personality, or somatoform disorder; current opioid or cannabis use; implanted metal or electrical devices in the head (not including dental implants); implanted neurostimulators, surgical clips (above the shoulder line), or any medical pumps; pregnancy, nursing, or planned pregnancy; unwillingness to use a medically acceptable method of contraception; participation in a clinical study within the previous 3 months; prior experience with the Relivion®MG device; or a head circumference < 51 cm or > 60 cm.
The Relivion®MG System
Relivion®MG is a noninvasive system for external concurrent occipital and trigeminal neurostimulation (eCOT-NS). It is US FDA-cleared and CE-approved for the acute treatment of migraine. A similar device has been developed for the treatment of major depression (Relivion®DP) [30].
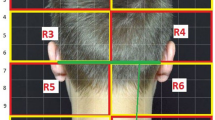
The eCOT-NS device is an ergonomic headset designed to externally stimulate pericranial nerves in the head region. The novel design enables accurate and repeatable positioning of the embedded electrode array over the target nerves. The headset integrates three pairs of output electrodes, which contact the scalp at the forehead (two pairs) and occiput (one pair). The frontal electrodes stimulate the trigeminal supraorbital and supratrochlear nerve branches bilaterally, and the posterior electrodes bilaterally stimulate the greater occipital nerve branches (Fig. 1). All nerve branches were stimulated concurrently. The device is adjustable to fit scalp anatomic variations and to ensure the six electrodes are accurately positioned over the underlying nerves each time the headset is worn. The device incorporates a simple on-board interface, which allows the user to activate/deactivate the device and to adjust the stimulation intensity. It provides visual and auditory indications such as whether the device is active and its battery status.

Relivion®MG system. The eCOT-NS device provides noninvasive neuromodulation by concurrently stimulating the two primary nerve pathways in the brain associated with migraine, precisely targeting six nerve branches across the occipital and trigeminal nerves. The device is connected to a custom mobile application designed to upload treatment data to a cloud database. Treatment reports and metrices are available to the physician over a web-based interface. TCC, trigeminocervical complex; Channel 1, occipital stimulation channel; Channel 2 and Channel 3, trigeminal (supraorbital/supratrochlear) stimulation channels; numbers 1–6 represent the six electrodes
A technical log is produced by the system after each use enabling full quantification of the treatment course. To enable remote treatment monitoring, the Relivion®MG system is connected to a custom mobile application, used by the patient, designed to continuously record data and to securely upload it to a cloud database. Treatment reports and metrices are available over a web-based physician interface (Fig. 1).
The following parameters were used for active stimulation: symmetrical biphasic waveform, phase width 400 µs, pulse frequency 80 Hz, trigeminal stimulation intensity up to 6 mA, and occipital stimulation intensity up to 12 mA. The following parameters were used for sham stimulation: symmetrical biphasic waveform, phase width 100 µs, pulse frequency 0.33 Hz, trigeminal stimulation intensity up to 5 mA, and occipital stimulation intensity up to 7 mA.
Study Design
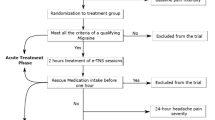
Following eligibility and health screening, enrolled subjects were randomized 1:1 in double-blind manner, stratified by gender, to receive either active (n = 27) or sham (placebo) stimulation (n = 28) with a similar-appearing device (Fig. 2). Subjects were then trained on use of the eCOT-NS device and received an electronic or hard copy diary. Subjects were instructed to practice using the device and diary in their home environment twice, not during a migraine episode, for a duration of 20–60 min. Device logs were downloaded by the research clinician to ensure completion of at least one successful 20–60 min home self-practice session.

Consort flow diagram. Among the randomized subjects (N = 55), the subjects completing the study in the active treatment group (n = 25) and sham group (n = 26) formed the modified intent-to-treat population (mITT)
Subjects were instructed to treat a single migraine, initiating treatment within 45 ± 15 min of migraine headache onset, and not to have used analgesics within 4 h before treatment. Subjects used the personal electronic or hard copy diary to record pain intensities, use of analgesics, and adverse events (AEs). A baseline (t0) outcome measure of perceived pain intensity, recorded on a visual analog scale (VAS), was documented in the study diary prior to treatment initiation and after treatment was initiated. Treatments with a stimulation time of > 30 min were counted as a completed treatment. The subject then recorded the pain VAS score again at 1 h (t1, end of treatment), 2 h (t2), and 24 h (t3) after treatment initiation. If necessary, rescue drugs were permitted 2 h after treatment initiation.
Subjects documented AEs and medication use in the study diaries. Upon completion of one treatment of a migraine headache, subjects returned the device to the clinic. Device logs were downloaded, reviewed, and documented to ascertain that the subject completed the treatment successfully. A successful treatment was defined as a recorded treatment with total stimulation time of more than 30 min and pass status (a minimum of 2 mA stimulation intensity) in the current device log. If the treatment was not successful, the subject was withdrawn from the analysis.
Precautions were taken to avoid compromising subject blinding, including concealed allocation, use of an identical sham and active devices, and the same treatment protocol in the preliminary test stimulation and treatment sessions. Additionally, sham stimulation was set to a level well above sensory threshold to further enhance subject blinding.
Outcome Measures
Pain intensity was recorded prior to treatment and 1, 2, and 24 h after treatment initiation. The primary endpoint was the change in mean baseline pain intensity based on VAS pain scores 1 h after treatment initiation and defined as relative (percent) change. The change in VAS score for each subject was calculated using the formula:
If the subject used a rescue medication prior to one of the 1-, 2-, or 24-h time points, the last pain score prior to the rescue medication was carried forward. If rescue therapy was used before the 1-h assessment, the baseline VAS score was carried forward.
Secondary outcomes were the mean change in baseline pain VAS scores at 2 h after starting treatment (if a rescue medication was not used); mean change in baseline pain VAS score at 24 h after starting treatment (if a rescue medication was not used); the proportion of subjects not requiring a rescue medication at 2 h; the proportion of subjects not requiring a rescue medication within 24 h of starting treatment; the proportion of subjects who were pain-free at 1, 2, and 24 h after starting treatment (if a rescue medication was not used); the proportion of treatment responders, defined as subjects with a ≥ 50% decrease in baseline pain VAS score at 1, 2, and 24 h after starting treatment (if a rescue medication was not used). An additional ad hoc analysis was headache relief, defined as improvement from baseline moderate or severe pain to mild or no pain. Standard pain categories were derived based on VAS scores: (0–1, no pain; 2–3, mild pain; 4–6, moderate pain; 7–10, severe pain) [31,32,33].
Statistical Analysis
Statistical analyses were performed on the intent-to-treat (ITT) population comprised of all randomized subjects and on the modified intent-to-treat (mITT) population. The mITT set included all randomized subjects who completed Visit 1, received the device for home use, had baseline and 1-h VAS data reported not out of window, a baseline VAS score of ≥ 2, and achieved a minimal effective level of stimulation per the protocol. If a rescue medication was used, the previous VAS score was carried forward. The ITT analysis served as the main analysis set for all safety evaluations, and the mITT set served for efficacy analysis (active vs. sham stimulation). The time post-treatment initiation [t0, baseline; t1, 1 h (end of treatment); t2, 2 h; t3, 24 h] and group (sham vs. active stimulation) were independent factors.
Data were summarized with descriptive statistics and are presented in tables and figures. Continuous variables were summarized by a mean, standard deviation, minimum, median, and maximum, and categorical variables by count and percentage. For comparison of means (continuous variables), the two-sample t-test or the Wilcoxon rank sum test was used, as appropriate. The percentage decrease from baseline of the VAS was analyzed with repeated measures ANCOVA. The decrease was modeled as a function of group, time points (entered as categorical variable), and the group by time interaction term; baseline VAS score was entered as a covariate. For comparison of proportions (categorical variables), the chi-squared test or Fisher’s exact test was used, as appropriate. The overall significance level for this study was 5% using two-tailed tests. Secondary endpoints were exploratory in nature; therefore, per secondary endpoints, nominal p-values are presented. Where confidence limits are appropriate, the confidence level was set at 95%. Data were analyzed using commercial statistical software (SAS® version 9.4, SAS Institute, Inc., Cary, NC).
Results
This study was conducted between February 8, 2018, and November 11, 2018. The mean (SD) age of enrolled subjects was 30 (8.34) (range, 20–58) years, and most (n = 45, 82%) were female. The mean age of migraine onset was 16.2 (4.7) (range, 4 to 30) years. The mean minimum and maximum monthly migraine attacks (SD) (see Table 2) were 3.8 (2.8) and 4.6 (3.1), respectively. Most subjects (n = 33; 60%) had migraine without aura, and few (n = 4, 7.3%) had chronic migraine. There were no significant group differences in demographics or baseline characteristics. The demographics of study subjects are summarized in Table 1, and their migraine history is summarized in Table 2.
Efficacy Outcomes
Baseline pain scores on the continuous (VAS) and categorical scale are shown in Table 3. There were no significant group differences in baseline pain VAS scores or migraine history. The between-group differences in percentage decrease in baseline VAS scores were significant at all time points with the decrease significantly greater in the active treatment group. The group difference at 1 h was 42.8% (95% CI 20.57%, 62.18%; p = 0.0002), at 2 h was 34.7% (95% CI 2.88%, 62.80%; p = 0.0324), and at 24 h was 37.0% (95% CI 5.46%, 66.96%; p = 0.0220). The change in mean pain intensity VAS scores is summarized in Table 4, and the percent reduction in mean baseline VAS scores is shown in Fig. 3.

Change in percent pain intensity. The therapeutic gain in pain intensity based on visual analog scale (VAS) scores was 42.8% at 1 h post-treatment (p = 0.0002), 34.7% at 2 h (p = 0.0324), and 37.0% at 24 h (p = 0.0220)
The proportion of pain-free subjects was numerically higher in the active treatment group at 1 h (29.2% vs. 16.0%), 2 h (41.7% vs. 20.0%), and 24-h (65% vs. 40.0%).
Although the between-group difference was not statistically significant, the number of pain-free subjects was more than twice as great among subjects in the active treatment group at 2 h (Table 5A, Fig. 4A). The pain freedom between-group difference 2-h post treatment among subjects with severe or moderate baseline pain was significantly higher in the active treatment group (42.86% vs. 10.53%, p = 0.02; Table 5b, Fig. 4B).

Proportion of pain-free subjects, modified intent-to-treat population. The proportion of subjects who were pain-free at 1, 2, and 24 h without requiring rescue medications was greater in the active stimulation group. Overall, the proportion of pain-free subjects was numerically superior in the active treatment group at 2 h (41.7% vs. 20.0%) (A). Among subjects with baseline moderate or severe pain, the difference was even greater (42.9% vs. 10.5%, p = 0.0217) (B)
Responder rates of subjects achieving ≥ 50% improvement of baseline VAS scores were significantly higher in the active treatment group at 1 h (66.7% vs. 20.0%; p = 0.0014), 2 h (66.7% vs. 32.0%; p = 0.0227), and 24 h (78.3% vs. 48.0%; p = 0.0401). Responder rate results are presented in Fig. 5.

Proportion of subjects responding to treatment, modified intent-to-treat population. The number of subjects achieving ≥ 50% improvement of baseline VAS pain scores (responders) was significantly higher in the active treatment group at 1 h (66.7% vs. 20.0%; p = 0.0014), 2 h (66.7% vs. 32.0%; p = 0.0227), and 24 h (78.3% vs. 48.0%; p = 0.0401)
Headache relief, defined as improvement from severe or moderate baseline pain to mild or no pain, was significantly greater among active treatment subjects at 1 h (66.7% vs. 26.3%; p = 0.0140) and 2 h (76.2% vs. 31.6%; p = 0.0100) and approached significance at 24 h (80% vs. 52.6%; p = 0.0958). Headache relief results are presented in Fig. 6. No difference in rescue medication intake was demonstrated between active and sham groups.

Subjects with headache relief following treatment. The proportion of subjects experiencing headache relief, defined as improvement from severe or moderate baseline pain to mild or no pain, was significantly greater among active treatment subjects at 1 h (66.7% vs. 26.3%; p = 0.0140) and 2 h (76.2% vs. 31.6%; p = 0.0100) and approached significance at 24 h (80% vs. 52.6%; p = 0.0958)
Adverse events (AEs) were reported by 14 subjects in the active treatment group (n = 6) and sham group (n = 8). There was no statistically significant difference between the groups in incidence of AEs. Seven reported AEs among subjects receiving active treatment were headache (n = 2), numbness/ paresthesia (n = 2), skin irritation (n = 1), nausea (n = 1), and itching scalp (n = 1). All were mild in severity except one reported headache, which was of moderate severity and considered as possibly related to the device. All events resolved spontaneously. Nine reported AEs among eight sham-treated subjects were nausea (n = 3), vomiting (n = 2), tiredness (n = 1), palpitation (n = 1), pressure in the head (n = 1), and photophobia (n = 1). All were mild in severity except one report of vomiting, which was of moderate severity. Tiredness and palpitation were considered to be possibly treatment related. All AEs resolved spontaneously.
Discussion
The objective of this study was to determine whether a self-administered stimulation protocol using the eCOT-NS device can provide a safe, effective, and fast-acting reduction of migraine-related pain. As hypothesized, the device was shown to be significantly effective across multiple clinically relevant outcome measures. At 1 h post-treatment, the active stimulation group showed a mean 53.1% reduction in pain severity compared to a mean 10.3% decrease among sham-treated subjects, a difference of 42.8%. Pain reduction was maintained at 2 h with 52% and 17.3% decreases in pain severity, respectively, a difference of 34.7%. The treatment effect is also durable, as the proportion of pain reduction among active treatment subjects was 71.3% at 24 h vs. 34.3% for the sham group. The responder rate was also significantly higher in the active treatment group at all time points, and the proportion of pain-free subjects at 2 h post-treatment was more than two-fold higher in the active group (41.7% vs. 20%). Although not statistically significant, it suggests a clinically meaningful response. At a post hoc pain-free assessment analysis of a subgroup of subjects with moderate or severe baseline headache pain, the between-group difference was in favor of the active group again at 1 and 2 h. These results support the safety and efficacy of the eCOT-NS, eventually leading to its clearance of for the acute treatment of migraine.
These clinical results were substantially higher in aborting migraine compared to a 60-min session of single-channel non-invasive trigeminal neurostimulation [16]. In that study, treated subjects achieved a net response rate of 29%, 18%, and 17% at 1, 2, and 24 h, respectively, compared to a net response of 42.8%, 34.7%, and 37% in the present study with eCOT-NS. In another study, subjects implanted with an occipital nerve stimulator achieved a responder rate, defined as percent reduction in monthly headache days, of 39% vs. 6% among sham-treated subjects, a net difference of 33%. Subjects receiving active stimulation in the current study achieved a responder rate, defined as at least a 50% reduction in pain intensity, of 66.7% vs. 20.0% among sham-treated subjects at 1 h post-treatment, a difference of 46.7%. Comparing the result of the current study with other clinical trials should be done with caution as the selection criteria and outcome measures may vary; nevertheless, the results of the current study support the efficacy of combined stimulation of both the trigeminal and occipital nerves for the acute treatment of migraine.
Enrolled subjects in this trial had one to six monthly migraine episodes similar to subjects enrolled in large-scale triptan trials [34] and are representative of migraine patients in the general population in need of migraine-abortive treatment [2]. The results achieved with eCOT-NS appear to be comparable to oral triptans, as demonstrated by 76% headache relief 2 h post treatment in our study compared to 42–76% reported following oral triptans [34]. Recent studies of new medications for the acute treatment of migraine, including ubrogepant [35] and rimegepant [36], appear to have more modest pain relief efficacy compared to eCOT-NS (60.7% and 59.3% at 2 h, respectively).
An additional benefit of the Relivion®MG system is that it utilizes a custom mobile application designed for continuously recording data and securely uploading it to a cloud database to enable remote treatment monitoring by the patient physician. Treatment reports are available to the treating physicians over a web-based interface. By remote monitoring, physicians can assess patient adherence to self-administered treatments and success of aborting or minimizing migraine severity. Using the aggregated data presented on the physician interface, the physician can optimize treatment by recommending treatment regimen adjustments, such as adjusting the stimulation time and intensity.
Treatment with eCOT-NS provided clinical benefits for migraine subjects with an acceptable safety profile. The reported AEs associated with the eCOT-NS device were relatively few, of mild severity, and resolved spontaneously with no intervention. This compares favorably with surgical risks associated with electrode implants or systemic AEs associated with drug administration [2]. The use of eCOT-NS might enable migraine patients to discontinue current migraine medications.
Limitations of the study include the modest number of subjects, which did not permit subgroup analysis based on headache severity. Another shortcoming was the efficacy assessment based on a single 60-min stimulation session, which limited assessment of long-term efficacy.
Conclusions
These results suggests that many migraine patients may achieve clinically significant pain relief and pain freedom with eCOT-NS, a non-invasive, home-based, self-administered neuromodulation of the trigeminal-occipital neural network. These beneficial effects appear to demonstrate comparable or higher efficacy compared to existing migraine treatments, without the adverse effects associated with medications or the surgical risks of invasive electrode procedures. eCOT-NS, with its physician remote monitoring capabilities, may optimize migraine treatment care, patient adherence, and a migraine patient’s quality of life.
References
Migraine Research Foundation 2021. Available: https://migraineresearchfoundation.org/about-migraine/migraine-facts/. Accessed: December 20, 2021.
Lipton RB, Bigal ME, Diamond M, Freitag F, Reed ML, Stewart WF, et al. Migraine prevalence, disease burden, and the need for preventive therapy. Neurology. 2007;8:343–9.
Pescador Ruschel MA, De Jesus O. Migraine headache. Treasure Island: StatPearls Publishing; 2021.
Mayans L, Walling A. Acute migraine headache: treatment strategies. Am Fam Physician. 2018;97:243–51.
Jamieson DG. The safety of triptans in the treatment of patients with migraine. Am J Med. 2002;112:135–40.
Tepper SJ, Millson D. Safety profile of the triptans. Expert Opin Drug Saf. 2003;2:123–32.
Loder E. Triptan therapy in migraine. N Engl J Med. 2010;363:63–70.
Robbins MS, Lipton RB. Transcutaneous and percutaneous neurostimulation for headache disorders. Headache. 2016;57:4–13.
Miller S, Watkins L, Matharu M. Long-term outcomes of occipital nerve stimulation for chronic migraine: a cohort of 53 patients. J Headache Pain. 2016;17:68.
Palmisani S, Al-Kaisy A, Arcioni R, Smith T, Negro A, Lambru G, et al. A six year retrospective review of occipital nerve stimulation practice–controversies and challenges of an emerging technique for treating refractory headache syndromes. J Headache Pain. 2013;14:67.
Silberstein SD, Dodick DW, Saper J, Huh B, Slavin KV, Sharan A, et al. Safety and efficacy of peripheral nerve stimulation of the occipital nerves for the management of chronic migraine: results from a randomized, multicenter, double-blinded, controlled study. Cephalalgia. 2012;32:1165–79.
Serra G, Marchioretto F. Occipital nerve stimulation for chronic migraine: a randomized trial. Pain Physician. 2012;15:2.
Saper JR, Dodick DW, Silberstein SD, McCarville S, Sun M, et al. Occipital nerve stimulation for the treatment of intractable chronic migraine headache: ONSTIM feasibility study. Cephalalgia. 2011;31:271–85.
Dodick DW, Silberstein SD, Reed KL, Deer TR, Slavin KV, Huh B, et al. Safety and efficacy of peripheral nerve stimulation of the occipital nerves for the management of chronic migraine: long-term results from a randomized, multicenter, double-blinded, controlled study. Cephalalgi. 2015;35:344–58.
Schoenen J, Vandersmissen B, Jeangette S, Herroelen L, Vandenheede M, Gérard P, et al. Migraine prevention with a supraorbital transcutaneous stimulator: a randomized controlled trial. Neurology. 2013;80:697–704.
Chou DE, Shnayderman Yugrakh M, Winegarner D, Rowe V, Kuruvilla D, Schoenen J. Acute migraine therapy with external trigeminal neurostimulation (ACME): a randomized controlled trial. Cephalalgia. 2019;39:3–14.
Magis D, Sava S, d’Elia TS, Baschi R, Schoenen J. Safety and patients’ satisfaction of transcutaneous supraorbital neurostimulation (tSNS) with the Cefaly® device in headache treatment: a survey of 2,313 headache sufferers in the general population. J Headache Pain. 2013;14:95.
Ashkan K, Sokratous G, Göbel H, Mehta V, Gendolla A, Dowson A, et al. Peripheral nerve stimulation registry for intractable migraine headache (RELIEF): a real-life perspective on the utility of occipital nerve stimulation for chronic migraine. Acta Neurochir (Wien). 2020;162:3201–11.
Reed KL, Will KR, Conidi F, Bulger R. Concordant occipital and supraorbitaln eurostimulation therapy for hemiplegic migraine; initial experience; a case series. Neuromodulation. 2015;18:297–304.
Hann S, Sharan A. Dual occipital and supraorbital nerve stimulation for chronic migraine: a single-center experience, review of literature, and surgical considerations. Neurosurg Focus. 2013;35:E9.
Reed KL, Black SB, Banta CJ, Will KR. Combined occipital and supraorbital neurostimulation for the treatment of chronic migraine headaches: initial experience. Cephalalgia. 2010;30:260–71.
Reed KL. Peripheral neuromodulation and headaches: history, clinical approach, and considerations on underlying mechanisms. Curr Pain Headache Rep. 2013;17:305.
Reed KL, Will KR, Chapman J, Richter E. Combined occipital and supraorbital neurostimulation for chronic migraine headaches: an extended case series. Cephalalgia. 2011;31:98.
Jiang JF, Diaz AN, Campbell M, Boulis NM, Keifer OP Jr. Supraorbital occipital circumferential stimulation (SOCS) for the treatment of refractory chronic primary headache: a case series. World Neurosurg. 2019;S1878–8750(18):32931.
Ahmed HE, White PF, Craig WF, Hamza MA, Ghoname ES, Gajraj NM. Headache. 2000;40:311–5.
Mueller O, Hagel V, Wrede K, Schlamann M, Hohn HP, Sure U, et al. Stimulation of the greater occipital nerve: anatomical considerations and clinical implications. Pain Physician. 2014;16:E181–9.
Tubbs RS, Salter EG, Wellons JC, Blount JP, Oakes WJ. Landmarks for the identification of the cutaneous nerves of the occiput and nuchal regions. Clin Anat. 2007;20:235–8.
General Assembly of the World Medical Association. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. J Am Coll Dent. 2014;81:14–8.
Headache Classification Committee of the International Headache Society (IHS). The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33:629–808.
Oved D, Sharon R, Tepper SJ. A device review of Relivion®: an external combined occipital and trigeminal neurostimulation (eCOT-NS) system for self-administered treatment of migraine and major depressive disorder. Expert Rev Med Devices. 2021;18:333–42.
Yarnitsky D, Dodick DW, Grosberg BM, Burstein R, Ironi A, Harris D, et al. Remote electrical neuromodulation (REN) relieves acute migraine: a randomized, double-blind, placebo-controlled, multicenter trial. Headache. 2019;59:1240–52.
Rapoport AM, Bonner JH, Lin T, Harris D, Gruper Y, Ironi A, et al. Remote electrical neuromodulation (REN) in the acute treatment of migraine: a comparison with usual care and acute migraine medications. J Headache Pain. 2019;20:83.
Yarnitsky D, Volokh L, Ironi A, Weller B, Shor M, Shifrin A, et al. Nonpainful remote electrical stimulation alleviates episodic migraine pain. Neurology. 2017;88:1250–5.
Cameron C, Kelly S, Hsieh SC, Murphy M, Chen L, Kotb A, et al. Triptans in the acute treatment of migraine: a systematic review and network meta-analysis. Headache. 2015;55:221–35.
Dodick DW, Lipton RB, Ailani J, Lu K, Finnegan M, Trugman JM, et al. Ubrogepant for the treatment of migraine. N Engl J Med. 2019;381:2230–41.
Croop R, Goadsby PJ, Stock DA, Conway CM, Forshaw M, Stock EG, et al. Efficacy, safety, and tolerability of rimegepant orally disintegrating tablet for the acute treatment of migraine: a randomised, phase 3, double-blind, placebo-controlled trial. Lancet. 2019;394:737–45.
Acknowledgements
Funding
Sponsorship for this study and Rapid Service Fee were funded by Neurolief Ltd, Netanya, Israel. Editorial assistance in the preparation of this article was provided by Carl S. Hornfeldt, PhD, Apothekon, Inc. Support for this assistance was funded by Neurolief Ltd. The authors thank the subjects in this study for their enthusiastic involvement in the study.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
All authors contributed to the study conception and design. Dr. Oved Daniel was the study principal investigator and enrolled all subjects, Dr. Lisa Deutsch performed the study statistical design and performed all statistical analysis, Dr. Stewart Tepper consulted during study design and manuscript preparation, and Dr. Roni Sharon acted as medical advisor and reviewed all adverse events.
Disclosures
Drs. Roni Sharon and Stewart Tepper are advisors to Neurolief Ltd, and both have served as clinical investigators to Neurolief Ltd. The other authors declare they have nothing to disclose. Dr. Lisa Deutsch is a biostatistician for hire and receives financial compensation for performing statistical analyses for companies in the medical device industry.
Compliance with Ethics Guidelines
This study was performed in accordance with the Helsinki Declaration of 1964 and subsequent amendments. All subjects provided informed consent to participate in the study prior to participating in any study-related activities. The study protocol and related materials were approved by the Institution Ethics Committee of the Laniado Medical Center; Netanya, Israel.
Data Availability
The datasets generated and analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Daniel, O., Tepper, S.J., Deutsch, L. et al. External Concurrent Occipital and Trigeminal Neurostimulation Relieves Migraine Headache: A Prospective, Randomized, Double-Blind, Sham-Controlled Trial. Pain Ther 11, 907–922 (2022). https://doi.org/10.1007/s40122-022-00394-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-022-00394-w