
Abstract
Introduction
The phase 3 PROVENT and STORM CHASER studies evaluated AZD7442 (tixagevimab/cilgavimab) for pre-exposure and post-exposure prophylaxis of symptomatic coronavirus disease 2019 (COVID-19). We report the final 15-month results of both studies.
Methods
In PROVENT, participants were randomized 2:1 to receive 300 mg AZD7442 (n = 3460) or placebo (n = 1737). In STORM CHASER, participants were enrolled within 8 days of exposure to a severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2)-infected individual and randomized 2:1 to receive 300 mg AZD7442 (n = 749) or placebo (n = 372).
Results
In PROVENT, the relative risk reduction (RRR) in symptomatic COVID-19 for AZD7442 versus placebo was 76.7% at primary analysis [95% confidence interval (CI) 46.1, 90.0; p < 0.001], 83.0% at day 183 (95% CI 67.3, 91.2; nominal p < 0.001), and 46.3% at day 366 (95% CI 23.1, 62.4; nominal p < 0.001). Severe/critical COVID-19 was reduced by 91.4% with AZD7442 versus placebo by day 366 (95% CI 61.3, 98.1; nominal p < 0.0001). Adverse events (AEs) occurred in 58.2% and 58.0% of participants administered AZD7442 or placebo, respectively; serious AEs (SAEs) occurred in 6.2% and 5.6%, respectively.
In STORM CHASER, the RRR in symptomatic COVID-19 for AZD7442 versus placebo was 33.3% at primary analysis (95% CI − 25.9, 64.7; p = 0.212), 43.3% at day 183 (95% CI 1.4, 67.4; nominal p = 0.044) and 3.4% at day 366 (95% CI − 35.6, 31.2; nominal p = 0.842). Severe/critical COVID-19 did not occur in participants receiving AZD7442 versus 0.5% of participants receiving placebo by day 366. AEs occurred in 46.5% and 51.9% of participants administered AZD7442 or placebo, respectively; SAEs occurred in 2.7% and 4.3%, respectively. In both studies, serum concentration–time profiles over 457 days were similar for tixagevimab and cilgavimab and consistent with the extended half-life reported for AZD7442 (approximately 90 days).
Conclusion
This analysis provides proof of concept supporting long-term safety of intramuscularly administered AZD7442 for prevention of symptomatic/severe COVID-19.
A graphical abstract is available with this article.
Clinicaltrials.gov Identifiers
PROVENT (NCT04625725) and STORM CHASER (NCT04625972).
Graphical abstract

Plain Language Summary
Antibodies are proteins produced by the body’s immune system to specifically target foreign substances, such as viruses. AZD7442 is made up of an antibody pair (tixagevimab and cilgavimab) that specifically bind and neutralize severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the virus causing coronavirus disease 2019 (COVID-19). AZD7442 was designed to give several months of protection against the virus. These antibodies were tested in two clinical trials to see if they could either protect people from getting COVID-19 (PROVENT trial) or prevent people already exposed to SARS-CoV-2 from getting COVID-19 (STORM CHASER trial). In the two trials, approximately 6000 adults received AZD7442 or placebo (injections that look exactly like AZD7442 but contain no medicine). Protection against COVID-19 was monitored for up to 1 year, and safety for up to 15 months. The percentage of trial participants who reported side effects was similar in the AZD7442 and placebo groups, in both trials. The PROVENT trial showed that AZD7442 reduced the risk of getting COVID-19 up to 6 months and protected against severe COVID-19 for up to 1 year. In STORM CHASER, participants were treated after SARS-CoV-2 exposure but before a positive COVID-19 test. Some participants were already infected with SARS-CoV-2 at the start of the trial, others were not. STORM CHASER showed that AZD7442 protected people against COVID-19 for up to 6 months if they were not already infected at the start. The results of these trials provide proof of concept to support the long-term safety of AZD7442 for the prevention of COVID-19.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Why carry out this study? |
Long-acting monoclonal antibodies may provide protection for individuals who respond suboptimally to coronavirus disease 2019 (COVID-19) vaccines and are at risk of severe disease, hospitalization, and death. |
What did the study ask? |
The PROVENT study assessed the efficacy and safety of pre-exposure prophylaxis with a single intramuscular injection of 300 mg AZD7442 (tixagevimab/cilgavimab) versus placebo in adults without prior severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection who were at appreciable risk of exposure to a SARS-CoV-2-infected individual and/or likely to respond suboptimally to vaccination. |
The STORM CHASER study assessed the efficacy and safety of post-exposure prophylaxis with a single intramuscular injection of 300 mg AZD7442 versus placebo in adults without prior SARS-CoV-2 infection who had reported exposure to a SARS-CoV-2-infected individual in the previous 8 days, and were therefore at appreciable risk of developing COVID-19. |
What was learned from the study? |
The final 15-month analysis of the PROVENT study confirmed the efficacy of AZD7442 in reducing the risk of symptomatic COVID-19 over 6 months, and also suggested a reduction in severe and critical COVID-19 through 12 months. |
The final 15-month analysis of STORM CHASER confirmed the findings of the primary analysis; although the study did not meet its primary efficacy endpoint of reducing symptomatic COVID-19 following post-exposure prophylaxis with AZD7442, a reduction in symptomatic COVID-19 was observed in participants who had SARS-CoV-2 reverse transcription-polymerase chain reaction status negative or missing at baseline. |
In the final analysis of both trials, the overall safety profile of AZD7442 was consistent with that observed in previous studies with shorter follow-up time. |
These analyses provide a proof of concept supporting the long-term safety of intramuscularly administered AZD7442 for the prevention of symptomatic and severe COVID-19. |
Digital Features
This article is published with digital features, including a graphical abstract, to facilitate understanding of the article. To view digital features for this article, go to https://doi.org/10.6084/m9.figshare.25499122.
Introduction
AZD7442 is a combination of two long-acting monoclonal antibodies (mAbs), tixagevimab and cilgavimab, developed to provide protection against coronavirus disease 2019 (COVID-19) by neutralizing severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) [1, 2]. The progenitor mAbs were isolated from individuals with prior SARS-CoV-2 infection and their fragment crystallizable (Fc) regions were enhanced with YTE and TM amino acid modifications to extend their serum half-lives and reduce Fc effector functions, respectively [1, 2].
AZD7442 was assessed for pre-exposure and post-exposure prophylaxis of COVID-19 in the PROVENT (clinicaltrials.gov identifier NCT04625725) and STORM CHASER (clinicaltrials.gov identifier NCT04625972) randomized, controlled, phase 3 clinical trials [3, 4]. Primary results from these trials, conducted prior to the emergence of Omicron and subvariants, supported the Emergency Use Authorization of AZD7442 for pre-exposure prophylaxis of COVID-19 in the USA [5]; AZD7442 received similar authorizations or approvals in other countries and regions. Furthermore, real-world data have shown that AZD7442 is effective at preventing COVID-19 caused by the Omicron BA1, BA2, BA4, and BA5 variants in immunocompromised individuals [6]. However, BQ1.1 and XBB subvariants have demonstrated reduced susceptibility to neutralization by tixagevimab and cilgavimab in vitro [7,8,9], and authorization for use in the USA has been withdrawn [10].
Previously, primary safety and efficacy results after approximately 6 months of data collection were reported for PROVENT and STORM CHASER [3, 4]. However, both trials were designed to run for 15 months to collect longer-term safety data, as these were the first studies using long-acting antibodies incorporating the YTE and TM modifications in large human populations. Here, we report data from the final 15-month analyses of the PROVENT and STORM CHASER studies.
Methods
Trial Overviews
PROVENT (NCT04625725) and STORM CHASER (NCT04625972) were phase 3, double-blind, placebo-controlled, multicenter studies, full details of which have been reported [3, 4]. In PROVENT, adults without prior SARS-CoV-2 infection who were at appreciable risk of exposure to a SARS-CoV-2-infected individual and/or likely to respond suboptimally to vaccination received pre-exposure prophylaxis with AZD7442. In STORM CHASER, adults without prior SARS-CoV-2 infection who had reported exposure to a SARS-CoV-2-infected individual in the previous 8 days and were therefore at appreciable risk of developing COVID-19 received post-exposure prophylaxis with AZD7442. In both studies, participants were randomized 2:1 to receive 300 mg AZD7442 (intramuscular injections of 150 mg tixagevimab and 150 mg cilgavimab) or placebo. The final analyses were performed on participants who remained in the studies and completed follow-up through day 457 (approximately 15 months).
Ethical Approval
These studies adhered to Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable laws and regulations. The protocols were approved by the institutional review boards of the coordinating centers for PROVENT (WIRB Copernicus) and STORM CHASER (Advarra IRB) as well as the institutional review boards or ethics committees at all other participating centers (Supplementary Table S1). All participants provided informed, written consent.
Endpoints
The primary efficacy endpoint in both trials was the incidence of the first SARS-CoV-2 reverse transcription-polymerase chain reaction (RT-PCR)-positive symptomatic illness occurring through day 183. In PROVENT, participants who received a COVID-19 vaccine or who left the main study to enroll in a separate phase 3, open-label substudy (evaluating repeat doses of AZD7442) were censored at the date of unblinding. Secondary endpoints included the incidence of severe or critical COVID-19 and the single-dose pharmacokinetics of AZD7442. Evaluations of the primary efficacy and secondary endpoints were repeated as previously described [3, 4] to account for the final follow-up of all participants. Also evaluated were incidences and serum titers of treatment-emergent antidrug antibodies (ADAs) in participants receiving AZD7442, and the impact of treatment emergent ADAs (TE-ADAs) on AZD7442 pharmacokinetics. ADA positivity was defined by ADA titers ≥ 160 for tixagevimab or ≥ 80 for cilgavimab (titer thresholds were determined as two times the limit of detection). ADAs were defined as treatment emergent if they occurred post-dose in participants who were ADA negative at baseline, or if they occurred in participants who were ADA positive at baseline but who experienced an increase in ADA titers of ≥ fourfold over the course of the study. Exploratory efficacy endpoints included the incidence of the first case of SARS-CoV-2 RT-PCR-positive symptomatic illness occurring through day 366, and genotypic and biochemical/susceptibility analysis of SARS-CoV-2 variants through day 366.
Safety was evaluated on the basis of incidences of adverse events (AEs), serious AEs (SAEs), medically attended AEs (MAAEs), and AEs of special interest (AESIs). AESIs included anaphylaxis and other serious hypersensitivity reactions (including immune complex disease), and injection site reactions. In PROVENT, the study protocol was amended so that cardiac ischemia, cardiac failure, and thrombotic events were classified as AESIs, and a cardiovascular event adjudication committee was commissioned to independently validate the diagnosis of all cardiovascular events, including cardiac ischemia, cardiovascular death, heart failure, stroke, and thrombotic events, as well as all COVID-19-related deaths.
Statistical Analyses
In PROVENT, efficacy was evaluated using the pre-exposure full analysis set, comprising all participants who were randomized, received at least one of the two antibody injections, and who did not have an RT-PCR-confirmed SARS-CoV-2 infection at baseline. In STORM CHASER, efficacy was evaluated using the full analysis set, comprising all participants who were randomized and received at least one injection of study drug. Safety in both studies was evaluated using the safety analysis set, comprising all participants who received at least one injection of study drug. In both studies, the single-dose pharmacokinetics of AZD7442 were evaluated using the pharmacokinetics analysis set, comprising all participants who received AZD7442 and had at least one quantifiable serum pharmacokinetic observation post-dose. TE-ADAs and their impact on the pharmacokinetics of AZD7442 were evaluated using the AZD7442 ADA set, comprising all participants in the safety analysis set who received AZD7442 and were assessed for ADAs at baseline and at least one post-baseline visit.
Results
PROVENT
Trial Population
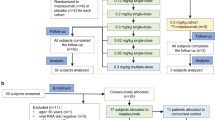
Of 5254 randomized participants, 3460 and 1737 were randomly assigned to receive AZD7442 or placebo, respectively. One participant was randomly assigned to placebo but incorrectly received AZD7442, and was therefore analyzed in the AZD7442 group for safety. In total, 3669 (69.8%) participants completed the study, 1082 (20.6%) discontinued, and 503 (9.6%) left the main study to be enrolled in a separate phase 3, open-label substudy evaluating repeat doses of AZD7442 (Supplementary Fig. S1a). Median (range) follow-up was 456 (1–605) days in the AZD7442 group and 455 (3–587) days in the placebo group (follow-up was greater than the 457 days of the study in some participants because of delayed final visits). By the end of the study, 1697 (49.3%) participants in the AZD7442 group and 880 (50.8%) in the placebo group had elected to be unblinded, and 1851 (53.8%) in the AZD7442 group and 1058 (61.1%) in the placebo group received a COVID-19 vaccine.
Baseline characteristics, which were previously reported, were generally similar between the groups [3]. Briefly, mean age was 53.5 (standard deviation 15.0) years, 43.4% of participants were aged ≥ 60 years, 53.9% were male, 73.0% reported their race as White, 79.7% reported their ethnicity as not Hispanic or Latino, and 77.7% had one or more of the following comorbidities considered risk factors for progression to severe COVID-19: history of obesity (42.4%), obesity at enrollment (41.7%), hypertension (36.4%), current smoker (21.0%), or diabetes (14.3%); in addition, 3.1% were receiving immunosuppressive treatment and 0.5% had immunosuppressive disease.
Efficacy
In the primary analysis of PROVENT, pre-exposure prophylaxis with AZD7442 reduced the risk of developing symptomatic COVID-19 versus placebo, with a relative risk reduction (RRR) of 76.7% [95% confidence interval (CI) 46.1, 90.0; p < 0.001); after 6 months of follow-up, the RRR was 82.8% [95% CI 65.8, 91.4) [3]. The result of the primary analysis was confirmed at the final analysis (follow-up to day 183), with an RRR of 83.0% (95% CI 67.3, 91.2; nominal p < 0.001). By day 366, the RRR was 46.3% (95% CI 23.1, 62.4; nominal p < 0.001). Kaplan–Meier and Cox proportional hazards analysis of the time to first incidence of SARS-CoV-2 RT-PCR-positive symptomatic illness through day 366 support the primary endpoint analyses (Fig. 1a). Severe or critical symptomatic COVID-19 occurred in 0 of 3441 (0%) AZD7442 participants and 6 of 1731 (0.3%) participants in the placebo group by day 183. Severe or critical symptomatic COVID-19 through day 366 occurred in two of 3441 (0.1%) participants in the AZD7442 group versus 11 of 1731 (0.6%) in the placebo group, corresponding to an RRR of 91.4% (95% CI 61.3, 98.1; nominal p < 0.0001) (Fig. 1b).

a Time to first SARS-CoV-2 RT-PCR-positive symptomatic illness and b severe or critical COVID-19 in PROVENT over 366 days. CI confidence interval, HR hazard ratio, RT-PCR reverse transcription-polymerase chain reaction, SARS-CoV-2 severe acute respiratory syndrome coronavirus 2. aIndicates the percentage of events within each group (AZD7442 and placebo) using the group N values as denominators, rather than the total population N for the cumulative incidence shown on the Y-axis
After a median 6 months of follow-up, the efficacy of AZD7442 was found to be consistent regardless of age (RRR of 87.8% in participants aged ≥ 60 years) or comorbidities, including obesity, hypertension, diabetes, smoking, or receipt of immunosuppressive treatment (RRR of 71.7–82.9%) [3]. Although the medical histories of some participants had been updated by the time of the final analysis, the efficacy of AZD7442 remained largely unchanged in participants aged ≥ 60 years (RRR of 88.0%) or with comorbidities, including obesity, hypertension, diabetes, smoking, or receipt of immunosuppressive treatment (RRR of 70.2–83.8%).
At day 366, sequencing data were available for 26 participants in the AZD7442 and 16 in the placebo group with symptomatic COVID-19. The most common variants detected were Delta (15 events), Omicron (14 events), and Alpha (five events). Within this small sample of participants with breakthrough infections there was no evidence of resistance to AZD7442, and substitutions in the AZD7442 binding site were balanced between the AZD7442 and placebo groups.
Safety
AEs occurred in 2016 (58.2%) participants in the AZD7442 group and 1007 (58.0%) in the placebo group. In both groups, the most common AEs (Medical Dictionary for Regulatory Activities Preferred Terms) were COVID-19, headache, cough, and fatigue (Table 1). Lower respiratory tract infection occurred in 28 (0.8%) participants in the AZD7442 group and 20 (1.2%) in the placebo group, and COVID-19 pneumonia occurred in four (0.1%) participants in the AZD7442 group and 21 (1.2%) in the placebo group (Supplementary Table S2). Most AEs were mild to moderate in severity. Grade 3–4 AEs occurred in 256 (7.4%) participants in the AZD7442 group and 125 (7.2%) in the placebo group. SAEs occurred in 215 (6.2%) participants in the AZD7442 group and 97 (5.6%) in the placebo group (Table 1).
No discernable pattern in SAEs was reported (Supplementary Table S3). Two participants in the AZD7442 group had SAEs considered possibly related to study drug by the investigator: one participant had chronic myeloid leukemia and one participant, with a history of diverticulitis and previous partial colectomy, had mesenteric artery thrombosis. There were no notable differences or trends between the AZD7442 and placebo groups in the maximum severity of the SAEs reported. AESIs occurred in 103 (3.0%) participants in the AZD7442 group and 43 (2.5%) in the placebo group (Table 1). There was no discernable pattern in AESIs reported (Supplementary Table S4). Cardiovascular AESIs occurred in 19 (0.5%) participants in the AZD7442 group and seven (0.4%) in the placebo group. Of the 88 (2.5%) and 39 (2.2%) participants in the AZD7442 and placebo groups, respectively, who had cardiovascular events evaluated by the cardiovascular event adjudication committee, 40 (1.2%) and 12 (0.7%) participants were adjudicated as having had true cardiovascular events. All participants with cardiac disorder SAEs during the study had cardiac risk factors and/or a history of cardiovascular disease at baseline, and there was no clear temporal or clinical pattern.
Twenty-two (0.6%) participants died in the AZD7442 group between study days 6 and 503, and 10 (0.6%) participants died in the placebo group between study days 13 and 313 (Table 1). The most common fatal AEs in each group were cardiac disorders (Supplementary Table S5).
STORM CHASER
Trial Population
Of 1131 randomized participants, 749 and 372 received AZD7442 or placebo, respectively. In total, 928 (82.1%) participants completed the study and 203 (17.9%) discontinued (Supplementary Fig.S1b). Median (range) follow-up was 455 (5–573) days in the AZD7442 group and 455 (11–527) days in the placebo groups. By the end of the study, 140 (18.5%) participants in the AZD7442 group and 98 (26.1%) in the placebo group had elected to be unblinded, and 347 (45.9%) in the AZD7442 group and 181 (48.3%) in the placebo group received a COVID-19 vaccine.
Baseline characteristics were generally similar between the groups [4]. Briefly, mean age was 46.4 (standard deviation 15.9) years, 20.0% of participants were aged ≥ 60 years, 50.3% were male, 84.1% reported their race as White, 57.5% reported their ethnicity as Hispanic or Latino, and 65.0% had one or more of the following comorbidities considered risk factors for progression to severe COVID-19: history of obesity (40.1%), obesity at enrollment (39.7%), hypertension (24.1%), current smoker (19.2%), or diabetes (11.6%); in addition, 0.7% were receiving immunosuppressive treatment. Most participants were SARS-CoV-2 RT-PCR negative (87.2%) or their SARS-CoV-2 RT-PCR status was missing (8.5%), and 4.3% were SARS-CoV-2 RT-PCR positive.
Efficacy
At the primary analysis of STORM CHASER, the RRR in symptomatic COVID-19 prior to day 183 was 33.3% (95% CI − 25.9, 64.7) for AZD7442 compared with placebo, which was not statistically significant (p = 0.212) [4]. However, a marginally significant RRR of 73.2% (95% CI 27.1, 90.1) was observed in an ad hoc analysis of the subgroup of participants with a negative or missing SARS-CoV-2 RT-PCR status at baseline. In the final analysis of the primary efficacy endpoint, the RRR in symptomatic COVID-19 was 43.3% at day 183 (95% CI 1.4, 67.4; nominal p = 0.044), while in the ad hoc analysis among participants with a negative or missing SARS-CoV-2 RT-PCR status at baseline, the RRR was 70.2% (95% CI 36.6, 86.0). The RRR in symptomatic COVID-19 in the primary analysis at day 366 was 3.4% (95% CI − 35.6, 31.2; nominal p = 0.842). Kaplan–Meier and Cox proportional hazards analysis of the time to first incidence of SARS-CoV-2 RT-PCR-positive symptomatic illness through day 366 are presented in Fig. 2. Severe or critical symptomatic COVID-19 did not occur in participants in the AZD7442 group and occurred in two (0.5%) participants in the placebo group through day 366.

Time to first symptomatic COVID-19 event in STORM CHASER over 366 days. HR are from a proportional hazards model with Efron method, with p values from log-rank tests. The + symbols indicate a censored observation. CI confidence interval, HR hazard ratio. aIndicates the percentage of events within each group (AZD7442 and placebo) using the group N values as denominators, rather than the total population N for the cumulative incidence shown on the Y-axis
At day 366, sequencing data were available for isolates from 31 participants in the AZD7442 group and 22 in the placebo group with symptomatic COVID-19. The most common variants detected were Omicron (18 events), Alpha (13 events), and Delta (10 events). There was no evidence of resistance to AZD7442 within these isolates from participants with breakthrough infections and substitutions in the AZD7442 binding site were balanced between the AZD7442 and placebo groups. When the 2:1 randomization sequence was considered, the proportion of participants experiencing breakthrough infections was similar between the AZD7442 and placebo groups.
Safety
AEs occurred in 348 (46.5%) participants in the AZD7442 group and 193 (51.9%) in the placebo group (Table 1). The most common AEs in both groups were COVID-19, headache, cough, and fatigue (Supplementary Table S6). Most AEs were mild to moderate in severity. Grade 3–4 AEs occurred in 30 (4.0%) participants in the AZD7442 group and 26 (7.0%) in the placebo group. SAEs occurred in 20 (2.7%) participants in the AZD7442 group and 16 (4.3%) in the placebo group (Table 1). There was no discernable pattern in SAEs reported (Supplementary Table S7), no notable differences or trends between the AZD7442 and placebo groups in the maximum severity of SAEs reported, and no SAEs were considered possibly related to study drug by the investigator. AESIs occurred in four (0.5%) participants in the AZD7442 group and four (1.1%) in the placebo group (Table 1). There was no discernable pattern in AESIs reported (Supplementary Table S8). Cardiovascular events were not categorized as AESIs in STORM CHASER, but cardiac disorder SAEs were balanced across treatment groups [two (0.3%) and two (0.5%) participants in the AZD7442 and placebo groups, respectively] (Supplementary Table S8). Three (0.4%) participants died in the AZD7442 group between study days 131 and 335, and two (0.5%) participants died in the placebo group between study days 187 and 364 (Table 1; Supplementary Table S9).
Pharmacokinetics and ADAs
In both PROVENT and STORM CHASER, serum concentration–time profiles for tixagevimab and cilgavimab were similar and consistent with these antibodies having an extended duration of protection (Fig. 3). Incidence and serum titers of TE-ADAs in participants receiving AZD7442 are summarized in Supplementary Table S10. Geometric mean serum concentrations of AZD7442 were similar among participants with TE-ADAs versus those without TE-ADAs, indicating that ADAs had no apparent effect on the pharmacokinetics of AZD7442 (Fig. 3).

AZD7442 serum concentrations by participant ADA status in a PROVENT and b STORM CHASER. ADA antidrug antibody, %CV percent geometric coefficient of variation, TE-ADA treatment-emergent ADA
Discussion
In the final analysis of the PROVENT study, AZD7442 significantly reduced the risk of symptomatic COVID-19 by 83.0% through day 183, confirming the efficacy of AZD7442 as pre-exposure prophylaxis reported by the primary analysis [3]. Furthermore, a protective effect against severe or critical COVID-19 was observed over 12 months. In the final analysis of STORM CHASER, AZD7442 reduced the risk of SARS-CoV-2 RT-PCR-positive symptomatic COVID-19 through day 183 by 43.3%. Consistent with the primary analysis [4], this result confirms that the study did not meet its primary efficacy endpoint of post-exposure prophylaxis of COVID-19. However, efficacy was again observed in participants whose SARS-CoV-2 RT-PCR status was negative or missing at baseline, with an RRR of 70.2% supporting the outcome of AZD7442 pre-exposure prophylaxis observed in PROVENT. Exploratory data from PROVENT show a waning in efficacy against symptomatic COVID-19 between 6 months and 1 year after dosing. This is expected on the basis of the known half-life of AZD7442, 12 months of pharmacokinetic data, preclinical data, plus in vitro virus neutralization data of plasma from convalescent patients with COVID-19 [1]. Moreover, AZD7442 reduced the risk of severe or critical COVID-19 through day 366 by 91.5%, suggesting that the dose required to protect against severe COVID-19 is lower than that required to prevent symptomatic COVID-19.
In these final 15-month analyses of PROVENT and STORM CHASER, the safety profiles of the single intramuscular dose of 300 mg AZD7442 were found to be consistent with the respective primary analyses [3, 4], with no new safety signals observed. SAEs, AESIs, and deaths occurred with similar frequency in the AZD7442 and placebo groups. Cardiovascular AESIs occurred infrequently in PROVENT, with no imbalance observed between the groups. Cardiovascular AESIs were not assessed in STORM CHASER.
Tixagevimab and cilgavimab demonstrated similar serum concentrations over 366 days, consistent with an extended half-life of approximately 90 days reported from phase 1 pharmacokinetic analyses [1, 11]. TE-ADAs were found to have no apparent effect on AZD7442 pharmacokinetics in PROVENT or STORM CHASER.
Immunocompromised individuals who respond suboptimally to vaccination are most likely to benefit from additional passive protection against COVID-19. The small number of immunocompromised individuals enrolled in the PROVENT study is therefore a study limitation, but the safety and effectiveness of AZD7442 in this vulnerable population is supported by real-world evidence [6, 12]. The efficacy portions of PROVENT and STORM CHASER were conducted before the emergence of Omicron subvariants that are resistant to tixagevimab and/or cilgavimab, and robust genotypic and biochemical/susceptibility analyses of SARS-CoV-2 variants to AZD7442 were limited by small sample sizes. Furthermore, while these trials were designed to evaluate a 300-mg dose of AZD7442, the recommended pre-exposure prophylaxis dose was doubled to 600 mg (with redosing introduced after 6 months) in 2022 to counter the emergence of Omicron variants less susceptible to neutralization by cilgavimab and/or tixagevimab [5, 13]. However, the 600 mg intramuscular dose and intravenous doses of 1000 mg and 3000 mg have been found to be well tolerated with favorable safety profiles in other studies [11, 14, 15].
These final analyses of the PROVENT and STORM CHASER studies provide a proof of concept for the long-term safety of AZD7442 for the prevention of symptomatic COVID-19 and add to the growing body of evidence supporting the safety of long-acting mAbs enhanced with YTE and TM modifications [16,17,18,19]. New long-acting mAbs that can neutralize Omicron subvariants are urgently needed to provide protection for vulnerable individuals who respond suboptimally to vaccination and are at high risk of severe COVID-19 and death.
Data Availability
Data underlying the findings described in this manuscript may be requested in accordance with AstraZeneca’s data sharing policy described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
AstraZeneca Group of Companies allows researchers to submit a request to access anonymized subject-level clinical data, aggregate clinical or genomics data (when available), and anonymized clinical study reports through the Vivli web-based data request platform.
References
Loo YM, McTamney PM, Arends RH, et al. The SARS-CoV-2 monoclonal antibody combination, AZD7442, is protective in nonhuman primates and has an extended half-life in humans. Sci Trans Med 2022;14(635):eabl8124.
Zost SJ, Gilchuk P, Case JB, et al. Potently neutralizing and protective human antibodies against SARS-CoV-2. Nature. 2020;584(7821):443–9.
Levin MJ, Ustianowski A, De Wit S, et al. Intramuscular AZD7442 (tixagevimab-cilgavimab) for prevention of Covid-19. N Engl J Med. 2022;386(23):2188–200.
Levin MJ, Ustianowski A, Thomas S, et al. AZD7442 (tixagevimab/cilgavimab) for post-exposure prophylaxis of symptomatic COVID-19. Clin Infect Dis. 2023;76(7):1247–56.
United States Food and Drug Administration. Fact sheet for healthcare providers: emergency use authorization for Evusheld™ (tixagevimab co-packaged with cilgavimab). 2023. https://www.fda.gov/media/154701/download. Accessed Feb 19 2024.
Suribhatla R, Starkey T, Ionescu MC, Pagliuca A, Richter A, Lee LYW. Systematic review and meta-analysis of the clinical effectiveness of tixagevimab/cilgavimab for prophylaxis of COVID-19 in immunocompromised patients. Br J Haematol. 2023;201(5):813–23.
Imai M, Ito M, Kiso M, et al. Efficacy of antiviral agents against Omicron subvariants BQ.1.1 and XBB. N Engl J Med. 2023;388(1):89–91.
Arora P, Kempf A, Nehlmeier I, et al. Omicron sublineage BQ1.1. resistance to monoclonal antibodies. Lancet Infect Dis. 2022;23(1):S1473–3099(22)00733–2.
Wang Q, Li Z, Ho J, et al. Resistance of SARS-CoV-2 omicron subvariant BA.4.6 to antibody neutralisation. Lancet Infect Dis. 2022;22(12):1666–8.
United States Food and Drug Administration. FDA announces Evusheld is not currently authorized for emergency use in the U.S. 2023. https://www.fda.gov/drugs/drug-safety-and-availability/fda-announces-evusheld-not-currently-authorized-emergency-use-us. Accessed Feb 19 2024.
Forte-Soto P, Albayaty M, Brooks D, et al. Safety, tolerability and pharmacokinetics of half-life extended severe acute respiratorysyndrome coronavirus 2 neutralizing monoclonal antibodies AZD7442 (tixagevimab-cilgavimab) in healthy adults. J Infect Dis. 2023;227(10):1153–63.
Ustianowski A. Tixagevimab/cilgavimab for prevention and treatment of COVID-19: a review. Expert Rev Anti Infect Ther. 2022;20(12):1–11.
Clegg LE, Stepanov O, Schmidt S, et al. 394. Consistency of AZD7442 (cilgavimab/tixagevimab) pharmacokinetics across prophylaxis and treatment and adult and pediatric participants: application of population pharmacokinetics to enable rapid decision-making during the COVID-19 pandemic. Open Forum Infect Dis. 2023;10(Suppl_2):ofad500.464.
Okada H, Ishikawa K, Itoh Y, et al. Safety, tolerability, and pharmacokinetics of half-life extended SARS-CoV-2-neutralizing monoclonal antibodies AZD7442 (tixagevimab/cilgavimab) in healthy Japanese adults. J Infect Chemother. 2023;29(11):1061–7.
Montgomery H, Hobbs FDR, Padilla F, et al. Efficacy and safety of intramuscular administration of tixagevimab-cilgavimab for early outpatient treatment of COVID-19 (TACKLE): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Respir Med. 2022;10(10):985–96.
ClinicalTrials.gov. Study Understanding Pre-Exposure pRophylaxis of NOVel Antibodies (SUPERNOVA). NCT05648110. 2022. https://clinicaltrials.gov/ct2/show/NCT05648110. Accessed Feb 19 2024.
Robbie GJ, Criste R, Dall’acqua WF, et al. A novel investigational Fc-modified humanized monoclonal antibody, motavizumab-YTE, has an extended half-life in healthy adults. Antimicrob Agents Chemother. 2013;57(12):6147–53.
Yu XQ, Robbie GJ, Wu Y, et al. Safety, tolerability, and pharmacokinetics of MEDI4893, an investigational, extended-half-life, anti-Staphylococcus aureus alpha-toxin human monoclonal antibody, in healthy adults. Antimicrob Agents Chemother. 2017;61(1):e01020-e1116.
Hammitt LL, Dagan R, Yuan Y, et al. Nirsevimab for prevention of RSV in healthy late-preterm and term infants. N Engl J Med. 2022;386(9):837–46.
Acknowledgements
The authors would like to thank the participants, their families, and all investigators involved in this study. The sponsor was involved in the study design, collection, analysis, and interpretation of data, as well as data checking of information provided in the manuscript. However, ultimate responsibility for opinions, conclusions, and data interpretation lies with the authors.
Medical Writing/Editorial Assistance.
Medical writing support was provided by Matthew Young, DPhil, Matt Brownsword, PhD, and Lorna Forse, PhD, and editorial support was provided by Jess Galbraith, BSc, all of Core (a division of Prime, London, UK), supported by AstraZeneca according to Good Publication Practice guidelines.
Funding
This analysis was funded by AstraZeneca and includes data from the PROVENT and STORM CHASER trials that were funded by AstraZeneca and the US Government. The journal’s Rapid Service fee was funded by AstraZeneca.
This project has been supported in whole or in part with federal funds from the Department of Health and Human Services, Administration for Strategic Preparedness and Response, Biomedical Advanced Research and Development Authority in partnership with the Department of Defense, and Joint Program Executive Office for Chemical, Biological, Radiological and Nuclear Defense, under Contract No. W911QY-21–9-0001. The findings and conclusions in this presentation have not been formally disseminated by the Department of Health and Human Services and should not be construed to represent any agency determination or policy.
Author information
Authors and Affiliations
Contributions
Myron J. Levin was international coordinating investigator for the trials. Katie Streicher and Mark T. Esser contributed to study conceptualization. Mark T. Esser was responsible for funding acquisition. Seth Seegobin, Rohini Beavon, Karen A. Near, and Mark T. Esser contributed to study design. Myron J. Levin and Mark T. Esser supervised the trials. Myron J. Levin and Mark T. Esser contributed to project administration. Kanika Dey provided Clinical Operations Oversight for the trials. Andrew Ustianowski, Stephane De Wit, Karen A. Near, and Rohini Beavon collected the data. Katie Streicher validated the data. Jesse Thissen and Seth Seegobin conducted the statistical analysis. Myron J. Levin, Andrew Ustianowski, Stephane De Wit, Rohini Beavon, Jesse Thissen, Seth Seegobin, Kanika Dey, Karen A. Near, Katie Streicher, Alexandre Klazand, and Mark T. Esser had access to all the data in this study, contributed to data interpretation and writing and editing of the manuscript, and reviewed and approved the manuscript for submission.
Corresponding author
Ethics declarations
Conflict of Interest
Myron J. Levin reports research support from Johnson & Johnson, Novavax, Moderna, and GSK; consultancy for Merck & Co., GSK, Pfizer, Dynavax and Seqirus; and data safety monitoring/advisory board for GSK. Andrew Ustianowski reports honoraria/speaker fees from AstraZeneca, Sanofi, Merck, Janssen, GSK, and Gilead, and advisory boards for Gilead, Merck, Pfizer, and ViiV Healthcare/GSK. Stephane De Wit reports financial support from AstraZeneca for the conduct of this study. Myron J. Levin, Stephane De Wit, Rohini Beavon, Jesse Thissen, Seth Seegobin, Kanika Dey, Karen A. Near, Katie Streicher, Alexandre Klazand, and Mark T. Esser are employees of, and may hold stock and/or stock options in, AstraZeneca.
Ethical Approval
These studies adhered to Good Clinical Practice guidelines and the Declaration of Helsinki, Council for International Organizations of Medical Sciences International Ethical guidelines, applicable International Conference on Harmonisation Good Clinical Practice guidelines, and all applicable laws and regulations. The protocols were approved by the institutional review boards of the coordinating centers for PROVENT (WIRB Copernicus) and STORM CHASER (Advarra IRB) as well as the institutional review boards or ethics committees at all other participating centers (Supplementary Table S1). All participants provided informed, written consent.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Prior Presentation: This work was partly presented in poster form at IDWeek, Boston, MA, USA, 11–15 October 2023.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Levin, M.J., Ustianowski, A., De Wit, S. et al. Efficacy, Safety, and Pharmacokinetics of AZD7442 (Tixagevimab/Cilgavimab) for Prevention of Symptomatic COVID-19: 15-Month Final Analysis of the PROVENT and STORM CHASER Trials. Infect Dis Ther 13, 1253–1268 (2024). https://doi.org/10.1007/s40121-024-00970-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-024-00970-x