
Abstract
Introduction
Back and neck pain are common musculoskeletal disorders. Topical non-steroidal anti-inflammatory drugs (NSAIDs) are frequently used to reduce pain and inflammation with fewer systemic side effects and drug interactions compared with oral NSAIDs. This study assessed efficacy and tolerability of a topical combination of capsaicin + diclofenac to treat acute back/neck pain.
Methods
In a randomized, double-blind, controlled, multicenter, parallel group trial, 746 patients were treated twice-daily for 5 days with diclofenac 2% + capsaicin 0.075%, diclofenac 2%, capsaicin 0.075% or placebo. Efficacy assessments included change and area under the curve in pain on movement for the worst procedure (POMWP), change in pressure algometry, and number of patients with decrease in POMWP of ≥ 30% and ≥ 50%. Adverse events (AEs) were recorded.
Results
Change in POMWP between baseline and day 2 evening, 1 h after drug application, demonstrates superiority of the combination (− 3.05 cm) versus diclofenac alone (− 2.33 cm) and placebo (− 2.45 cm), but not capsaicin alone (− 3.26 cm). AEs were consistent with known safety profiles.
Conclusion
Capsaicin alone and capsaicin + diclofenac showed superior benefit compared with placebo. However, diclofenac alone demonstrated efficacy comparable with placebo, and therefore its addition to capsaicin added no increased pain relief over capsaicin alone.
Trial registration
ClinicalTrials.gov identifier; NCT02700815.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Topical non-steroidal anti-inflammatory drugs (NSAIDs) are frequently used to reduce pain and inflammation in the treatment of common recurring pain conditions (such as acute back or neck pain), with fewer systemic side effects and drug interactions compared with oral NSAIDs. |
Diclofenac and capsaicin have different pharmacodynamic properties and may have complementary mechanisms of action, although a combination product has not yet been tested. |
This double-blind clinical study assessed efficacy and tolerability of a topical combination of capsaicin + diclofenac versus gels with diclofenac alone, capsaicin alone, and placebo in the treatment of acute lower back or neck pain. |
What was learned from the study? |
The change in the level of pain between baseline and day 2 evening, 1 h after drug application, demonstrates superiority of the combination (− 3.05 cm) versus diclofenac alone (− 2.33 cm) and placebo (− 2.45 cm), but not capsaicin alone (− 3.26 cm). |
Capsaicin alone and capsaicin + diclofenac showed superior benefit compared with placebo. |
However, diclofenac alone demonstrated efficacy comparable with placebo, and therefore its addition to capsaicin added no increased pain relief over capsaicin alone. |
Introduction
Oral analgesics are commonly prescribed for the treatment of acute and chronic pain, but these agents often produce adverse systemic effects, which sometimes are severe. Topical analgesics offer the potential to provide pain relief with minimal adverse systemic effects [1]. This may be of particular relevance when treating frequently recurring pain conditions such as acute back or neck pain [1].
Lower back pain is a common disorder with an estimated global point prevalence between 1980 and 2009 of 11.9 ± 2.0%, for activity-limiting low back pain lasting more than one day [2]. In most cases specific causes cannot be identified (i.e. “nonspecific back pain”) [3]. Usually, the condition is self-limiting, and approximately 90% of patients experience resolution of their pain within 6 weeks [3]. Musculoskeletal pain in the back area can cause a non-physiological posture, which in turn causes further pain. Only around 2% of patients have a serious or systemic disorder, such as systemic inflammatory disorders, infections, spinal malignancy or spinal fracture. In approximately 5–10% of these, the pain may be associated with radicular features with or without neurological deficit, which can be linked to underlying pathology, such as disc prolapse, lateral recess and canal stenosis or advanced grade spondylolisthesis [4].
Neck pain, specifically uncomplicated neck pain (i.e., not attributable to a specific disease or disorder, absence of fracture, no concurrent shoulder pain or nerveroot symptoms, etc.), is a common, mostly musculoskeletal disorder in primary care [5, 6]. Neck pain can be defined as pain in the neck, with or without pain referred into one or both upper limbs that lasts for at least 1 day [7]. The point prevalence of neck pain ranges from 0.4% to 41.5% (mean: 14.4%); 1-year prevalence ranges from 4.8% to 79.5% (mean: 25.8%). The prevalence of neck pain is generally higher in women, high-income countries and more urban areas [8].
NSAIDs, such as ibuprofen, diclofenac or naproxen and paracetamol, have been used for decades to rapidly and effectively reduce back and neck discomfort, often as over the counter analgesics. A systematic review concluded that NSAIDs are effective for short-term symptomatic relief of acute and chronic lower back pain without sciatica. However, it must be noted that effect sizes were small in the studies investigated [9, 10]. Additionally, NSAIDs are not well tolerated by many patients, especially those who are sensitive to gastrointestinal side effects.
Other treatment options are local heat [11] and hyperemization-inducing agents [12], which are both believed to be beneficial in the treatment of musculoskeletal conditions [13, 14] and have been shown to augment blood flow and hemoglobin oxygenation in the skin and muscles [15, 16]. Furthermore, in a clinical trial, a hyperemization-inducing agent has been shown to reduce back/neck pain with low incidence of side effects [17,18,19,20]. Importantly, topically applied remedies have a low potential for systemic side effects and interactions with other (including systemic) medications.
Diclofenac is a non-steroidal drug with anti-inflammatory and analgesic activity. It inhibits the enzyme cyclooxygenase, and so directly inhibits the biosynthesis of prostaglandins and thromboxanes from arachidonic acid. The topical usage of diclofenac in acute pain conditions is considered well-established and is associated with an improved safety profile due to low systemic plasma concentrations [21].
Capsaicin, or the structurally closely related nonivamide, is a selective agonist for the transient receptor potential vanilloid 1 (TRPV1) receptor. In the nociceptive (mostly C- and some Aδ-fibers) nerve endings which selectively express TRPV1, capsaicin-mediated depolarization results in warming, burning, stinging or itching sensations. Initial excitation of the neurons and a short period of hypersensitivity is followed by reduced sensitivity and, after repeated applications, persistent desensitization to pain. There is also evidence that capsaicin treatment may interfere with substance P synthesis [22, 23]. Topical capsaicin acts in the skin to attenuate cutaneous hypersensitivity and reduces pain by a process best described as ‘defunctionalization’ of nociceptor fibers. Capsaicin formulations are widely used to manage pain and have been shown to be effective and safe against various pain syndromes, including post-herpetic neuralgia, diabetic neuropathy, and chronic musculoskeletal pain [23]. Two trials investigating nonivamide-containing topicals showed pronounced efficacy for the treatment of acute lower back pain [19, 20].
Both diclofenac and capsaicin have different pharmacodynamic properties and are assumed to most probably have complementary mechanisms of action [21, 23, 24]. Whether the addition of capsaicin 0.075% to a gel containing diclofenac 2% in the treatment of acute pain increases efficacy compared to diclofenac alone, has not yet been investigated. Therefore, this double-blind clinical study aimed to compare the efficacy and safety of a topical diclofenac + capsaicin gel combination versus gels with diclofenac alone, capsaicin alone, and placebo in the treatment of acute lower back or neck pain.
Methods
Study Population
The study population included male and female patients, aged 18 years and over, suffering from acute back pain or neck pain for at least 24 h, but less than 21 days, diagnosed as pain on movement (POM) ≥ 5.0 cm [on a Visual Analog Scale (VAS) ranging from 0–10 cm] for at least one POM procedure out of five standardized procedures [5]. The POM measurement with the highest pain response determined if the patient had back or neck pain. Patients must also have had an algometric pressure trigger point with a pain pressure threshold of ≤ 25 N/centimeter2 (N/cm2).
Patients were excluded if they had experienced three or more episodes of back or neck pain in the last 6 months, had surgery due to back or neck pain in the previous 12 months, if the pain was attributable to an organic disease (e.g. prolapsed disc, inflammatory arthritis, neurological diseases, etc.) or experienced trauma or strains of the back or neck muscles. Concomitant use of anti-inflammatory drugs, heparinoids, muscle relaxants, analgesics or non-pharmacological treatment, (e.g. heat treatment) 3 days before visit 1, or meeting any other criteria defined in the study protocol (such as known intolerance or hypersensitivity to the active ingredients or any excipients) were also grounds for exclusion.
Study Design and Treatments
This was a prospective, randomized, double-blind, parallel-group, phase 3 study conducted in two countries.
To obtain a minimum total of 700 evaluable patients, 746 patients received one of the following four topical gels, of which 2 g was applied twice daily, with patients allowing for 12 ± 4 h between applications: diclofenac + capsaicin gel: 2% diclofenac and 0.075% capsaicin, diclofenac gel (2%), capsaicin gel (0.075%) and placebo gel.
Patients were randomized to the treatment groups diclofenac + capsaicin, diclofenac, capsaicin, and placebo in a 3:3:3:1 ratio in order to achieve 700 patients eligible for the assessment of the primary endpoint. Randomization was stratified according to application site (back or neck) and country. Patients were blinded to their treatment and all participants were instructed to wear disposable gloves when applying the trial medication.
After a screening evaluation (visit 1), eligible patients were randomized on the same day into the double-blind treatment period of the trial. After administration of the first treatment, patients administered the second application of the trial medication in the evening at home on day 1. They returned to the trial site for four more visits (visits 2–5) during the treatment period as follows: in the morning and in the evening of the following day (day 2) and in the morning of days 3 and 4. At scheduled visits, patients were dosed while at the site. Evening applications on day 3 and day 4 were administered by the patients at home. Thereafter, the treatment was administered at home for one more day (day 5). The patients then returned to the trial site for final assessments (visit 6) in the morning of day 6. Depending on the severity of a patient’s condition and on a patient’s availability, the treatment period at home (day 5) could be skipped or treatment could be extended for 1 or 2 days. Visit 6 was then to be performed on the day immediately following the shortened or extended treatment period. Trial participation was concluded with a telephone call (visit 7 T) for follow-up on adverse events (AEs).
The interval between the two daily applications of the trial medication was approximately 12 h, which could be shortened or extended by 4 h as long as no more and no less than two applications were administered on each treatment day. Treatments were always to be applied by the patient (if possible).
If required, rescue medication (paracetamol, 500 mg tablets) was provided by the sponsor using locally sourced, commercially available products.
One global protocol amendment led to a global protocol revision, which was implemented in June 2016, shortly after the first patient entered the study. These amendments included the trigger point for algometric pressure being revised from ≤ 2.5 to ≤ 25 N/cm2. An analysis of the primary endpoint, including an additional variable for analgesic use was also added.
Endpoints
The primary endpoint was the change in POM for the worst procedure (the movement with maximum pain when assessed at baseline; POMWP) between baseline and day 2 evening, 1 h after drug application. POM was assessed by patients assisted by adequately trained personnel, on performance of standardized, muscle-group-specific movements measured using a VAS (0–10 cm) ranging from 0 = no pain to 10 = worst pain possible. The change in POM was calculated as POM at a given time point subtracted by the POM at baseline.
Key secondary variables were area under the curve (AUC) of POMWP calculated until day 4 morning, i.e. 72 h after start of treatment (POMWP AUC72), and POMWP AUC120 (AUC of POMWP calculated until day 6 morning, i.e. 120 h after start of treatment). Other POM-related endpoints were: the number of patients with a decrease in POMWP of at least 30% and 50% from baseline on day 2 evening, 1 h after drug application, and the change in POMWP between baseline and the morning of day 6.
Change in pressure algometry (PA) between baseline and the morning of day 6 was also measured as a secondary endpoint. PA was determined by the investigator at baseline on day 1 and at defined time points between days 2 and 6 as the pressure value (N/cm2) at a defined trigger point located in the area of POMWP. The pain reaction was determined by increasing the pressure via an algometer on the most tender position (always done at the same position) within the painful area until the patient asked not to increase the pressure anymore. The corresponding pressure value was documented. The change in PA between baseline and day 2 evening (before drug application) and between baseline and the morning of day 6 was compared between treatment groups.
Exposure to rescue medication (days) was analyzed descriptively. Rescue medication was defined as study-dispensed paracetamol.
Safety endpoints included skin reactions using a numerical dermal response score (DRS; categorized as positive for scores ≥ 3 and negative for scores < 3), incidence and intensity of AEs, and changes in safety laboratory parameters, vital signs, and patient and investigator assessment of tolerability.
Statistical Analysis
The sample size for this study was based on an anticipated treatment difference of 1.2 cm on a 0–10 cm VAS and a common standard deviation (SD) of 3 cm, yielding a standardized treatment difference of 0.4 concerning the primary endpoint. The allocation ratio to the treatment groups diclofenac + capsaicin, diclofenac, capsaicin and placebo was planned to be 3:3:3:1.
A total sample size of 700 patients (210 patients each treated with diclofenac + capsaicin, diclofenac, and capsaicin, and 70 patients with placebo) was calculated to have 98% power to detect a difference of 1.2 cm on a 0–10 cm VAS for the primary endpoint between treatment effects of diclofenac + capsaicin versus diclofenac and capsaicin, respectively (3:3 allocation ratio) and 82% power to detect the same difference between diclofenac + capsaicin and placebo (3:1 allocation ratio). This assumes a common SD of 3 cm and uses a 0.05 two-sided significance level.
The primary endpoint was analyzed using a restricted maximum likelihood (REML)-based repeated measures approach using all available longitudinal POMWP observations at the assessment times up to day 2 evening, 1 h after drug application. The statistical model, applied to the analysis of change from baseline in POMWP, included the fixed categorical effects of treatment, country, application site (back/neck), time and treatment-by-time interaction, as well as the continuous fixed covariates of baseline POMWP and baseline-by-time interaction.
The key secondary endpoints POMWP AUC72 and POMWP AUC120 were analyzed using analysis of covariance (ANCOVA) including treatment, country, and application site (back/neck) as fixed effects and baseline POMWP as a continuous covariate. The key secondary endpoints were analyzed hierarchically (POMWP AUC72 first) in a confirmatory way, only if statistical significance was achieved for the primary endpoint. Therefore, no alpha-adjustment for multiple endpoint testing was applied.
All other secondary endpoints were considered as supportive only. The change in PA between baseline and day 2 evening before drug application, and the morning of day 6, respectively, were analyzed using an REML approach analogous to the primary analysis. The numbers of patients with a decrease in POMWP of at least 30% or 50%, respectively, from baseline until day 2 evening, 1 h after drug application, were analyzed by a logistic regression model, including the factor of treatment and the categorical stratification factors of country and application site (back/neck). No interim analysis was planned or performed.
Compliance with Ethics Guidelines
The clinical study (ClinicalTrials.gov identifier: NCT02700815) was conducted in compliance with the clinical trial protocol, the principles laid down in the Declaration of Helsinki, in accordance with ICH-GCP, and was based on the Guideline on Clinical Development of Fixed Combination Medicinal Products of the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) current at trial initiation [33]. The study was done to support the registration of a new product so the respective guidelines had to be followed in addition to the specific requests from the German Federal Institute for Drugs and Medical Devices. The study protocol (EudraCT number 2015-000404-25), protocol amendment and associated documents were reviewed by the Independent Ethics Committees and/or Institutional Review Boards (IECs/IRBs) of the participating centers; the master ethics committee is Ethik-Kommission der Bayrischen Landesärztekammer, München, Germany (Ethic Committee 16004) and further details of local committees can be found in Supplementary Table S1. Informed consent was obtained from each patient in writing before randomization and the rights of patients were protected.
Results
Patient Disposition and Demographics
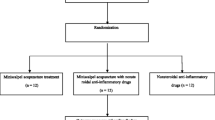
In total, 757 patients were screened and 746 patients were randomized from 18 centers within two different countries (14 in Germany and four in Russia) between May 2016 and July 2017, when the last patient completed the study. All 746 randomized patients were treated with study medication (Fig. 1). A total of 19 patients (2.5%) prematurely discontinued study medication, and the frequencies were comparable across the four treatment groups. The most frequent reasons for discontinuation were AEs [n = 11 (1.5%)], lack of efficacy [n = 3 (0.4%)], lost to follow-up [n = 2 (0.3%)], and refusal to continue taking trial medication [n = 2 (0.3%)]. AEs leading to discontinuation of study medication were reported for patients on active treatment only [capsaicin: n = 6 (2.7%), diclofenac: n = 2 (0.9%), diclofenac + capsaicin: n = 3 (1.3%)].

Disposition of patients by treatment
Analgesic concomitant therapies including dispensed rescue medication, during the treatment period, were reported for 65 patients (8.7%). The frequencies across treatment groups ranged from 17 patients (7.6%) in the diclofenac + capsaicin group to eight patients (10.7%) in the placebo group.
The demographic data were well balanced across the four treatment groups and are shown in Table 1. The overall mean duration of exposure was 5.2 days (SD 1.38; median 5 days) and exposure was well balanced across the four treatment groups.
Baseline disease characteristics were well balanced across the four treatment groups. In total, at baseline, mean PA was 15.65 (SD 5.372) N/cm2, mean pain intensity at rest was 6.0 (SD 1.62) on a numerical rating scale (NRS), mean average pain intensity (pain over the last 24 h) on an NRS was 6.4 (SD 1.48), and mean highest pain on movement score (VAS) was 7.26 cm (SD 1.198).
Pain on Movement Endpoints
The analysis of change in POMWP between baseline and day 2 evening, 1 h after drug application was based on the full analysis set (FAS), which slightly deviated from the treated set in that one patient from the capsaicin group and one patient from the diclofenac group was not included due to no recorded post-baseline POMWP value. From baseline to day 2 evening, 1 h after drug application, POMWP was most effectively lowered by capsaicin alone (− 3.26 cm), followed by the combination therapy diclofenac + capsaicin (− 3.05 cm), placebo (− 2.45 cm), and diclofenac alone (− 2.33 cm). Diclofenac + capsaicin was superior to placebo and to diclofenac alone, but superiority of diclofenac + capsaicin versus capsaicin alone could not be demonstrated (Table 2). The change over time from baseline to 1 h after drug application on the evening of day 2 is illustrated by treatment group in Fig. 2. Sensitivity analyses of the primary endpoint using the per-protocol set, and after adjusting for the use of rescue medication, supported the results of the primary analyses.

POMWP (cm) adjusted mean (SE) change from baseline to day 2 evening, 1 h after drug application—FAS
Exploratory analyses of key secondary parameters POMWP AUC(0–72h) and POMWP AUC(0–120h) supported the results of the POMWP from baseline to day 2 evening: the effect sizes of the combination therapy diclofenac + capsaicin and capsaicin alone in POMWP AUC(0–72h) (Table 2) and in POMWP AUC(0–120h) (Table 3) were similar. The effect sizes of diclofenac alone and placebo were also similar (Fig. 2).
Post-hoc analysis with the same data set and model, using capsaicin as a reference, revealed a distinct treatment effect for capsaicin, when change from baseline at day 2 evening, 1 h after drug application for POMwp was compared with placebo (P = 0.0035).
The highest percentage of patients with a decrease in POMWP of at least 30% and 50% from baseline until day 2 evening, 1 h after drug application was achieved with capsaicin alone, with 150 (67.3%) and 95 (42.6%) patients, respectively. The combination achieved a higher percentage of patients with at least a 30% and 50% POMWP decrease than diclofenac alone and placebo, with 134 (59.6%) and 85 (37.8%) patients, respectively (Table 5). By the morning of day 6, 190 (84.4%) and 159 (70.7%) patients in the diclofenac + capsaicin group experienced at least a 30% or 50% reduction in pain, respectively. This is higher than diclofenac alone and only slightly lower than the capsaicin group, in which 189 (84.8%) and 167 (74.9%) patients achieved a reduction in pain of at least 30% or 50%, respectively.
Consistent with the primary endpoint (change in POMWP between baseline and day 2 evening) and key exploratory secondary endpoints [POMWP AUC(0–72h) and AUC(0–120h)], the change from baseline in POMWP at day 6 in the morning showed no relevant difference between the combination therapy diclofenac + capsaicin and capsaicin alone [adjusted mean difference: 0.20, 95% confidence interval (CI) − 0.24, 0.64]. However, diclofenac + capsaicin was better than diclofenac alone (adjusted mean difference: − 1.12; 95% CI − 1.56, − 0.68) and placebo (adjusted mean difference: − 1.05; 95% CI − 1.67, − 0.44).
Pressure Algometry Endpoints
At day 2 evening, the pressure on the trigger point in the area of the worst procedure site could be increased by at least 3 N/cm2 in all treatment groups. Change from baseline in PA at day 2 evening, before drug application, showed only small differences between all treatment arms; capsaicin alone (adjusted mean difference: 0.31 N/cm2, 95% CI − 0.87, 1.49), diclofenac alone (adjusted mean difference: 0.76 N/cm2, 95% CI − 0.42, 1.95) and placebo (adjusted mean difference: − 0.13 N/cm2, 95% CI − 1.78, 1.53), compared with diclofenac + capsaicin.
Similarly, change from baseline in PA at day 6 morning is shown in Table 6. Only small differences were reported between diclofenac + capsaicin and capsaicin alone (adjusted mean difference: 0.28 N/cm2, 95% CI − 1.58, 2.15); however, diclofenac + capsaicin was superior to diclofenac alone (adjusted mean difference: 2.02 N/cm2, 95% CI 0.15, 3.88). No difference was seen between diclofenac + capsaicin and placebo (adjusted mean difference: 1.65 N/cm2, 95% CI − 0.98, 4.27). The changes in PA (N/cm2) over time to day 6 morning are illustrated in Fig. 3.

Adjusted mean (SE) change from baseline in pressure algometry (N/cm2) to day 6 morning—TS
Safety
Of the 746 treated patients, a total of 146 patients (19.6%) reported AEs (Table 7). The overall percentage of patients with AEs was 21.3% (n = 48) in the diclofenac + capsaicin group, 26.5% (n = 59) for capsaicin alone, 12.1% (n = 27) for diclofenac alone and 16.0% (n = 12) in the placebo treatment group. The type, incidence, and severity of AEs reported in patients in each of the treatment groups were consistent with known profiles for capsaicin and diclofenac. As per preferred term, the most frequent AEs for all patients were burning sensation [n = 29 (3.9%)], skin burning sensation [n = 21 (2.8%)], nasopharyngitis [n = 20 (2.7%)], headache [n = 20 (2.7%)], and application-site pain [n = 17 (2.3%)]. The incidences of AEs related to the application site (burning sensation, skin burning sensation and application-site pain) were more frequent in the diclofenac + capsaicin [n = 12 (5.3%), n = 12 (5.3%) and n = 7 (3.1), respectively] and capsaicin alone [n = 16 (7.2%), n = 7 (3.1%) and n = 10 (4.5%), respectively] treatment groups than the diclofenac alone [n = 1 (0.4%), n = 2 (0.9%) and n = 0 (0.0%), respectively] or placebo (n = 0, 0.0% for each AE) treatment groups.
The incidence of AEs leading to discontinuation of study medication was low in all active treatment groups [capsaicin alone: n = 5 (2.2%); diclofenac alone: n = 3 (1.3%); diclofenac + capsaicin: N = 3 (1.3%) and placebo: n = 0 (0.0%)]. Diarrhea and vomiting led to the discontinuation of two patients (0.9%) from the capsaicin-alone treatment group. Other causes for discontinuation were (skin) burning sensation [n = 2 (0.3%)], pruritus [n = 1 (0.1%)], gastroenteritis [n = 1 (0.1%)], nasopharyngitis [n = 1 (0.1%)], dizziness [n = 1 (0.1%)] and paresthesia [n = 1 (0.1%)].
Overall, the incidences of drug-related AEs were higher in the diclofenac + capsaicin [n = 39 (17.3%)] and capsaicin alone [n = 43 (19.3%)] treatment groups than in the diclofenac alone [n = 7 (3.1%)] and placebo [n = 2 (2.7%)] treatment groups. The incidences of drug-related AEs by system organ class (SOC) and preferred term followed the same pattern as for all AEs. There were no serious adverse events (SAEs) in any of the treatment groups; no relevant changes in safety laboratory parameters (hematology or biochemistry) or vital signs over time were observed in any treatment group.
The incidence of all skin irritations was reflected by the use of the dermal response score. The percentages of patients without skin irritation at 1 h after dosing on day 1 were higher for the diclofenac alone [n = 204 (91.5%)] and placebo [n = 65 (86.7%))] treatment groups than the diclofenac + capsaicin [n = 136 (60.4%)] and capsaicin alone [n = 113 (50.7%)] treatment groups. However, these skin irritations were confined to barely perceptible or readily visible erythema in almost all cases [diclofenac + capsaicin group (39.1%, one patient had minimal edema or papular response), capsaicin group (49.3%)]. Similar results were reported at evaluations on days 2–4. At day 6, the percentage of patients with no irritation from the diclofenac + capsaicin and capsaicin-alone treatment groups were comparable with the diclofenac alone and placebo treatment groups [diclofenac + capsaicin: n = 193 (87.3%); capsaicin alone: n = 198 (89.2%); diclofenac alone: n = 211 (95.5%); placebo: n = 72 (97.3%)].
Overall tolerability, as assessed by patients and investigators, was very good or good in the majority of patients in all treatment groups. In the diclofenac + capsaicin treatment group, overall tolerability was assessed as very good or good in 75.5% and 81.3% by patients and investigators, respectively (capsaicin alone: 77.1% and 81.6%, diclofenac alone: 91.9% and 92.8%, placebo: 94.7% and 96.0%, respectively).
Discussion
Diclofenac is an established active pharmaceutical ingredient found within topical formulations worldwide, with diclofenac salts available in concentrations of between 1 and 4%. Gels, creams, lotions, sprays or patches are mainly used for local symptomatic treatment of pain and inflammation [25]. Capsaicin is also widely used to manage neuropathic and musculoskeletal pain, and has been available in topical formulations (generally 0.025–0.1% by weight) in most countries since the 1980s [22]. The topical combination of diclofenac plus capsaicin has not yet been assessed in clinical studies. The different mechanisms of action and therapeutic effects associated with diclofenac and capsaicin and the reported role of capsaicin as a penetration enhancer for topical drugs (e.g. for indomethacin [26] and naproxen [27]) could increase both the exposure to and the therapeutic effect of topical diclofenac and thus result in an additive or synergistic effect when used in combination.
In this study, patients were suffering from low back and/or neck pain. In the diclofenac + capsaicin arm, 59.6% of patients exhibited a decrease in their pain of at least 30% by the evening of day 2, compared with 67.3% and 48% in the capsaicin and diclofenac-alone arms, respectively. The gel containing diclofenac in combination with capsaicin provided no additional pain relief when compared with capsaicin alone in analyses of the primary endpoint, key secondary endpoints, and all other efficacy endpoints, and was superior to diclofenac alone and placebo. The change from baseline in POMWP at day 2 in the evening, 1 h after drug application, diclofenac + capsaicin was significantly superior to diclofenac alone (adjusted mean difference: − 0.72, 95% CI − 1.10, − 0.33; P = 0.0003) and placebo (adjusted mean difference: − 0.60, 95% CI − 1.15, − 0.06; P = 0.0303). However, there was no significant difference demonstrated between diclofenac + capsaicin and capsaicin alone (adjusted mean difference: 0.21, 95% CI − 0.18, 0.60; P = 0.2886).
Results from the analyses of the primary endpoint were confirmed by the data from all secondary efficacy endpoints, including POMWP AUC(0–72h) and POMWP AUC(0–120h), which did not detect any relevant differences between the combination therapy diclofenac + capsaicin and capsaicin alone.
These results were supported in sensitivity analyses based on the per protocol set and on the FAS (OC-R), an observed case approach in which any value after rescue medication was excluded. These findings were consistent across all subgroups, with no significant interactions in any subgroup analyses based on application site, age, sex, and country.
The results for all other efficacy endpoints were supportive of the conclusion that diclofenac + capsaicin combination therapy was comparable with capsaicin alone in terms of efficacy in this patient population and superior to diclofenac alone and placebo. The use of rescue medication was infrequent in all treatment groups.
The similar efficacy of diclofenac + capsaicin and capsaicin alone in this treatment indication is corroborated by two randomized, double-blind, parallel group phase 3 studies [19, 20] which assessed efficacy and safety of multiple doses of topically applied capsacinoids [nonivamide (with or without the nicotinic acid ester nicoboxil)] at different strengths over a 4-day treatment period. The hyperemization-inducing ointments were applied on defined skin areas of patients suffering from acute low back pain. Comparable to capsaicin in this study, nonivamide/nicoboxil ointment was shown to be an effective and well-tolerated medication for the treatment of acute non-specific low back pain [20]. Somewhat surprisingly, analgesic efficacy of these topicals was higher compared to other treatments (including systemic analgesics and muscle relaxants) [10].
The gel preparation of diclofenac alone used in this study had limited effect, showing efficacy comparable with placebo, and therefore, unsurprisingly, its addition to capsaicin in the combination therapy gel added no benefit in terms of increased pain relief over capsaicin alone. The lack of efficacy of diclofenac versus placebo in this study was unexpected and not consistent with published findings. In a study in patients with acute neck pain, for example, Predel et al. [5] showed that a 1.16% diclofenac gel significantly reduced POM by 75% versus 23% compared with placebo at 48 h after application, and this change represented clinically relevant pain relief. In addition, all POM scores were significantly reduced versus placebo from 1 h after application [5]. In this study, however, diclofenac was formulated as ethanolamine salt, and therefore may have exposed different skin absorption properties compared with the formulation used in our study (diclofenac sodium) which could have contributed to the apparent lack of efficacy of diclofenac alone. In a phase 1 study comparing the bioavailability of diclofenac in a diclofenac + capsaicin combination gel to a reference product of diclofenac gel (Voltarol® 2.32% gel) in healthy volunteers, the diclofenac exposure following application of the topical gel combination product was less than half from the reference product (NCT03074162) [28, 29].
The authors acknowledge that the warming effect of the topical application of capsaicin could potentially have compromised the blinding of treatment assignments in the study. The difficulty of treatment blinding due to the skin warming sensation associated with capsaicin application is a recognized limitation for randomized controlled trials [30]. In addition, two of the four treatment groups included capsaicin, resulting in a low likelihood of inadvertent unblinding.
Although incidences of drug-related AEs were higher in the diclofenac + capsaicin (17.3%) and capsaicin alone (19.3%) treatment groups than in the diclofenac alone (3.1%) and placebo (2.7%) treatment groups, all topical treatments applied in this study were well tolerated. Overall, the majority of both, patients and investigators, assessed tolerability of all study treatments as good or very good. There were no fatal AEs, SAEs, or adverse events of special interest, and only a few AEs led to discontinuation of study medication.
The type and incidence of AEs occurring in patients on active treatment in this study were consistent with the known safety profiles for these topical medications [31]. The higher occurrence of skin irritations in the capsaicin-exposed treatment groups was attributable to barely perceptible or readily visible erythema in almost all cases. Localized hyperemia with erythema of the skin and a sensation of warmth can be attributed to the normal pharmacodynamic effect of this medicinal product [32]. When erythema was included in the assessment of skin reactions for the gels used in this study, the incidence of skin irritation was initially higher in the diclofenac + capsaicin and capsaicin-alone treatment groups than in the diclofenac alone and placebo treatment groups. The responder analysis of the DRS scores for skin reactions showed that the exclusion of erythema at all assessments and in all treatment groups resulted in a negative response for almost all patients.
Conclusion
Capsaicin alone and the combination therapy diclofenac + capsaicin were superior to placebo and to diclofenac alone, but the combination provided no additional pain relief when compared with capsaicin alone in analyses of change in POMWP between baseline and evening of day 2, POMWP over 72 and 120 h and for all other key efficacy endpoints.
The topical treatments applied in this study were well tolerated, with the type and incidence of AEs occurring in patients on active treatment consistent with the known safety profiles for these topical medications.
The reliability of the conclusions on the efficacy of the combination versus capsaicin alone can be challenged because the gel preparation of diclofenac alone in this study showed efficacy comparable with placebo. Therefore, unsurprisingly, its addition to capsaicin in the combination therapy gel added no benefit in terms of increased pain relief over capsaicin alone. As other topical diclofenac formulations have been proven to be effective for the treatment of lower back pain/neck pain, the principle of enhancing the effectiveness of the single ingredients by a combined topical formulation containing capsaicin and diclofenac may still be proven by further research.
References
Argoff CE. Topical analgesics in the management of acute and chronic pain. Mayo Clin Proc. 2013;88:195–205.
Hoy D, Bain C, Williams G, et al. A systematic review of the global prevalence of low back pain. Arthritis Rheum. 2012;64:2028–37.
Casser HR, Seddigh S, Rauschmann M. Acute lumbar back pain. Investigation, differential diagnosis, and treatment. Dtsch Arztebl Int. 2016;113:223–34.
O’Sullivan P, Lin E. Acute low back pain: beyond physical therapies. Pain Manag Today. 2014;1:8–13.
Predel HG, Giannetti B, Pabst H, Schaefer A, Hug AM, Burnett I. Efficacy and safety of diclofenac diethylamine 1.16% gel in acute neck pain: a randomized, double-blind, placebo-controlled study. BMC Musculoskelet Disord. 2013;14:250–9.
Ferrari R, Russell AS. Regional musculoskeletal conditions: neck pain. Best Pract Res Clin Rheumatol. 2003;17:57–70.
Hoy D, March L, Woolf A, et al. The global burden of neck pain: estimates from the global burden of disease 2010 study. Ann Rheum Dis. 2014;73:1309–15.
Hoy DG, Protani M, De R, Buchbinder R. The epidemiology of neck pain. Best Pract Res Clin Rheumatol. 2010;24:783–92.
Roelofs PDDM, Deyo RA, Koes BW, Scholten RJ, van Tulder MW. Non-steroidal anti-inflammatory drugs for low back pain. Cochrane Database Syst Rev. 2008;1:CD000396.
Keller A, Hayden J, Bombardier C, van Tulder M. Effect sizes of non-surgical treatments of non-specific low-back pain. Eur Spine J. 2007;16:1776–888.
French SD, Cameron M, Walker BF, Reggars JW, Esterman AJ. Superficial heat or cold for low back pain. Cochrane Database Syst Rev. 2006;1:CD004750.
Higashi Y, Kiuchi T, Furuta K. Efficacy and safety profile of a topical methyl salicylate and menthol patch in adult patients with mild to moderate muscle strain: a randomized, double-blind, parallel-group, placebocontrolled, multicenter study. Clin Ther. 2010;32:34–433.
French SD, Cameron M, Walker BF, Reggars JW, Esterman AJ. Superficial heat or cold for low back pain. Cochrane Database Syst Rev. 2006;2006:Cd004750.
Garra G, Singer AJ, Leno R, et al. Heat or cold packs for neck and back strain: a randomized controlled trial of efficacy. Acad Emerg Med. 2010;17:484–9.
Okada K, Yamaguchi T, Minowa K, Inoue N. The influence of hot pack therapy on the blood flow in masseter muscles. J Oral Rehabil. 2005;32:480–6.
Warnecke JM, Wendt T, Schak M, Schak M, Schiffer T, Kohl-Bareis M. Evaluation of haemoglobin changes of skin and muscle tissue of the calf induced by topical application of a nonivamide/nicoboxil cream [abstract]. Naunyn Schmiedebergs Arch Pharmacol. 2011;383(Suppl 1):P312.
Chrubasik S, Weiser T, Beime B. Effectiveness and safety of topical capsaicin cream in the treatment of chronic soft tissue pain. Phytother Res. 2010;24:1877–85.
Derry S, Moore RA. Topical capsaicin (low concentration) for chronic neuropathic pain in adults. Cochrane Database Syst Rev. 2012;9:CD010111.
Blahova Z, Holm JC, Weiser T, Richter E, Trampisch M, Akarachkova E. Nicoboxil/nonivamide cream effectively and safely reduces acute nonspecific low back pain—a randomized, placebo-controlled trial. J Pain Res. 2016;9:1221–300.
Gaubitz M, Schiffer T, Holm C, Richter E, Pisternick-Ruf W, Weiser T. Efficacy and safety of nicoboxil/nonivamide ointment for the treatment of acute pain in the low back—a randomized, controlled trial. Eur J Pain. 2016;20:263–73.
Zacher J, Altman R, Bellamy N, et al. Topical diclofenac and its role in pain and inflammation: an evidence-based review. Curr Med Res Opin. 2008;24:925–50.
O'Neill J, Brock C, Olesen AE, Andresen T, Nilsson M, Dickenson AH. Unravelling the mystery of capsaicin: a tool to understand and treat pain. Pharmacol Rev. 2012;64:939–71.
Anand P, Bley K. Topical capsaicin for pain management: therapeutic potential and mechanisms of action of the new high-concentration capsaicin 8% patch. Br J Anaesth. 2011;107:490–502.
Capsaicin and diclofenac (topical). 2019. https://www.drugs.com/mtm/capsaicin-and-diclofenac-topical.html. Accessed Jan 2020.
Diclofenac (2016). In: Sweetman SC, editor. Martindale: the complete drug reference. [Online]. London: Pharmaceutical Press; 2016. https://www.micromedexsolutions.com/micromedex2/librarian. Accessed January 2020.
Fang JY, Fang CL, Hong CT, Chen HY, Lin TY, Wei HM. Capsaicin and nonivamide as novel skin permeation enhancers for indomethacin. Eur J Pharm Sci. 2001;12:195–203.
Degim IT, Uslu A, Hadgraft J, Atay T, Akay C, Cevheroglu S. The effects of azone and capsaicin on the permeation of naproxen through human skin. Int J Pharm. 1999;179:21–5.
Clinical trial: NCT 03074162. https://clinicaltrials.gov/ct2/show/NCT03074162. Accessed Jan 2020.
Boehringer Ingelheim. Clinical study synopsis for public discosure. 2018. https://trials.boehringer-ingelheim.com/public/trial_results_documents/1358/_english_13582c9774171pdf.pdf#page=1. Accessed Jan 2020.
Guedes V, Castro JP, Brito I. Topical capsaicin for pain in osteoarthritis: a literature review. Reumatología Clínica (English Edition). 2018;14:40–5.
Investigator’s Brochure diclofenac and capsaicin for the short-term treatment of pain in the neck and back area associated with tense muscle. Version 1. 2015.
ABC Heat Cream. Summary of product characteristics. 2013.
European Medicines Agency (EMEA) Committee for medicinal products for human use (CHMP): guideline on clinical development of fixed combination medicinal products (London, 19 February 2009, doc. ref. CHMP/EWP/240/95 rev. 1). https://www.emea.europa.eu. Accessed Jan 2020.
Acknowledgements
We would like to thank the participants of the study and Erika Richter who provided statistical support for the manuscript.
Funding
This study was sponsored by Boehringer Ingelheim Pharma GmbH & Co. KG, Ingelheim am Rhein, Germany. The development of this manuscript was supported by the current marketing authorisation holder Sanofi-Aventis Group. The Rapid Service Fee was also funded by Sanofi-Aventis group.
Medical Writing Assistance
The authors would like to acknowledge Bernward Fladung, a freelancer working for Sanofi-Aventis and Claire Lydon of iMed Comms, an Ashfield Company, part of UDG Healthcare plc for medical writing support that was funded by Sanofi-Aventis in accordance with Good Publications Practice (GPP3) guidelines (https://www.ismpp.org/gpp3).
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authorship Contributions
H-GP worked on trial preparation, conduct and reporting and supported the manuscript writing with critical review. CE-B made substantial contributions to the analysis and interpretation of the work, critically revised important intellectual content and finally approved the version to be published. BP worked on trial preparation, conduct and reporting, was involved in the statistical analysis and supported the manuscript writing with critical review. TWW made substantial contributions to the conception and interpretation of the work, critically revised important intellectual content finally approved the version to be published and agrees to be accountable for all aspects of the work. RL reviewed and approved the trial protocol, had the medical oversight during trial conduct, evaluated the trial results and reviewed and approved the trial report.
Disclosures
Caty Ebel-Bitoun, Thomas W. Weiser and Robert Lange are employees of Sanofi-Aventis. Barbara Peil is an employee of Boehringer Ingelheim, who sponsored the study. Hans-Georg Predel has received study grants, honoraria for speaking at congresses and consultancy fees from Boehringer Ingelheim and Sanofi-Aventis.
Compliance with Ethics Guidelines
The clinical study (ClinicalTrials.gov identifier: NCT02700815) was conducted in compliance with the clinical trial protocol, the principles laid down in the Declaration of Helsinki, in accordance with ICH-GCP, and was based on the Guideline on Clinical Development of Fixed Combination Medicinal Products of the European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) current at trial initiation [33]. The study was done to support the registration of a new product so the respective guidelines had to be followed in addition to the specific requests from the German Federal Institute for Drugs and Medical Devices. The study protocol (EudraCT number 2015-000404-25), protocol amendment and associated documents were reviewed by the Independent Ethics Committees and/or Institutional Review Boards (IECs/IRBs) of the participating centers; the master ethics committee is Ethik-Kommission der Bayrischen Landesärztekammer, München, Germany (Ethic Committee 16004) and further details of local committees can be found in Supplementary Table S1. Informed consent was obtained from each patient in writing before randomization and the rights of patients were protected.
Data Availability
Qualified researchers may request access to patient level data and related study documents including the clinical study report, study protocol with any amendments, blank case report form, statistical analysis plan, and dataset specifications. Patient level data will be anonymized and study documents will be redacted to protect the privacy of trial participants. Further details on Sanofi’s data sharing criteria, eligible studies, and process for requesting access can be found at: https://www.clinicalstudydatarequest.com.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.11914479.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Predel, HG., Ebel-Bitoun, C., Peil, B. et al. Efficacy and Safety of Diclofenac + Capsaicin Gel in Patients with Acute Back/Neck Pain: A Multicenter Randomized Controlled Study. Pain Ther 9, 279–296 (2020). https://doi.org/10.1007/s40122-020-00161-9
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40122-020-00161-9