
Abstract
Osteoarthritis (OA) is a leading cause of disability among older adults worldwide. Treatment aims are to alleviate inflammatory pain and improve physical function through non-pharmacological and pharmacological interventions. Non-steroidal anti-inflammatory drugs (NSAIDs) are recommended as first-line therapy. However, selection is challenged by patient age, comorbidities and polypharmacy, and by the drug’s benefit/risk balance, all of which together influence the risk of cardiovascular (CV), gastrointestinal (GI) and renal adverse events (AEs). While the efficacy profile of the various NSAIDs is delineated, the differences in their safety profile are not straightforward. This narrative review provides practical indications by a multidisciplinary Italian expert panel for general practitioners and specialists managing OA patients with chronic inflammatory pain; the goal is to maximize therapy efficacy while reducing untoward effects caused by inappropriate NSAID use. The discussion on the best approach to NSAIDs spanned the following topics: (1) patient evaluation: investigate pain origin, duration and components together with possible risk factors for CV, GI and renal AEs; (2) non-pharmacological interventions: the physiatrist provides a person-centered, holistic approach accounting for all patient aspects; (3) pharmacological interventions: patient profile and drugs’ pharmacological properties affect NSAID selection, which drugs to be used in combination or to be avoided, formulation and therapy duration; (4) the pharmacologist’s, general practitioner’s and pain therapist’s points of view; (5) NSAID safety: the individual baseline risk and the drug’s safety profile are major determinants of CV, GI and renal risk; consider possible drug–drug interactions; (6) periodical re-evaluation of treatment response and adherence, using scales to assess pain and function.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The goal of treatment in osteoarthritis is to alleviate inflammatory pain and improve physical function through non-pharmacological and pharmacological interventions. |
Therapy selection is hampered by older age, comorbidities and polypharmacy. |
This narrative review addresses all phases of the patient journey from a multidisciplinary perspective, focusing on the safety of non-steroidal anti-inflammatory drugs. |
We provide practical indications for general practitioners and specialists managing patients with osteoarthritis who suffer from chronic inflammatory pain. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14308292.
Introduction
Osteoarthritis (OA) is the most frequent form of arthritis worldwide and a leading cause of disability among older adults [1]. In Italy, its prevalence is 24.9% in women and 16% in men and is highest in persons aged > 85 years (63.0% in women and 50.9% in men) [2, 3]. After hypertension, it is the second most common chronic disease managed by general practitioners (GPs) [2].
The main risk factors for OA are age, gender, obesity and adverse mechanical factors [4]. Knees, hips and hands are the most commonly affected appendicular joints, and patients often suffer from pain, stiffness, swelling and loss of normal joint function, with a negative impact on their quality of life and a relevant socioeconomic burden.
The goal of treatment in OA is to reduce pain intensity and improve function and quality of life through a combination of non-pharmacological and pharmacological interventions [5, 6]. As first-line therapy, guidelines [4,5,6,7,8] recommend the non-steroidal anti-inflammatory drugs (NSAIDs), a chemically heterogeneous group of agents that inhibit the production of prostaglandins (PG) and thromboxane A through the blockade of cyclooxygenase (COX). Traditional NSAIDs (tNSAIDs), which target the COX-1 and COX-2 isozymes to varying degrees, have a consolidated role in the symptomatic treatment of pain in musculoskeletal disorders [9,10,11], but their long-term use is limited by toxicity, mainly cardiovascular (CV), gastrointestinal (GI) and renal toxicities. Although COX-2-selective NSAIDs (coxibs) were initially introduced as a safer alternative to tNSAIDs, their use has been associated to a high risk of CV events [12].
The frequent, inappropriate use of over-the-counter NSAIDs is a matter of concern as it rises the risk of untoward events [13,14,15]. According to a recent Italian long-term active pharmacovigilance study, NSAIDs are responsible for 8.4% of the emergency department visits and 24.4% of emergency department visits resulting in hospitalizations [16].
In practice, both drugs’ and patients’ characteristics influence the choice of therapy. The efficacy profile of NSAIDs has been delineated by meta-analyses of randomized controlled trials (RCTs) [17,18,19,20,21,22,23]. Among these, the network meta-analysis by da Costa and colleagues, comparing the effectiveness of various NSAIDs, paracetamol and placebo on pain and physical function improvement, included the highest number of preparations and doses and provided also information on the dose–response relation [21]. It included 74 RCTs, for a total of 58,556 OA patients. Overall, there was not enough statistical evidence to support the superiority of diclofenac 70 mg/day, naproxen 750 mg/day and ibuprofen 1200 mg/day over placebo for pain and physical function improvement. In contrast, for pain reduction, diclofenac 150 mg/day and etoricoxib given at 30 mg/day, 60 mg/day and 90 mg/day had a probability of reaching the minimum clinically important difference compared to placebo of ≥ 95%, reaching 100% only in the case of diclofenac 150 mg/day and etoricoxib 60 mg/day. Notably, a significant linear dose–effect response was found only for celecoxib (P = 0.030), diclofenac (P = 0.031) and naproxen (P = 0.026). As for the physical function improvement, a minimum clinically important treatment effect was observed solely for diclofenac 150 mg/day. The authors concluded that diclofenac at 150 mg/day is the best NSAID in terms of both pain and function amelioration in OA, superior to the maximum doses of frequently used NSAIDs, including ibuprofen, naproxen and celecoxib. Albeit etoricoxib at the maximum dose of 60 mg/day was as effective as diclofenac 150 mg/day for the treatment of pain, its effect estimates on physical disability remain unclear. Finally, paracetamol had no clinical effect and should not be recommended for the symptomatic treatment of OA. This study demonstrates that the same NSAID at different doses has different effects and provides important information on the minimal effective dosages of a number of compounds [21].
While the efficacy profile of the various NSAIDs is clear, the differences in their safety profile are not straightforward and are affected by individual characteristics [20, 24]. In the last 10 years, several meta-analyses of RCTs and observational studies have compared the safety profile of these drugs [20, 25,26,27,28,29,30,31,32,33]. Yet, study design and endpoints are heterogeneous [24], and data are biased, for instance, by the fact that they often rely on prescriptions rather than on actual administrations (i.e. no consideration of the exposure duration and dose) without accounting for the reason for the prescription, nor for concomitant diseases and risk factors. For some compounds, the lack of robust data from large cohort studies may be mistakenly regarded as a guarantee of safety.
To support healthcare providers in the optimization of OA patient management, i.e. trying to maximize therapy efficacy while reducing untoward effects caused by inappropriate NSAID use [14], a multidisciplinary expert panel (i.e., 1 GP, 1 pharmacologist, 1 pain therapist, 1 cardiologist, 1 gastroenterologist, 1 nephrologist and 1 physiatrist) thoroughly discussed the best approach in this complex setting. To inform the group’s discussion, a literature search was performed via PubMed using the following items as the main keywords: “NSAID,” “osteoarthritis,” “chronic pain,” “effectiveness,” “efficacy,” “safety,” “cardiovascular,” “gastrointestinal” and “renal.” We limited the search to articles in English. Electronic Supplementary Material (ESM) Table 1 presents the systematic reviews and/or meta-analyses of RCTs and observational studies on NSAID efficacy and safety published in the past 10 years and included in the present work. This work is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
This narrative review summarizes the main messages and practical indications for GPs and specialists.
Chronic Pain
Definition of Chronic Pain
The term “chronic” refers to a pain persisting over time (according to the International Association for the Study of Pain [IASP], chronic pain either persists or recurs for > 3 months [34]) but provides no details on whether the stimuli persist or if other pathogenetic mechanisms intervene. Certainly, pain persistence affects patients’ life and complicates their clinical status.
For some investigators, “chronic” implies the involvement of the central nervous system, where pathogenetic mechanisms able to maintain chronicity even in the absence of peripheral stimuli develop. Others believe that “chronic” relies on the occurrence of nervous mechanisms typical of neuropathic pain and refer to it as to a mixed or neuropathic-like pain.
There are degenerative and neurological disorders characterized by chronic injuries and in which pain is induced chronically (erythromelalgia, fibromyalgia, deafferentation pain, OA, rheumatoid arthritis [RA], etc.). Many conditions are accompanied by episodes of pain of variable persistence that affects distinct parts of the body over time.
Finally, pain could be defined as chronic if it is not effectively treated or if it is related to undiagnosed diseases.
Types of Chronic Pain
The IASP describes three types of pain: nociceptive, neuropathic and nociplastic [34]. Nociceptive pain is of inflammatory or degenerative origin depending on the presence or absence of a mechanism of nociceptor sensitization. Inflammatory pain starts in the tissue nociceptive nerve endings and represents the type of chronic pain experienced by patients with OA. The responsible mechanism, i.e. peripheral sensitization, consists in a threshold reduction at the peripheral ends of the sensory nerve fibers, which become responsive to low-intensity stimuli (i.e. allodynia) or may even become spontaneously active. Peripheral sensitization depends on biochemical modifications of nociceptive fibers triggered by mediators of inflammation, such as PGs and cytokines. If the sensitizing agents are removed, the biochemical processes revert, and the normal threshold is re-established.
Neuropathic pain is classified as peripheral or central, based on the site of injury and, thus, of the ectopic activity: the site of pain origin is along the somatosensory pathway affected by a disease or a lesion (from peripheral nociceptors to central neurons). Other definitions, such as neuropathic-like pain, neuropathic component and mixed pain, are frequently associated to nociceptive pain to underline a central component of pain (spinal cord sensitization) that becomes responsible for neuropathic symptoms. It cannot be considered a real neuropathic pain because of the lack of neurological deficit signs.
Patient Evaluation
The Diagnostic Work-Up
Osteoarthritis is a heterogeneous disease with distinct phenotypes [35]. Before commencing a therapy with NSAIDs, it is fundamental to
-
(1)
Collect all relevant clinical information to define the disease characteristics, clinical status and possible risk factors for OA, with particular attention to pain description, psychosocial aspects, comorbidities and risk of CV, GI and renal complications.
-
(2)
Perform the I- and II-level assessments as per current guidelines [4].
Pain Assessment
As a first step in pain assessment, the GP must define the pain type, as NSAIDs are effective against inflammatory nociceptive pain but not against non-inflammatory mechanical–structural pain (occurring in approximately 10–15% of OA patients). To infer the type of pain (i.e. somatovisceral or neurological condition), symptoms should be measured using the scales for neuropathic pain, keeping in mind that many symptoms are typical of both types of pain. The most used tools to discriminate neuropathic pain from non-neuropathic pain in clinical settings include painDETECT [36], the Leeds assessment of neuropathic symptoms and signs Pain Scale [37] and the Douleur Neuropathique 4 questions [38]; all of these tools rely on the description of pain and on the bedside examination of sensory dysfunction. Moreover, painDETECT has been recommended by the Initiative on Methods, Measurement, and Pain Assessment in Clinical Trials to screen for neuropathic pain phenotypes [39]. Although these scales are easy to use and allow a preliminary clinical assessment, they are considered to be useful screening tools and cannot replace a thorough clinical assessment and their accuracy varies across different populations [40, 41].
When considering the pathogenetic mechanisms, it is appropriate to investigate whether the pain is localized, evoked, radiating or referred (by identifying the pain area and inspecting it, evoking pain with non-painful stimuli and testing skin sensitivities), as well as the negative symptoms, which predominate at the sites of neuropathic pain. The following tools should be employed: the generic and unidimensional pain assessment tools Visual Analogue Scale (VAS) and Numeric Rating Scale (NRS), to rapidly and easily measure pain intensity [42]; the Western Ontario and McMaster Universities Arthritis Index (WOMAC), to measure arthritis symptoms, pain and physical functional disability specifically in patients with OA of the knee and the hip [43]. Both the VAS and NRS are self-administered and can detect changes over time [42]. The NRS may be preferred over the VAS because of its simpler score calculation and because it may be administered both verbally and in writing, while the VAS can be administered only in writing. However, due to their nature, they do not provide a comprehensive pain evaluation in patients with rheumatic disease [42]. As for the WOMAC, it is one of the most appropriate patient-reported outcome measures to be employed in trials of knee and hip OA; however, interpretation of the results and comparisons among studies are frequently challenged by the different versions available (Likert, VAS or NRS) and by the wide variation in its use and analysis [44, 45].
A definitive diagnosis is made by combining the clinical and neurophysiological evaluations with the diagnostic nerve block test.
Often, in a context of degenerative pain, patients experience periods of inflammation. The test of the response to NSAIDs (acting on PGs) and cortisone (acting on cytokines) may be helpful, but it must follow—not replace—the clinical evaluation. PGs and cytokines act by sensitizing the peripheral nociceptive endings, i.e. increasing their responsiveness to stimuli below the normal threshold. PGs are the first mediators of inflammation released during the inflammatory process that follows the injury, while cytokines are released later on. When performing the test, it is important to keep in mind that the central analgesic activity of certain NSAIDs may interfere with the result.
Function and patient global assessment (PGA) of disease severity have to be assessed as well. The most frequently used tools are the WOMAC, VAS or Likert scales and global function score for function and the VAS or 5-point Likert scale for PGA of disease severity.
Atypical Presentation
In case of atypical presentation, imaging is recommended to confirm the diagnosis of OA and/or make alternative or additional diagnoses [4]. Radiological imaging allows potential changes in bone, cartilage and inflammation to be monitored [35] and includes cartilage evaluation to verify possible interjoint space reduction, increased density of subchondral bone and abnormal reactive growth of the bone at the edge of joint (osteophytes). Conventional radiography is the gold standard.
Differential Diagnosis
It is important to exclude RA and other types of chronic arthritis in OA patients. According to the updated recommendations of the Italian Society of Rheumatology, laboratory tests (blood count, inflammation, urinalysis or synovial fluid) should be performed for OA patients with marked inflammatory symptoms and/or signs, especially when atypical sites are involved, for differential diagnostic purposes, particularly to exclude chronic or crystal-induced arthropathies [4].
Comorbidities and Risk Assessment
During the visit and before starting the chosen therapy with NSAIDs, GPs should consider the conditions at the highest risk of potental complications upon NSAID treatment and verify the presence of a number of important factors that increase the risk of CV, GI, and renal adverse events (AEs) (Table 1):
-
(1)
Prior CV events (major acute myocardial infarction [AMI], stroke, peripheral venous and arterial thrombosis). The CV risk should be calculated using the European Society of Cardiology score [46]; however, in outpatient practice, it is rarely calculated and is frequently overlooked, although it is a key determinant of the choice of the most appropriate treatment option. Patients are considered at high risk when the score of the 10-year fatal CV disease (CVD) risk is 5–10% or if they have familial dyslipidemia, severe hypertension, diabetes without CV risk factors and organ damage or moderate chronic renal failure [46, 47].
-
(2)
GI intolerance (abdominal pain, constipation, diarrhea, dyspepsia and nausea) and major GI events (e.g. perforation and bleeding, which depends on age and comorbidities [48]).
-
(3)
Kidney function: diseases like acute kidney failure, interstitial nephritis and chronic kidney failure should be considered. A thorough screening and a complete laboratory work-up should be undertaken for each patient at risk of renal AEs based on age, comorbidities such as diabetes and chronic renal failure and concurrent antihypertensive therapies (anti-angiotensin [ANG] II, anti-aldosterone treatment).
The patient must be educated on lifestyle and prevention.
The panelists agreed that the ideal pathway for OA patients suffering from chronic inflammatory pain is the process illustrated in Fig. 1.

The ideal pathway for osteoarthritis patients suffering from chronic inflammatory pain. GP General practitioner, NSAIDs non-steroidal anti-inflammatory drugs
When to Consult the Pain Therapist
It is necessary to refer a patient to a pain center if:
-
(1)
The origin of pain was not identified (lack of pathogenetic diagnosis).
-
(2)
The pharmacological treatment was not successful. The pain therapist should be consulted before referring the patient to specialists and before administering opioids.
-
(3)
Treatment reduced pain but not disability.
-
(4)
The patient presented intolerance or contraindications to NSAIDs.
Practical Indications
-
(1)
During the visit, pain must be thoroughly evaluated, considering:
-
pain origin and duration
-
component (inflammatory or degenerative)
-
NSAID activity (peripheral or central)
-
-
(2)
Consider possible factors that increase the risk of CV, GI and renal AEs.
-
(3)
Calculate the CV risk.
-
(4)
Prescribe a complete laboratory work-up, including serum chloride measurement, to test the kidney function.
Non-pharmacological Therapy
Orthopedic Treatment, Supplements, and Physiotherapy
In the setting of OA, beyond pharmacological therapy, it is important to consider specific orthopedic treatments (surgical and non-surgical), use of dietary supplements and physiatrist assessment, especially for rehabilitative interventions and supplemental physical therapies.
The specialist in orthopedics and traumatology is a surgeon and, as such, should be consulted to determine whether a patient suffering from OA may benefit from a surgical approach. In all the other cases, the reference specialist is the physiatrist, who takes charge of OA patients and makes a prospective evaluation of their functional needs. Once the patient has been evaluated, the physiatrist makes an individual rehabilition plan to estimate the functional aspects as well as rehabilitative prognosis.
Role of the Physiatrist
In first place is the GP, who prescribes I-level instrumental/imaging examinations (i.e. radiography and musculoskeletal ultrasound) for OA patients before a surgical consultation with the orthopedic surgeon. The physiatrist will then take charge of the patient and design the individual rehabilitation project (i.e. “the set of propositions, elaborated by the rehabilitation team, coordinated by the medical doctor specialist”) [49]. This represents the basis of a person-centered, holistic approach that accounts for the individual conditions globally: indeed, it includes the main scales of motor, cognitive and social assessments characterizing the clinical history of the patient. The physiatrist is the leader of the rehabilitation team that takes charge of patients undergoing or not undergoing surgery.
Based on the individual rehabilitation project, the physiotherapist sets up the individual rehabilitation program aimed at achieving the therapeutic objectives established in the individual rehabilitation project.
Rehabilitation Therapies and Physical Exercises
The prerequisite to optimization of the process of care for OA patients is pain reduction/treatment. Physical antalgic therapies, similar to minimally invasive interventional treatments (i.e. intra- or extra-articular injections), represent an integral part of the rehabilitative approach, with its rationale built into the individual rehabilitation project.
OA causes a reduction in the mechanical functioning and overall clinical status. Most patients suffering from OA are frail with several comorbidities, and thus the physiatrist consults with other specialists, including neurologists, geriatrists, anesthesiologists, internists, among others. Sarcopenia and frailty increase the risk of falls, leading to the need for a broader range of therapeutic strategies, including adequate diet and exercise, to support OA patients.
Practical Indications
-
(1)
Use functional scales for the clinical assessment.
-
(2)
Consider consulting a physiatrist during the post-operative rehabilitation.
-
(3)
Account for all patient aspects.
-
(4)
Pay close attention to prevent falls and the sequelae of reduced mobility caused by OA.
Pharmacological Therapy: NSAIDs
The Pharmacology of NSAIDs
Rationale for the Use of NSAIDs in OA
In case of inflammation, NSAIDs can switch off peripheral sensitization by inhibiting a relevant amount of PGs. Thus, their use as first-line therapy aimed at treating inflammatory nociceptive pain is virtually always appropriate; however, the feasibility of such strategy depends on the condition of the patient.
Conversely, the use of paracetamol (very common in OA although it is not an NSAID) is inappropriate in inflammatory pain, since it is a weak inhibitor of COX-1 and a very weak inhibitor of COX-2 and, as such, it does not interfere with peripheral sensitization. In addition, in tissues with inflammation, the free radicals inactivate paracetamol, abolishing any action on COX-2 [50]. In line with these observations, its analgesic effect cannot depend on COX inhibition. Paracetamol is actually a central analgesic with multiple effects, the main one being the stimulation of the endogenous cannabinoid system [51]. Hence, paracetamol has a lower efficacy than NSAIDs in reducing inflammatory pain [21] and its central analgesic efficacy is also lower than that of opioids.
Are All NSAIDs Equal?
All NSAIDs have an inhibitory activity on COX-1 and COX-2 but there are several differences among NSAIDs (for details on the mechanisms of action of the most common NSAIDs refer to [52,53,54]) that impact on their efficacy and safety [54]. These include:
-
Chemical similarity
-
COX isoform selectivity and potency [55]. NSAIDs comprise non-selective drugs, such as ibuprofen and naproxen, and selective COX-2 inhibitors (i.e. coxibs), such as etoricoxib and celecoxib. Potency is not a synonym of selectivity and cannot be used to predict dosages: a drug is potent if it inhibits 50% of available COX-1 and COX-2 at low dose. For example, etoricoxib is a selective inhibitor of COX-2 but it is less potent than diclofenac which, together with ketorolac (which has no indication in OA treatment), is the most potent inhibitor of COX-2 [55].
Importantly, the kinetics of COX-1 and COX-2 inhibition are different (non-linear and linear, respectively [56]).
In clinical practice, to achieve a significant anti-thrombotic effect through the blockade of thromboxane A synthesis, 95–97% of platelet COX-1 must be inhibited. If 90% of the enzyme is blocked, no anti-thrombotic effect occurs. The only NSAID able to inhibit 95% of COX-1 is acetylsalicylic acid (ASA), which irreversibly blocks the enzyme and, if administered at the dose of 100 mg per day every day, maintains this level of inhibition. No other NSAID is able to produce this effect, with the exception of naproxen but at non-conventional doses and regimens. Thus, it is not completely true that NSAIDs alone interfere with platelet function. Certainly, there are COX-1-independent antiplatelet effects that may play a role. NSAIDs induce mostly GI bleeding as they block COX-1 in surface epithelial cells. Given in concomitance with other drugs, such as the selective serotonin reuptake inhibitors, the antiplatelet effects of NSAIDs are enhanced.
-
Plasma half-life. This feature impacts on the occurrence of AEs. Indeed, the NSAIDs inhibiting gastric COX-1 for a longer time are more harmful for the stomach. For example, piroxicam and diclofenac have a half-life of about 60 and 1 h, respectively, but the latter is a more potent inhibitor of COX-1 and is associated to a relative risk of gastric bleeding of 3.61 compared to 8.00 for piroxicam [57].
-
Interference with ASA. Not all NSAIDs interfere with the cardioprotective effects of ASA. Diclofenac, ketorolac and etoricoxib do not, but they are the most potent inhibitors of COX-2 and, thus, of the endothelial prostacyclin (PGI2) production. Upon ASA treatment, the levels of thromboxane A drop and only the endogenous PGI2 remain, leading to a “thrombotic equilibrium.” If COX-2 is blocked, the equilibrium is impaired again. Ibuprofen, but not etoricoxib or diclofenac, seems to interfere with the capability of ASA to irreversibly acetylate platelet COX-1. This might reduce the protective effect of ASA against the risk of atherothrombotic events. Notably, combining ASA (required to prevent CV events) with a coxib may enhance the protective effect of COX-2 inhibition toward the gastric mucosal and prolong the time to recover from gastric mucosal injury [58].
According to the pharmacologist, patients on ASA must not take any NSAID. In particular cases, such as of a gout flare or of a renal colic, they may take such therapy for 1–2 days.
-
Penetration into the synovial liquid. Not all NSAIDs adequately penetrate into the synovial liquid (e.g. ibuprofen does not while diclofenac does), so even in the case of a short half-life, the higher the absorption at the synovial site, the longer the pharmacological effect [59].
-
Passage through the blood–brain barrier. This aspect related to the central action of NSAIDs may be of interest when selecting the most appropriate drug. Some compounds, such as diclofenac, pass through the barrier and reach the spinal cord, where the PGs produced by neurons and astroglia play a role in central sensitization. Therefore, at this site, inhibition of COX-1 and COX-2 adds to the peripheral effect (possible synergism) so that the analgesic activity resulting from the anti-inflammatory action adds to a central analgesic effect occurring when high drug doses reach the central nervous system.
Factors Influencing the Individual Response to NSAIDs
Several players affect the inter-patient variability observed in the response to NSAID therapy:
-
(1)
Genetic variations in the enzymes that metabolize NSAIDs (cytochrome P450 2C9 [CYP2C9] in many cases) and COXs.
-
(2)
The microbiota, for its capability to inactivate drugs. However, data in this regard are scarce.
-
(3)
The possibility of phenotyping OA (e.g. coxarthrosis vs. gonarthrosis), which is rather concrete [35] and may help to decide if and how to use an NSAID therapy—the choice should rely on the evidence from head-to-head comparisons or network meta-analyses [19,20,21].
-
(4)
Gender, which is responsible for relevant differences in the incidence, prevalence and prognosis of several immunoinflammatory diseases. Pre-clinical studies have demonstrated that the molecular mechanisms of inflammation and pain may differ between men and women. All of these differences provide a plausible background to understand why women use more NSAIDs than men. However, the pharmacological mechanisms underlying the gender-driven NSAID responses remain elusive [60].
By When Should We Expect the Response to NSAID treatment?
Usually, the maximum peak plasma concentration is reached within 2–3 h of administration, but the efficacy also depends on other factors (e.g. plasma protein binding and tissue distribution with particular regard to the inflammatory osteoarticular tissue). The rapid effect is pain reduction, which is achieved also through the central activity of NSAIDs; the delayed effect is the reduction of inflammation and thus the rise of the threshold; the variable effect is the improvement in disability. The analgesic effect occurs within about 1 week and the full anti-inflammatory effect is often achieved in 3 weeks (which questions the 3-day test validity, as the specificity is very low) [61]. A recent study has shown that the NSAID-induced improvement in pain and function peaks at 2 weeks and starts to decline by 8 weeks, while minor CV and GI AEs occur as early as 4 weeks after the initiation of NSAID treatment [62].
What is the Adequate Duration of NSAID Therapy?
In general, NSAIDs should be used for the shortest duration possible and at the lowest dose that guarantees both inflammation reduction and physical function improvement, as established in efficacy studies [21, 61]. Therapy duration must be tailored to the patient profile [61]. Usually, the treatment duration is at least 7–10 days, taking into account the time required to achieve both the analgesic and full anti-inflammatory effects [61]. If at the end of the 3-week period no result has occurred, a switch to another agent should be attempted [61].
Monotherapy or Combination Therapy?
It is possible to combine NSAIDs with central analgesics, such as paracetamol and opioids. By targeting different mechanisms, such combinations permit the dose to be limited, thus reducing the risk of AEs. In contrast, the combination of NSAIDs with steroids should be avoided: in fact, although they are very effective against inflammation and cause only marginal gastric erosion in subjects without risk factors, these drugs delay the healing of possible microulcers, highly enhancing the NSAID-induced gastric erosion. In this context, the number of administrations plays a central role.
General Considerations on the Different Formulations
Oral intake through the direct contact between drugs and the GI tract mucosa increases the likelihood of topical damage until absorption. Topical formulations are usually preferred over systemic treatments for safety reasons, such as in patients aged > 75 years [4, 5]. In patients with comorbidities, to favor compliance, formulations relying on one or few administrations (e.g. modified release) should be considered.
Practical Indications
-
(1)
Avoid the use of paracetamol in case of inflammatory pain.
-
(2)
NSAIDs should be used for the shortest duration and at the lowest dose that guarantees the effect on inflammation and improvement in physical function.
-
(3)
Define therapy duration based on the patient profile and avoid the on-demand use of NSAIDs: in the case of inflammatory pain, therapy must be administered for at least 10 days to achieve analgesia and for 3 weeks to achieve the full anti-inflammatory effect.
-
(4)
It is possible to combine NSAIDs with central analgesics such as paracetamol and opioids.
-
(5)
Avoid the combination of NSAIDs with steroids.
-
(6)
Consider formulations relying on one or few administrations to improve adherence.
Making Sense of NSAID Therapy: The Specialists’ Point of View
In clinical practice, in-depth knowledge of each NSAID’s efficacy and safety profile, together with the patient characteristics, is critical to define the benefit/risk balance of each compound for a specific individual and drive the therapeutic choice.
Table 2 summarizes the considerations made by the GP, the pharmacologist and the pain therapist of the multidisciplinary panel.
The Safety Profile of NSAIDs
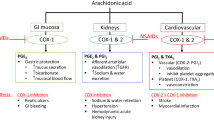
The main AEs that may occur upon NSAID therapy are illustrated in Fig. 2.

The main adverse events observed in osteoarthritis patients upon NSAID therapy (see text for discussion). AKI Acute kidney injury, AMI acute myocardial infarction, CKD chronic kidney disease, COX cyclooxygenase, coxibs COX-2 inhibitors, CV cardiovascular, GFR glomerular filtration rate, GI gastrointestinal, tNSAIDs traditional non-steroidal anti-inflammatory drugs, TXA2 thromboxane A2
NSAIDs and CV Risk
NSAID-Related CV AEs
The CV safety of NSAIDs is a very controversial matter. Following the observation that NSAIDs could increase the risk of CV events at therapeutic doses or higher, in 2005 the U.S. Food and Drug Administration added a black box warning to their use [63], while the European Medicines Agency decided to contraindicate coxibs (but not tNSAIDs [64]) in patients with coronary heart disease or stroke and to advise those at risk for coronary heart disease to use these agents with caution [65].
The possible mechanisms proposed to explain CV complications include (1) the unbalance between the vasodilator effect of PGI2 and PGE2 in favor of vasoconstriction by thromboxane A2 in the endothelium, which results in a prothrombotic effect; and (2) sodium and water retention promoted by COX inhibition, which worsens heart failure, hypertension and ventricular remodeling.
The Coxib and Traditional NSAID Trialists’ (CNT) Collaboration meta-analysis is the largest meta-analysis on NSAID safety, based on 639 RCTs in which tNSAIDs/coxibs were used for long periods [26]. It investigated the vascular effects of coxibs (celecoxib, etoricoxib and lumiracoxib) and high-dose tNSAIDs (diclofenac, ibuprofen and naproxen) in older patients with rheumatic diseases [26]. Coxibs, diclofenac and ibuprofen displayed a similar relative risk for CV events (range 1.37–2.49), whereas naproxen did not seem to increase it (range 0.39–1.87). Coxibs, diclofenac and ibuprofen also displayed a comparable annual absolute risk for major vascular events, which varied according to the baseline predicted risk: in low-risk subjects, the predicted absolute risk of major vascular events was low regardless of the NSAID administered (2 per 1000 in all cases for coxibs, diclofenac and ibuprofen; 0 per 1000 for naproxen); in high-risk patients, the risk increased and was similar for high-dose diclofenac and coxibs (8 per 1000 and 7 per 1000, respectively) and possibly ibuprofen (9 pe 1000), while it seemed to be lower for high-dose naproxen (− 1 per 1000). [26]. A subsequent network meta-analysis found no difference in the risk of major CV events with diclofenac, ibuprofen, naproxen, celecoxib and etoricoxib for the treatment of pain in patients with OA or RA [20].
The PRECISION trial, conducted in subjects with OA or RA at increased CV risk and treated with celecoxib, naproxen and ibuprofen, showed a similar number of CV-related deaths, nonfatal myocardial infarction (MI) or nonfatal stroke among the three groups of NSAIDs, but ibuprofen and naproxen had been used at doses and for periods not in line with guidelines [66].
The absolute risk for CV effects increases to a greater extent in patients with or at risk for active atherosclerotic processes (e.g. with recent bypass surgery, unstable angina or ischemic cerebrovascular events) receiving a COX inhibitor. The excess number of events depends on the underlying risk of the patient, the relative risk of the drug and the duration of the follow-up [58].
A recent meta-analysis of individual patient data in real-world settings [30] has shown that all traditional NSAIDs are associated with an increased risk of AMI, similar to that reported with celecoxib therapy. Using a high daily dose (celecoxib > 200 mg, diclofenac > 100 mg, ibuprofen > 1200 mg, naproxen > 750 mg) for 8–30 days was associated with the greatest risk, which did not increase further beyond the first 30 days. Based on these findings, prescribers should consider weighing the risks and benefits of NSAIDs before selecting the treatment, particularly for higher doses.
In patients with a prior MI, the excess risk of mortality has been estimated to be approximately six deaths per 100 person-years of treatment with a COX-2 inhibitor compared with no NSAID treatment [67]. A Danish large-scale study based on national administrative registers and conducted in healthy individuals demonstrated an increased risk for death/MI in diclofenac and celecoxib users (hazard ratio [95% confidence interval] vs. non-users: 1.63 [1.52–1.76] and 2.01 [1.78–2.27], respectively), which increased in a dose-dependent fashion [68]. Moreover, in low-risk patients, an increased risk of pooled CV events was found with lower doses of diclofenac versus paracetamol, ibuprofen and naproxen (which, however, varied based on the event considered) over 1 month; surprisingly, the relative risk decreased in patients at high CV risk [69], but the explanation remains unclear [24]. Finally, the SOS project, which included millions of Europeans, showed a similar modest increase in CV risk with diclofenac and other NSAIDs, compared to non-use [70]. Nonetheless, bias linked to the design of the available studies do not allow definitive conclusions to be drawn.
A recent study showed that patients on anticoagulant therapy with both vitamin K antagonists and dabigatran should avoid NSAIDs due to a greater risk of hemorrhage, especially GI bleeding, and more frequent complications (such as strokes and embolisms) [71]. The use of NSAIDs has always been discouraged in patients receiving antivitamin K therapy, but this is valid advice also for those who receive dabigatran and likely all direct anticoagulants. No specific data are currently available for rivaroxaban, edoxaban and apixaban.
Finally, it must be pointed out that the concomitant administration of certain NSAIDs weakens the protective CV effects of ASA [72,73,74]. Co-administration of ibuprofen in patients with documented CVD on low-dose ASA therapy significantly increased the risk of all-cause and CV mortality (hazard ratio [HR] 1.93, 95% confidence interval [CI] 1.30–2.87; HR 1.73, 95% CI 1.05–2.84, respectively) compared to ASA alone [72]. No difference was observed when diclofenac or other NSAIDs were used with ASA versus ASA alone [72].
In conclusion, the main determinant of the risk of AEs is the patient profile. A slight increase in CV risk occurs mainly in case of high doses and long-term use, which, however, are not recommended by current guidelines. The only indication to limit the CV risk is to adhere to the recommended dosages and duration and, possibly, undergo cycles of therapy with periodical interruptions.
Practical Indications
-
(1)
In patients with a prior MI, extra caution is needed in the use of NSAIDs/coxibs.
-
(2)
Use only the recommended doses and for the shortest period necessary to control or relieve symptoms.
-
(3)
Monitor renal function and blood pressure in NSAID/coxib users, especially if they present preexisting conditions such as hypertension, renal disease and heart failure [75].
NSAIDs and GI Risk
NSAID-Related GI AEs
Non-steroidal anti-inflammatory drugs-induced GI AEs are various and sometimes severe even though their prevalence is not high [16]. The most frequent GI AEs assocated with NSAID therapy are gastric injuries, which range from subjective manifestations, such as dyspepsia, to ulcers with complications. In elderly patients with arthritis, the incidence of GI intolerability AEs was reported to be significantly lower with celecoxib (16.7%) than with naproxen (29.4%; P < 0.0001), ibuprofen (26.5%; P = 0.0016) and diclofenac (21.0%; P < 0.0001). The discontinuation rate due to these AEs was similar for celecoxib (4.0%) and diclofenac (4.2%; P = 0.75) and significantly lower than for naproxen (8.1%; P < 0.0001) and ibuprofen (7.3%; P < 0.05) [76].
Up to 70% of NSAID users experience minimal mucosal lesions as early as within a few hours of intake [77]; these may indicate gastric mucosa frailty and the tendency to become real ulcers [78]. NSAID-induced ulcer, mainly gastric, is becoming more and more frequent due to the increased use of these drugs, especially in the elderly. As NSAID use and Helicobacter pylori are two independent determinants of ulcer development, they may have additive effects on the ulcer risk in the same subject. Thus, the most recent international guidelines recommend that patients be tested for the presence of the infection and, if present, to eradicate it in those who have to start a prolonged therapy with NSAIDs [79]. The most frequent ulcer complication is bleeding, with a rate ratio (RR) of 1–2% per year. The underlying disease seems to be important: for example, the rate of bleeding is 1.3–2% per year in RA patients and 0.7–10% per year in those with OA [80].
NSAID users may also experience intestinal disorders, including small bowel injuries [77], which may be caused by the mucosal inflammatory pathway triggered by microbiota changes [81].
Liver toxicity events are much less frequent than gastric injuries. Paracetamol used at high doses, at least 4 g per day, may damage the liver [82]. Other studies found that the RR of liver damage defined by hypertransaminasemia was higher for nimesulide (2.2) and sulindac (5) than for diclofenac (1.5) [82].
Most of the patients who develop a serious GI AE while on NSAID therapy are asymptomatic prior to the event [83], particularly the elderly. Among the risk factors for the onset of NSAID-associated ulcer complications (Table 1), advanced age is a primary risk factor for GI events [84]: indeed, NSAID users aged 75–89 years have a twofold higher risk of bleeding (RR 4.1) compared to users aged 60–74 years (RR 2.0) [85]. It is frequent to observe, in the emergency department, elderly patients who use NSAIDs chronically and present severe anemia with hemoglobin levels of 4–5 gr/dl without having ever experienced any dyspeptic symptom. Therefore, physicians must check their patients periodically for the presence of anemia (fecal occult blood test, hematocrit) and symptoms associated with this condition (headache, asthenia, dyspnea, etc.). Conversely, many patients with troublesome symptoms (e.g. epigastric pain and dyspepsia) may have a normal endoscopy at the upper GI tract [86]. As for steroids increasing the risk of complications, it must be pointed out that, when used alone, they do not represent an actual risk for ulcerogenesis [87].
The type of non-selective NSAID impacts on the frequency of GI damage. The results from two epidemiological studies have led to establish a scale of risk for different tNSAIDs (i.e. ibuprofen, diclofenac, naproxen, ketoprofen, indomethacin, piroxicam and azapropazone); azapropazone and piroxicam were associated to the highest risk of gastroduodenal bleeding (odds ratio [OR] 23.4–31.5 and 13.7–18, respectively) and diclofenac and ibuprofen with the lowest (OR 3.9–4.2 and 2.0–2.9, respectively) [88, 89]. Table 3 presents the results from two recent meta-analyses of RCTs that report the rate of risk for bleeding associated with tNSAIDs and coxibs versus placebo [26] and for major GI events associated with tNSAIDs and coxibs versus diclofenac [20]. In particular, the CNT meta-analysis reported that the annual absolute risk of upper GI complications for coxibs, diclofenac, ibuprofen and naproxen depended on the baseline risk [26]. Both in patients at low and high risk, diclofenac and coxibs yielded a similar risk (in low-risk patients: 2 per 1000; in high-risk patients: 6 per 1000, respectively) that was lower than that of ibuprofen and naproxen (in low-risk patients: 4 per 1000; in high-risk patients: 15 and 16 per 1000, respectively), in line with the results from previous epidemiological studies [88, 89].
Among the NSAID features that may impact on gastrolesivity, plasma half-life plays a major role. A study conducted in elderly subjects [90] evaluated the presence of gastroduodenal bleeding through the measurement of fecal blood loss and found that it was higher with drugs with a longer plasma half-life, such as naproxen (2.76 ml fecal blood loss) and piroxicam (1.16 ml), compared to diclofenac (0.53 ml), a NSAID with a shorter half-life, and placebo (0.28 ml). Other factors responsible for a different gastrolesive effect among NSAIDs are the level of pK (higher levels increase the toxic effect) and the dosage.
When is it Adequate to Use Proton Pump Inhibitors with NSAIDs in the Prevention of NSAID-Induced Damage?
Non-steroidal anti-inflammatory drug-induced GI damage can be significantly reduced by increasing the gastric pH through the administration of proton pump inhibitors (PPIs), which are the most potent acid inhibitors available. Unlike H2-antagonists that prevent only the onset of duodenal ulcers, PPIs can protect both the stomach, the main site of NSAID-induced damage, and the duodenum [91]. The protective action of PPIs depends on the fact that the weakening of the mechanisms of mucosal defense induced by NSAIDs implies that even a reduced amount of acid, such as in the case of the chronic gastritis that is always associated to gastric ulcers, may be dangerous [92]. A number of important risk factors must be considered due to the need to administer an appropriate prophylactic therapy with PPIs [93] (Table 4).
PPIs have to be administered throughout the period of NSAID use; even half the standard dose seems to be sufficient to achieve the benefit [94].
Practical Indications
-
(1)
There are no dietary or behavioral suggestions to prevent or reduce NSAID-induced GI lesions.
-
(2)
When selecting a NSAID, in high-risk patients or in case of prolonged therapy duration, compounds with the lowest risk of GI events should be preferred.
-
(3)
The optimal treatment duration depends on the disease and corresponds to the period of acute symptoms or of functional joint impairment.
-
(4)
Use PPIs in the presence of particular risk factors.
NSAIDs and Renal AEs
NSAID-Induced Renal and Reno-Vascular Events and Risk Factors
At the center stage of the untoward effects of NSAIDs is the inhibition of endogenous or inflammatory renal PGs, a subfamily of eicosanoids. Endogenous eicosanoids fine-tune renal microcirculation and water and electrolyte transport across renal tubules. PGs, such as PGE1 and 2 or PGF2α, control sodium reabsorption and the concentration/dilution mechanism. Likewise, endothelial PGI2 and platelet thromboxane A2 balance each other to control vascular tone in glomeruli and renal arterioles, including the vasa recta. As this counterbalance mechanism is marginal in the normal kidney, NSAID inhibitors of eicosanoid biosynthesis have very modest effects in the healthy kidney and/or younger individuals and are usually well tolerated in persons with normal renal function.
Elderly individuals or patients with chronic kidney disease (CKD) are likely to experience at least some mild AEs, ranging from local edema (e.g. hands, lower limbs, water retention with rapid weight gain) to worsening of glomerular filtration rate (GFR) and/or hyperkalemia. This is usually more frequent in patients with certain comorbidities: in particular, in hypertensive subjects NSAID therapy may lead to intensification of anti-hypertensive regimen [95]. The effects are usually reversible but tend to synergize with other agents affecting renal function, such as anti-hypertensive drugs. In selected circumstances, acute kidney injury (AKI) may occur with severe oligoanuria. A meta-analysis of observational studies [28] found a statistically significant elevated AKI risk in patients treated with indomethacin, piroxicam, ibuprofen, naproxen and sulindac versus non-users, with pooled RRs ranging from 1.58 to 2.11. In all other cases (i.e. diclofenac, meloxicam, and celecoxib), the increase in AKI risk was not significant. Another meta-analysis of observational studies [29] reported that, in the general population, the pooled OR of AKI for ongoing NSAID exposure was 1.73 (95% CI 1.44–2.07) and was higher in older people (OR 2.51, 95% CI 1.52–2.68); in people with CKD, it was 1.63 (95% CI 1.22–2.19) and ranged from 1.12 to 5.25. Notably, the risk was higher for NSAIDs with no COX-2 selectivity (OR 1.84, 95% CI 1.54–2.19) and decreased with increasing COX-2 selectivity (≥ 5-fold, OR 1.41, 95% CI 1.07–1.87).
Various NSAIDs have been implicated in glomerular disorders leading to proteinuria and/or the nephrotic syndrome, possibly due to some podocyte-specific type of injury [96]. In other instances, interstitial nephritis can occur bacause of NSAID immuno-allergic effects that are most likely unrelated to COX inhibition [97]. Under most circumstances, proteinuria or non-oliguric AKI rapidly disappear upon therapy discontinuation.
Another issue that may impact on the renal adverse effects of NSAIDs is the lack of apparent recognition of renal dysfunction by prescribing physicians. Notably, sudden changes of GFR may go unnoticed if serum creatinine, blood urea nitrogen or serum K+ are not measured during NSAIDs therapy. Thus, the real prevalence of renal untoward effects of NSAIDs may be largely underestimated. A recent systematic review [98] noted a cross-sectional point prevalence of NSAID use of between 8 and 21% in 49,209 patients with CKD, demonstrating that despite guidelines recommending against their use, a substantial proportion of CKD patients continue to receive NSAIDs.
NSAIDs and Arterial Pressure
The pro-hypertensive effects of NSAIDs are believed to stem from three major mechanisms [99]:
-
(1)
Na+ and Cl− retention and increased antidiuretic hormone-mediated water reabsorption at the distal collecting duct
-
(2)
Blockade of the vasodilator effects of PGE2 and PGI2 on the kidney microcirculation
-
(3)
Unbalanced activity of the renin/angiotensin/aldosterone axis, normally regulated by local vascular and tubular eicosanoid biosynthesis.
No effect on blood pressure (BP) has been observed in ASA [100,101,102] and coxib users [103, 104]. Among non-selective NSAIDs, ibuprofen and indomethacin—but not diclofenac—were shown to increase the risk of hypertension in arthritis patients [103]. In a meta-analysis of 19 RCTs including 45,000 patients with arthritis treated for > 4 weeks with COX-2 inhibitors, non-selective NSAIDs or placebo, coxibs caused a weighted mean difference point estimate increase in systolic and diastolic BP compared with placebo and non-selective NSAIDs, and were associated with a non-significantly higher RR of causing hypertension compared with placebo and non-selective NSAIDs [105]. Another meta-analysis of 49 RCTs with 130,000 patients—mostly with arthritis—found that coxibs caused greater hypertension than either non-selective NSAIDs or placebo after at least 4 weeks of treatment. However, the effect was heterogeneous, with a marked BP increase in etoricoxib users and a slight effect in users of celecoxib, valdecoxib and lumiracoxib [106]. The review did not report absolute risk changes or provide numbers needed to treat or harm.
Monitoring the Renal and Nephrovascular Effects of NSAID Therapy
We suggest that patients with cardio-renal risk factors receive a complete nephrological assessment, including calculation of the estimated GFR, age-adjusted renal function, urinalysis, electrolyte and acid–base profiling (acidosis/hyperkalemia), microalbuminuria, proteinuria (if any), concurrent anti-hypertensive therapy (anti-ANG II, anti-aldosterone treatment). Measurement of serum chloride is particularly useful [107]: at < 100 mmol/l, Cl− predicts a setting of metabolic alkalosis (diuretics, hyperaldosteronism); at > 105 mmol/l, it suggests a hyperchloremic metabolic acidosis (renal failure with normal anion gap). Failure of Cl− to increase in the presence of metabolic acidosis with low HCO3− levels implicates an elevated anion gap acidosis, resulting from an unmeasured anion (ketones, lactate, alcohol metabolites, salicylate or other intoxications, sepsis). Both alkalosis and acidosis usually drive significant changes of serum K+, potentially relevant to treatment with NSAIDs, which tend to increase K+ by interfering with prostacyclin-mediated K+ secretion in the distal tubule. If used in conjunction with an angiotensin-converting enzyme inhibitor (ACEi) or ANG II receptor antagonist in a diabetic patient, the risk of hyperkalemia is greatly increased.
Practical Indications
-
(1)
NSAIDs have very modest effects in the healthy kidney and/or younger individuals and are usually well tolerated in subjects with normal renal function.
-
(2)
Any patient with chronic renal disease should be warned against possible sideeffects of NSAIDs, both in terms of renal function and/or blood pressure control. Should a course of NSAIDs be deemed necessary, the following measures should be taken:
-
i.
Obtain a baseline measurement of renal function (i.e. estimated [e]GFR by Cockroft-Gault, CKD-EPI or MDRD equations) and serum K+.
-
ii.
Withdraw any concurrent anti-hypertensive therapy with ACEi or ANG II receptor blockers (known to decrease eGFR in elderly patients with widespread atherosclerotic vascular lesions).
-
iii.
Keep daily doses of the chosen NSAID to the lowest effective level, for no longer than 1 week to 10 days.
-
iv.
Avoid dehydration or concurrent diuretic therapy, unless mandatory.
-
v.
Monitor eGFR and serum K on weekly basis. Virtually all non-selective COX-1 and -2 inhibitors have the potential to induce or aggravate AKI; selective COX-2 inhibitors (rofecoxib, celecoxib) can also affect renal function, whereas NSAIDs with higher COX-2 selectivity (diclofenac, meloxicam) also have renal effects, however not statistically significant.
-
vi.
Closely monitor individuals with increased risk of AKI due to underlying comorbidities (arterial hypertension, diabetes, heart failure, stroke).
-
vii.
Withdrawal of NSAIDs is almost always followed by recovery of renal function, although not all cases of AKI are entirely reversible.
Tables 5 and 6 summarize the indications on the selection of the most adequate NSAID according to the CV, GI and renal risk (low vs. high).
Patient Management During NSAID Therapy
The management of pain relies on a sequential pharmacologic approach. Following the core principles of patient-centered care [108], the treatment plan must be periodically revised based on the assessment of response (in terms of both efficacy and tolerability) and adherence, taking into consideration the possibility to switch to other options in case of inefficacy or intolerance.
When is it Appropriate to Re-evaluate the Patient Receiving NSAID Therapy and What Aspects Should Be Re-evaluated?
In OA, an effective treatment improves both pain and physical function: as already stated, the same NSAID at different dosages exerts different effects and the minimum effective dose is defined in efficacy studies [21]. Therapy duration must be tailored to the patient profile [61] and the revision of the treatment plan must be periodic.
When revising the treatment plan, efficacy, tolerability, and adherence must be assessed.
A complete assessment should include the following: evaluation of pain through any of the available scales (VAS, NRS and WOMAC), impact on the quality of life, pain tolerability, functional recovery and therapy duration. Factors that may help increasing compliance are dosage, regimens and formulations.
Practical Indications
Treatment response and adherence should be periodically re-evaluated, using scales to assess pain and function.
Conclusions
This narrative review provides practical indications for GPs and specialists managing patients with OA who suffer from chronic inflammatory pain. Selection of the appropriate therapy is hampered by the patients often being elderly and burdened with comorbidities and polypharmacy. Thus, both patient and drug characteristics (i.e. pharmacology, interactions and benefit/risk balance) must be carefully evaluated, keeping in mind that the same NSAID at different doses has different effects on pain and physical function. To summarize:
-
During the first visit, the GP must investigate the origin, duration and component of pain, and collect information on possible risk factors for CV, GI and renal AEs, including comorbidities and concomitant therapies.
-
If a non-pharmacological intervention is planned, the physiatrist comes into play, providing a person-centered, holistic approach that accounts for the individual conditions globally.
-
If the patient has to receive a pharmacological intervention, the selection of the most appropriate NSAID, of possible drugs to be used in combination or to be avoided, of the formulation and of therapy duration must rely on both the patient profile and the drugs’ pharmacological properties (i.e., COX isoform selectivity and potency and plasma half-life):
-
in OA patients with inflammatory pain, the use of paracetamol must be avoided as it is ineffective.
-
the dose to be administered is the minimum effective dose as determined by available studies.
-
in low-risk patients, therapy must be administered for at least 10 days to achieve analgesia and 3 weeks to achieve the full anti-inflammatory effect.
-
-
NSAID safety: The main determinants of the risk of AEs are the individual baseline risk (in case of high risk, specific parameters should be monitored during therapy) and the drug’s safety profile. Possible drug–drug interactions must be considered.
-
to limit the CV risk, the only indication is to adhere to the recommended dosages and duration and, possibly, undergo cycles of therapy with periodical interruptions. The use of ASA limits the choice of NSAIDs.
-
NSAID-induced GI damage can be significantly reduced through the administration of PPIs in the presence of particular risk factors.
-
NSAIDs have very modest effects in the healthy kidney and/or younger individuals and are usually well tolerated in subjects with normal renal function. Adjust treatment to the individual needs, keeping it as short as possible, while monitoring key renal function parameters in elderly patients or subjects with known renal disease, reduced renal function, or high-risk conditions, including diabetic nephropathy or cardio-renal syndromes.
-
-
Periodically re-evaluate treatment response and adherence, using scales to assess pain and function.
References
Kloppenburg M, Berenbaum F. Osteoarthritis year in review 2019: epidemiology and therapy. Osteoarthr Cartil. 2020;28:242–8.
Rapporto Osservasalute. Osservatorio sulla Salute. 2019. https://www.osservatoriosullasalute.it/osservasalute/rapporto-osservasalute-2019. Accessed 14 Sept 2020.
XXX. Report_XII SIMG 2019.pdf. 2019. https://report.healthsearch.it/Report_XII.pdf. Accessed 14 Sept 2020.
Ariani A, Manara M, Fioravanti A, et al. The Italian Society for Rheumatology clinical practice guidelines for the diagnosis and management of knee, hip and hand osteoarthritis. Reumatismo. 2019;71:5–21.
Kloppenburg M, Kroon FP, Blanco FJ, et al. 2018 update of the EULAR recommendations for the management of hand osteoarthritis. Ann Rheum Dis. 2019;78:16–24.
Bruyère O, Honvo G, Veronese N, et al. An updated algorithm recommendation for the management of knee osteoarthritis from the European Society for Clinical and Economic Aspects of Osteoporosis, Osteoarthritis and Musculoskeletal Diseases (ESCEO). Semin Arthritis Rheum. 2019;49:337–50.
Bannuru RR, Osani MC, Vaysbrot EE, et al. OARSI guidelines for the non-surgical management of knee, hip, and polyarticular osteoarthritis. Osteoarthr Cartil. 2019;27:1578–89.
Kolasinski SL, Neogi T, Hochberg MC, et al. 2019 American College of Rheumatology/Arthritis Foundation Guideline for the Management of Osteoarthritis of the Hand, Hip, and Knee. Arthritis Care Res (Hoboken). 2020;72:149–62.
Derry S, Moore RA, Gaskell H, McIntyre M, Wiffen PJ. Topical NSAIDs for acute musculoskeletal pain in adults. Cochrane Database Syst Rev. 2015(6):CD007402.
Derry S, Wiffen P, Moore A. Topical nonsteroidal anti-inflammatory drugs for acute musculoskeletal pain. JAMA. 2016;315:813–4.
D’Arcy Y, McCarberg B. Managing patient pain: a focus on NSAID OTC formulations for relief of musculoskeletal and other common sources of pain. J Fam Pract. 2018;67:S67–72.
Mukherjee D, Nissen SE, Topol EJ. Risk of cardiovascular events associated with selective COX-2 inhibitors. JAMA. 2001;286:954–9.
Wilcox CM, Cryer B, Triadafilopoulos G. Patterns of use and public perception of over-the-counter pain relievers: focus on nonsteroidal antiinflammatory drugs. J Rheumatol. 2005;32:2218–24.
Ussai S, Miceli L, Pisa FE,et al. Impact of potential inappropriate NSAIDs use in chronic pain. Drug Des Devel Ther. 2015;9:2073–7.
Doomra R, Goyal A. NSAIDs and self-medication: a serious concern. J Fam Med Prim Care. 2020;9:2183–5.
Lombardi N, Crescioli G, Bettiol A, et al. Italian emergency department visits and hospitalizations for outpatients’ adverse drug events: 12-year active pharmacovigilance surveillance (The MEREAFaPS Study). Front Pharmacol. 2020;11:412.
Pavelka K. A comparison of the therapeutic efficacy of diclofenac in osteoarthritis: a systematic review of randomised controlled trials. Curr Med Res Opin. 2012;28:163–78.
Datto C, Hellmund R, Siddiqui MK. Efficacy and tolerability of naproxen/esomeprazole magnesium tablets compared with non-specific NSAIDs and COX-2 inhibitors: a systematic review and network analyses. Open Access Rheumatol. 2013;5:1–19.
Bannuru RR, Schmid CH, Kent DM, Vaysbrot EE, Wong JB, McAlindon TE. Comparative effectiveness of pharmacologic interventions for knee osteoarthritis: a systematic review and network meta-analysis. Ann Intern Med. 2015;162:46–54.
van Walsem A, Pandhi S, Nixon RM, Guyot P, Karabis A, Moore RA. Relative benefit-risk comparing diclofenac to other traditional non-steroidal anti-inflammatory drugs and cyclooxygenase-2 inhibitors in patients with osteoarthritis or rheumatoid arthritis: a network meta-analysis. Arthritis Res Ther. 2015;17:66.
da Costa BR, Reichenbach S, Keller N, et al. Effectiveness of non-steroidal anti-inflammatory drugs for the treatment of pain in knee and hip osteoarthritis: a network meta-analysis. Lancet. 2017;390:e21–33.
Kroon FPB, Carmona L, Schoones JW, Kloppenburg M. Efficacy and safety of non-pharmacological, pharmacological and surgical treatment for hand osteoarthritis: a systematic literature review informing the 2018 update of the EULAR recommendations for the management of hand osteoarthritis. RMD Open. 2018;4:
Jung S-Y, Jang EJ, Nam SW, et al. Comparative effectiveness of oral pharmacologic interventions for knee osteoarthritis: a network meta-analysis. Mod Rheumatol. 2018;28:1021–8.
Moore N. Coronary risks associated with diclofenac and other NSAIDs: an update. Drug Saf. 2020;43:301–18.
Castellsague J, Riera-Guardia N, Calingaert B, , et al. Individual NSAIDs and upper gastrointestinal complications: a systematic review and meta-analysis of observational studies (the SOS project). Drug Saf. 2012;35:1127–46.
Coxib and Traditional NSAID Trialists’ (CNT) Collaboration, Bhala N, Emberson J, et al. Vascular and upper gastrointestinal effects of non-steroidal anti-inflammatory drugs: meta-analyses of individual participant data from randomised trials. Lancet. 2013;382:769–79.
Varas-Lorenzo C, Riera-Guardia N, Calingaert B, et al. Myocardial infarction and individual nonsteroidal anti-inflammatory drugs meta-analysis of observational studies. Pharmacoepidemiol Drug Saf. 2013;22:559–70.
Ungprasert P, Cheungpasitporn W, Crowson CS, Matteson EL. Individual non-steroidal anti-inflammatory drugs and risk of acute kidney injury: a systematic review and meta-analysis of observational studies. Eur J Intern Med. 2015;26:285–91.
Zhang X, Donnan PT, Bell S, Guthrie B. Non-steroidal anti-inflammatory drug induced acute kidney injury in the community dwelling general population and people with chronic kidney disease: systematic review and meta-analysis. BMC Nephrol. 2017;18:256.
Bally M, Dendukuri N, Rich B, et al. Risk of acute myocardial infarction with NSAIDs in real world use: bayesian meta-analysis of individual patient data. BMJ. 2017;357:
Gunter BR, Butler KA, Wallace RL, Smith SM, Harirforoosh S. Non-steroidal anti-inflammatory drug-induced cardiovascular adverse events: a meta-analysis. J Clin Pharm Ther. 2017;42:27–38.
Martín Arias LH, Martín González A, Sanz Fadrique R, Vazquez ES. Cardiovascular risk of nonsteroidal anti-inflammatory drugs and classical and selective cyclooxygenase-2 inhibitors: a meta-analysis of observational studies. J Clin Pharmacol. 2019;59:55–73.
Curtis E, Fuggle N, Shaw S, et al. Safety of cyclooxygenase-2 inhibitors in osteoarthritis: outcomes of a systematic review and meta-analysis. Drugs Aging. 2019;36:25–44.
Treede R-D, Rief W, Barke A, et al. Chronic pain as a symptom or a disease: the IASP Classification of Chronic Pain for the International Classification of Diseases (ICD-11). Pain. 2019;160:19–27.
Van Spil WE, Kubassova O, Boesen M, Bay-Jensen A-C, Mobasheri A. Osteoarthritis phenotypes and novel therapeutic targets. Biochem Pharmacol. 2019;165:41–8.
Freynhagen R, Baron R, Gockel U, Tölle TR. painDETECT: a new screening questionnaire to identify neuropathic components in patients with back pain. Curr Med Res Opin. 2006;22:1911–20.
Bennett M. The LANSS Pain Scale: the Leeds assessment of neuropathic symptoms and signs. Pain. 2001;92:147–57.
Bouhassira D, Attal N, Alchaar H, et al. Comparison of pain syndromes associated with nervous or somatic lesions and development of a new neuropathic pain diagnostic questionnaire (DN4). Pain. 2005;114:29–36.
Edwards RR, Dworkin RH, Turk DC, et al. Patient phenotyping in clinical trials of chronic pain treatments: IMMPACT recommendations. Pain. 2016;157:1851–71.
Mathieson S, Maher CG, Terwee CB, Folly de Campos T, Lin C-WC. Neuropathic pain screening questionnaires have limited measurement properties. A systematic review. J Clin Epidemiol. 2015;68:957–66.
Patel KV, Amtmann D, Jensen MP, Smith SM, Veasley C, Turk DC. Clinical outcome assessment in clinical trials of chronic pain treatments. Pain Rep. 2021;6:
Hawker GA, Mian S, Kendzerska T, French M. Measures of adult pain: visual Analog Scale for Pain (VAS Pain), Numeric Rating Scale for Pain (NRS Pain), McGill Pain Questionnaire (MPQ), Short-Form McGill Pain Questionnaire (SF-MPQ), Chronic Pain Grade Scale (CPGS), Short Form-36 Bodily Pain Scale (SF-36 BPS), and Measure of Intermittent and Constant Osteoarthritis Pain (ICOAP). Arthritis Care Res (Hoboken). 2011;63(Suppl 11):S240–52.
Bellamy N, Buchanan WW, Goldsmith CH, Campbell J, Stitt LW. Validation study of WOMAC: a health status instrument for measuring clinically important patient relevant outcomes to antirheumatic drug therapy in patients with osteoarthritis of the hip or knee. J Rheumatol. 1988;15:1833–40.
Veenhof C, Bijlsma JWJ, van den Ende CHM, van Dijk GM, Pisters MF, Dekker J. Psychometric evaluation of osteoarthritis questionnaires: a systematic review of the literature. Arthritis Rheum. 2006;55:480–92.
Copsey B, Thompson JY, Vadher K, et al. Problems persist in reporting of methods and results for the WOMAC measure in hip and knee osteoarthritis trials. Qual Life Res. 2019;28:335–43.
Conroy RM, Pyörälä K, Fitzgerald AP, et al. Estimation of ten-year risk of fatal cardiovascular disease in Europe: the SCORE project. Eur Heart J. 2003;24:987–1003.
Agenzia Italiana del Farmaco (AIFA). Nota 13. https://aifa.gov.it/nota-13. Accessed 16 Sept 2020.
L’Agenzia Italiana del Farmaco (AIFA). Nota 01. https://aifa.gov.it/nota-01. Accessed 16 Sept 2020.
Zampolini M, Stucki G, Giustini A, Negrini S. The individual rehabilitation project: a model to strengthen clinical rehabilitation in health systems worldwide. Eur J Phys Rehab Med. 2020;56:1–4.
Bertolini A, Ferrari A, Ottani A, Guerzoni S, Tacchi R, Leone S. Paracetamol: new vistas of an old drug. CNS Drug Rev. 2006;12:250–75.
Ghanem CI, Pérez MJ, Manautou JE, Mottino AD. Acetaminophen from liver to brain: new insights into drug pharmacological action and toxicity. Pharmacol Res. 2016;109:119–31.
Cashman JN. The mechanisms of action of NSAIDs in analgesia. Drugs. 1996;52(Suppl 5):13–23.
Bertolini A, Ottani A, Sandrini M. Dual acting anti-inflammatory drugs: a reappraisal. Pharmacol Res. 2001;44:437–50.
Brunton LL, Hilal-Dandan R, Knollmann BC. Goodman & Gilman’s: The pharmacological basis of therapeutics, 13th edition. New York: McGraw-Hill Education; 2018.
Patrignani P, Patrono C. Cyclooxygenase inhibitors: from pharmacology to clinical read-outs. Biochim Biophys Acta. 2015;1851:422–32.
Brune K, Patrignani P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J Pain Res. 2015;8:105–18.
Massó González EL, Patrignani P, Tacconelli S, García Rodríguez LA. Variability among nonsteroidal antiinflammatory drugs in risk of upper gastrointestinal bleeding. Arthritis Rheum. 2010;62:1592–601.
Antman EM, Bennett JS, Daugherty A, et al. Use of nonsteroidal antiinflammatory drugs: an update for clinicians: a scientific statement from the American Heart Association. Circulation. 2007;115:1634–42.
Burton ME. Applied pharmacokinetics & pharmacodynamics: principles of therapeutic drug monitoring. Philadelphia: Lippincott Williams & Wilkins; 2006.
Demyanets S, Wojta J. Sex differences in effects and use of anti-inflammatory drugs. Handb Exp Pharmacol. 2012;443–72.
Agenzia Italiana del Farmaco. Nota 66. https://aifa.gov.it/nota-66. Accessed 15 Sept 2020.
Osani MC, Vaysbrot EE, Zhou M, McAlindon TE, Bannuru RR. Duration of symptom relief and early trajectory of adverse events for oral nonsteroidal antiinflammatory drugs in knee osteoarthritis: a systematic review and meta-analysis. Arthritis Care Res (Hoboken). 2020;72:641–51.
U.S. Food and Drug Administration. FDA releases black-box template for NSAIDs. 2005 https://www.fdanews.com/articles/73674-fda-releases-black-box-template-for-nsaids. Access 5 Oct 2020.
European Medicines Agency. European Medicines Agency review concludes positive benefit-risk balance for nonselective NSAIDs. 2006. https://www.ema.europa.eu/en/documents/press-release/european-medicines-agency-review-concludes-positive-benefit-risk-balance-non-selective-nsaids_en.pdf. Accessed 16 Nov 2020.
European Medicines Agency. European Medicines Agency concludes action on cox-2 inhibitors. 2005. https://www.ema.europa.eu/en/documents/press-release/european-medicines-agency-concludes-action-cox-2-inhibitors_en.pdf. Accessed 16 Nov 2020.
Nissen SE, Yeomans ND, Solomon DH, et al. Cardiovascular safety of celecoxib, naproxen, or ibuprofen for arthritis. N Engl J Med. 2016;375:2519–29.
Gislason GH, Jacobsen S, Rasmussen JN, et al. Risk of death or reinfarction associated with the use of selective cyclooxygenase-2 inhibitors and nonselective nonsteroidal antiinflammatory drugs after acute myocardial infarction. Circulation. 2006;113:2906–13.
Fosbøl EL, Gislason GH, Jacobsen S, et al. Risk of myocardial infarction and death associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs) among healthy individuals: a nationwide cohort study. Clin Pharmacol Ther. 2009;85:190–7.
Schmidt M, Sørensen HT, Pedersen L. Diclofenac use and cardiovascular risks: series of nationwide cohort studies. BMJ. 2018;362:k3426.
Masclee GMC, Straatman H, Arfè A, et al. Risk of acute myocardial infarction during use of individual NSAIDs: a nested case-control study from the SOS project. PLoS One. 2018;13:
Kent AP, Brueckmann M, Fraessdorf M, et al. Concomitant oral anticoagulant and nonsteroidal anti-inflammatory drug therapy in patients with atrial fibrillation. J Am Coll Cardiol. 2018;72:255–67.
MacDonald TM, Wei L. Effect of ibuprofen on cardioprotective effect of aspirin. Lancet. 2003;361:573–4.
Reed GW, Abdallah MS, Shao M, et al. Effect of aspirin coadministration on the safety of celecoxib, naproxen, or ibuprofen. J Am Coll Cardiol. 2018;71:1741–51.
Antman EM. The aspirin–NSAID interaction: more data, but a lack of clarity remains. J Am Coll Cardiol. 2018;71:1752–4.
Zhang J, Ding EL, Song Y. Adverse effects of cyclooxygenase 2 inhibitors on renal and arrhythmia events: meta-analysis of randomized trials. JAMA. 2006;296:1619–32.
Mallen SR, Essex MN, Zhang R. Gastrointestinal tolerability of NSAIDs in elderly patients: a pooled analysis of 21 randomized clinical trials with celecoxib and nonselective NSAIDs. Curr Med Res Opin. 2011;27:1359–66.
Scarpignato C, Bjarnason I. Drug-induced small bowel injury: a challenging and often forgotten clinical condition. Curr Gastroenterol Rep. 2019;21:55.
Hansen JM, Hallas J, Lauritsen JM, Bytzer P. Non-steroidal anti-inflammatory drugs and ulcer complications: a risk factor analysis for clinical decision-making. Scand J Gastroenterol. 1996;31:126–30.
Malfertheiner P, Megraud F, O’Morain CA, et al. Management of Helicobacter pylori infection—the Maastricht V/Florence Consensus Report. Gut. 2017;66:6–30.
Singh G, Triadafilopoulos G. Epidemiology of NSAID induced gastrointestinal complications. J Rheumatol Suppl. 1999;56:18–24.
Scarpignato C, Lanas A, Blandizzi C, et al. Safe prescribing of non-steroidal anti-inflammatory drugs in patients with osteoarthritis–an expert consensus addressing benefits as well as gastrointestinal and cardiovascular risks. BMC Med. 2015;13:55.
Laine L, White WB, Rostom A, Hochberg M. COX-2 selective inhibitors in the treatment of osteoarthritis. Semin Arthritis Rheum. 2008;38:165–87.
Singh G, Ramey DR, Morfeld D, Shi H, Hatoum HT, Fries JF. Gastrointestinal tract complications of nonsteroidal anti-inflammatory drug treatment in rheumatoid arthritis. A prospective observational cohort study. Arch Intern Med. 1996;156:1530–6.
Laine L, Bombardier C, Hawkey CJ, et al. Stratifying the risk of NSAID-related upper gastrointestinal clinical events: results of a double-blind outcomes study in patients with rheumatoid arthritis. Gastroenterology. 2002;123:1006–12.
Bombardier C, Laine L, Reicin A, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–8.
Paulus HE. FDA Arthritis Advisory Committee Meeting: serious gastrointestinal toxicity of nonsteroidal antiinflammatory drugs; drug-containing renal and biliary stones; diclofenac and carprofen approved. Arthritis Rheum.1988;31:1450–51.
Savarino V, Marabotto E, Zentilin P, et al. Proton pump inhibitors: use and misuse in the clinical setting. Expert Rev Clin Pharmacol. 2018;11:1123–34.
García Rodríguez LA, Jick H. Risk of upper gastrointestinal bleeding and perforation associated with individual non-steroidal anti-inflammatory drugs. Lancet. 1994;343:769–72.
Langman MJ, Weil J, Wainwright P, et al. Risks of bleeding peptic ulcer associated with individual non-steroidal anti-inflammatory drugs. Lancet. 1994;343:1075–8.
Scharf S, Kwiatek R, Ugoni A, Christophidis N. NSAIDs and faecal blood loss in elderly patients with osteoarthritis: is plasma half-life relevant? Aust N Z J Med. 1998;28:436–9.
Scheiman JM, Behler EM, Loeffler KM, Elta GH. Omeprazole ameliorates aspirin-induced gastroduodenal injury. Dig Dis Sci. 1994;39:97–103.
Savarino V, Mela GS, Zentilin P, et al. Effect of one-month treatment with nonsteroidal antiinflammatory drugs (NSAIDs) on gastric pH of rheumatoid arthritis patients. Dig Dis Sci. 1998;43:459–63.
Scarpignato C, Gatta L, Zullo A, et al. Effective and safe proton pump inhibitor therapy in acid-related diseases—a position paper addressing benefits and potential harms of acid suppression. BMC Med. 2016;14:179.
Yeomans ND, Tulassay Z, Juhász L, et al. A comparison of omeprazole with ranitidine for ulcers associated with nonsteroidal antiinflammatory drugs. Acid Suppression Trial: Ranitidine versus Omeprazole for NSAID-associated Ulcer Treatment (ASTRONAUT) Study Group. N Engl J Med. 1998;338:719–26.
Hwang AY, Dave CV, Smith SM. Use of prescription medications that potentially interfere with blood pressure control in new-onset hypertension and treatment-resistant hypertension. Am J Hypertens. 2018;31:1324–31.
Bakhriansyah M, Souverein PC, van den Hoogen MWF, de Boer A, Klungel OH. Risk of nephrotic syndrome for non-steroidal anti-inflammatory drug users. Clin J Am Soc Nephrol. 2019;14:1355–62.
Raghavan R, Shawar S. Mechanisms of drug-induced interstitial nephritis. Adv Chronic Kidney Dis. 2017;24:64–71.
Lefebvre C, Hindié J, Zappitelli M, Platt RW, Filion KB. Non-steroidal anti-inflammatory drugs in chronic kidney disease: a systematic review of prescription practices and use in primary care. Clin Kidney J. 2020;13:63–71.
Foy MC, Vaishnav J, Sperati CJ. Drug-induced hypertension. Endocrinol Metab Clin North Am. 2019;48:859–73.
Pope JE, Anderson JJ, Felson DT. A meta-analysis of the effects of nonsteroidal anti-inflammatory drugs on blood pressure. Arch Intern Med. 1993;153:477–84.
Johnson AG, Nguyen TV, Day RO. Do nonsteroidal anti-inflammatory drugs affect blood pressure? A meta-analysis. Ann Intern Med. 1994;121:289–300.
Hansson L. The Hypertension Optimal Treatment study and the importance of lowering blood pressure. J Hypertens Suppl. 1999;17:S9–13.
Morrison A, Ramey DR, van Adelsberg J, Watson DJ. Systematic review of trials of the effect of continued use of oral non-selective NSAIDs on blood pressure and hypertension. Curr Med Res Opin. 2007;23:2395–404.
Sherve K, Gerard CJ, Neher JO, St Anna L. Cardiovascular effects of NSAIDs. Am Fam Physician. 2014;90(4). https://www.aafp.org/afp/2014/0815/od2.html. Accessed 10 Nov 2020
Aw T-J, Haas SJ, Liew D, Krum H. Meta-analysis of cyclooxygenase-2 inhibitors and their effects on blood pressure. Arch Intern Med. 2005;165:490–6.
Chan CC, Reid CM, Aw T-J, Liew D, Haas SJ, Krum H. Do COX-2 inhibitors raise blood pressure more than nonselective NSAIDs and placebo? An updated meta-analysis. J Hypertens. 2009;27:2332–41.
Kishen R, Honoré PM, Jacobs R, Joannes-Boyau O, De Waele E, De Regt J, et al. Facing acid-base disorders in the third millennium—the Stewart approach revisited. Int J Nephrol Renovasc Dis. 2014;7:209–17.
Institute of Medicine (US) Committee on Quality of Health Care in America. Crossing the quality chasm: a new health system for the 21st century. Washington DC: National Academies Press; 2001. http://www.ncbi.nlm.nih.gov/books/NBK222274/. Accessed 25 Jun 2019.
Acknowledgements
Funding
This editorial project and sponsorship for the journal’s Rapid Service Fee were supported by Alfasigma SpA.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
AM: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. PA: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. CB: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. GM: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. PM: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. VS: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. DF: substantial contribution to the conception of the work; drafting and revising the work critically for important intellectual content; final approval of the version to be published; agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Medical Writing and Editorial Assistance
Medical writing support and editorial assistance were provided by Clara Ricci, PhD (Edra SpA, Milan, Italy), and unconditionally funded by Alfasigma SpA.
Disclosures
Alberto Magni has been a consultant for Angelini, Pfizer, Ibsa Farmaceutici, Grunenthal, Alfasigma and Alfasigma. Piergiuseppe Agostoni, Cesare Bonezzi, Giuseppe Massazza, Paolo Menè and Vincenzo Savarino have nothing to declare; Diego Fornasari has received fees from Abiogen, Alfasigma, Astellas, Bayer, Daiichi Sankyo, Grunenthal, Kyova Kirin, Lundbeck, Molteni, Recordati, Scharper, SPA and Zambon.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Magni, A., Agostoni, P., Bonezzi, C. et al. Management of Osteoarthritis: Expert Opinion on NSAIDs. Pain Ther 10, 783–808 (2021). https://doi.org/10.1007/s40122-021-00260-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-021-00260-1