
Abstract
Introduction
There is an urgent need to develop new drugs to treat malaria due to increasing resistance to first-line therapeutics targeting the causative organism, Plasmodium falciparum (P. falciparum). One drug candidate is DM1157, a small molecule that inhibits the formation of hemozoin, which protects P. falciparum from heme toxicity. We describe a first-in-human, phase 1 trial of DM1157 in healthy adult volunteers that was halted early because of significant toxicity.
Methods
Adverse events were summarized using descriptive statistics. We used pharmacokinetic modeling to quantitatively assess whether the DM1157 exposure needed for P. falciparum inhibition was achievable at safe doses.
Results
We found that there was no dose where both the safety and efficacy target were simultaneously achieved; conversely, the model predicted that 27 mg was the highest dosage at which patients would consistently maintain safe exposure with multiple dosing. By pre-defining dose escalation stopping rules and conducting an interim pharmacokinetic/pharmacodynamic analysis, we determined that the study would be unable to safely achieve a dosage needed to observe an anti-malarial effect, thereby providing strong rationale to halt the study.
Conclusion
This study provides an important example of the risks and challenges of conducting early phase research as well as the role of modeling and simulation to optimize participant safety (ClinicalTrials.gov, NCT03490162).
Similar content being viewed by others

Avoid common mistakes on your manuscript.
We describe a first-in-human, phase 1 trial of of an anti-malarial drug in healthy adult volunteers that was halted early because of significant toxicity |
By pre-defining dose escalation stopping rules and conducting an interim pharmacokinetic/pharmacodynamic analysis, we determined that the study would be unable to safely achieve a dosage needed to observe an anti-malarial effect |
This study provides an important example of the risks and challenges of conducting early phase research as well as the role of modeling and simulation to optimize participant safety |
Introduction
Malaria is a substantial global health crisis causing more than 400,000 deaths annually [1]. As a result of rising resistance to first-line therapeutics targeted at the most deadly causative organism, Plasmodium falciparum (P. falciparum), there is an urgent need to develop new treatments for malaria. One such drug candidate is DM1157, a small molecule that inhibits the formation of hemazoin, which protects P. falciparum from heme toxicity [2]. In addition, DM1157 has the additive advantage of competitively inhibiting the chloroquine resistance transporter (PfCRT), thereby potentially impairing drug efflux and overcoming common resistance mechanisms in P. falciparum. In vitro studies demonstrated that DM1157 had minimum 50% inhibitory concentrations (IC50) between 0.9 and 1.8 nM against laboratory strains of P. falciparum and was potent against 39 clinical isolates, most of which were resistant to chloroquine.
DM1157 is an analog of chloroquine bound to a dipyridinylmethylamino piperidinyl moiety. Preclinical pharmacokinetic (PK) and metabolism studies in rat and cynomolgus monkey indicate oral DM1157 is rapidly absorbed and has relatively high bioavailability. In these species, DM1157 accumulates in erythrocytes and undergoes hepatic metabolism, and concentrations increase in a greater-than-proportional manner relative to dose, suggesting non-linear kinetics. In a non-GLP (Good Laboratory Practice) microsomal stability study, the half-life for DM1157 in liver microsomes from rat, cynomolgus monkey and humans was > 60 min with an estimated intrinsic clearance of < 115.5 μl/min/mg. In a non-GLP in vitro metabolite identification study, DM1157 is metabolized to 18 metabolites in rat, cynomolgus monkey and human liver microsomes. All major metabolites produced by human liver microsomes are also produced by rat and cynomolgus monkey liver microsomes.
The half-life of DM1157 was 6.5–13.7 h in cynomolgus monkeys, supporting the testing of once daily dosing in humans. The preclinical toxicity studies demonstrated dose-limiting gastrointestinal intolerance, weight loss, lethargy, reversible elevation in liver enzymes and a systemic inflammatory response consisting of elevations in neutrophils, monocytes, fibrinogen and globulins. The inflammatory response, which occurred in animals at dosages of 10 mg/kg/day and 80 mg/kg/day, lacked microscopic correlates. Additionally, there was potential for QTc prolongation based on in vitro human ether-a-go-go (hERG) channel activation. Overall, the constellation of findings was similar to the known and expected class effects for chloroquine and related molecules. A no observed adverse event level dose was determined to be 9 mg/kg in rat and 10 mg/kg in cynomolgus monkey; standard conversion factors for human equivalent doses and a further safety factor of tenfold were applied to select a maximal clinical starting dose of 0.145 mg/kg for this study.
DM1157 was evaluated in a first-in-human, phase 1 trial that was terminated because of significant toxicity in healthy adult volunteers. Due to the need to raise awareness of drug toxicity that can occur during clinical trials, we aimed to present a study overview with an emphasis on safety outcomes for DM1157 and the use of PK/pharmacodynamics (PD) modeling and simulation to inform phase 1 clinical trial decision-making.
Methods
Study Synopsis
The study was a phase 1, randomized, double-blind, single site and placebo-controlled study of DM1157 in healthy adult volunteers (ClinicalTrials.gov, NCT03490162). Briefly, the study was designed in three parts; part 1 assessed the single ascending dose (SAD) DM1157 in seven cohorts (6 active drug and 2 placebo participants per cohort) at dosages ranging from 9 to 900 mg; part 2 was a multiple ascending dose (MAD) study in four cohorts (8 active drug and 2 placebo participants per cohort) at dosages ranging from 150–900 mg; and part 3 was a food effect study. The primary objective of the study was to assess the safety and tolerability of DM1157, and the secondary objective was to characterize DM1157 PK. Pre-defined study halting rules included any subject developing a serious adverse event (SAE) related to the study drug; two or more subjects in any cohort experiencing a grade 3 adverse event (AE) of the same type; or any subject developing an episode of anaphylaxis within 24 h of study drug administration. During the MAD arm, individual participants were to be halted if they experienced an SAE related to study drug or any other condition that unduly increased the risk to the subject at the site principal investigator’s discretion.
Ethics Approval
The study was approved by the Duke Institutional Review Board (Pro00091787), all subjects provided written consent, and the trial was conducted in accordance with the Declaration of Helsinki.
Definitions of Adverse Events
Using protocol-defined adverse event grading, a mild (grade 1) adverse event (AE) was defined as an event that required minimal or no treatment and did not interfere with a participant’s daily activities; a moderate (grade 2) AE was defined as that which resulted in a low level of inconvenience or concern with therapeutic intervention and may have caused some interference with daily activities; a severe (grade 3) AE interrupted the participant’s usual daily activities, may have required therapy or other treatment and was usually incapacitating.
Inclusion/Exclusion Criteria
Healthy male and non-pregnant females aged 18–45 years with body mass index 18–35 kg/m2 were eligible to participate. Subjects were excluded if they had clinically significant electrocardiogram abnormalities at baseline, used any prescription medication within 14 days prior to study drug (excluding oral contraceptives), used non-prescription medication within 7 days of study drug and/or had significant medical comorbidities or baseline laboratory abnormalities.
PK Sampling and Analysis
During the SAD, plasma and whole-blood samples were collected on days 1, 2, 3, 4, 5 and 6 at the following time points: pre-dose and at 0.5, 1, 2, 4, 6, 8, 10, 12, 16, 20, 24, 48, 72, 96 and 120 h post-dose. Samples from participants were included in the analysis if the participant had received a complete DM1157 dose without vomiting within 2 h and had at least one measureable concentration. PK samples were analyzed by Frontage Laboratories using liquid chromatography tandem-mass spectrometry (LC-MS/MS).
Noncompartmental analysis PK parameters were determined using Phoenix WinNOnlin (V8.1, Certara), including maximum drug concentration (Cmax), time to reach maximum concentration (Tmax), area under the concentration vs. time curve from dosing through the last measurable concentration (AUC0–last), area under the concentration vs. time curve from time 0 to infinity (AUC0–inf), apparent total clearance after oral administration (CL/F) and apparent volume of distribution during terminal phase (Vz/F). For noncompartmental analysis, values below the quantifiable limit were imputed as zero if there was no previous measurable concentration for the subject and otherwise treated as missing.
Population PK (PopPK) analysis was conducted as part of an ad hoc interim analysis after cohort 6 of the SAD. The analysis was conducted using Phoenix NLME (V8.1) to estimate PopPK parameters and conduct dosing simulations; 1-, 2- and 3-compartment models and a variety of approaches for modeling nonlinearity were explored. Results below the quantifiable limit (BQL) were included in the analysis dataset and imputed as one-half the limit of quantification. Modeling was conducted using total concentrations rather than unbound concentrations; accordingly, saturable protein binding was used empirically to predict nonlinearity. The first order conditional estimation- extended least square method was used for estimation, and the final model was selected based on the lowest negative log likelihood, Akaike information criterion and overall model fit. The final model was evaluated using prediction-corrected visual predictive checks. Thereafter, the final model was used to simulate 1000 time-concentration profiles for each dose. Pointwise prediction bands were plotted using the 2.5th and 97.5th percentiles for predicted concentration at each time point.
Correlations between whole-blood DM1157 exposure (AUC) and clinical laboratory results were explored using linear mixed effects models, and laboratory tests with significant (p < 0.01) main effects or time interactions with AUC were visualized graphically.
Efficacy and Safety Exposure Targets
For dosing simulations, AUC0–120 h, a measure of total exposure over three doses, was used to assess the safety of planned MAD dosing (three times daily for three consecutive days). Based on exposures at which severe AEs occurred in cohort 1–cohort 6, a safety target of AUC0–120 h < 5560 h ng/ml was used. For efficacy, in vivo experiments using whole blood from humanized mice suggested an AUC0–24 h of 9892 h ng/ml following five doses (given daily for five consecutive days) were required for parasite clearance of a chloroquine-resistant strain. Therefore, an efficacy target of day 5 AUC0-24 h > 9892 h ng/ml was used.
Statistics
All AEs were coded using MedDRA dictionary version 22.1. The number of AEs and number of subjects with an AE were summarized by dose group, MedDRA SOC and preferred term, maximum severity and relatedness to study product. Relatedness was categorized as related or not related. Summary statistics were computed for change from baseline in clinical laboratory tests by time point, severity and dose group. Summary statistics were also computed for change in baseline vital signs and electrocardiogram intervals. Clinical laboratory results, vital signs and electrocardiogram intervals were graded using a pre-defined toxicity table.
Results
Demographics and Study Disposition
A total of 121 subjects were screened, 50 were enrolled in the study, and 48 received study drug. Of 50 subjects enrolled, 60% were males and 40% were females. The majority of subjects were black or African American (48%) or white (32%), and most subjects were not Hispanic or Latino (94%). The mean (range) demographics were: age 30.7 years (19–45), weight 78.4 kg (50.4–110) and body mass index 26.4 kg/m2 (18.6–33).
An ad hoc interim analysis of safety and PK was conducted after dose escalation halting rules were met in cohort 6 of the SAD arm. After review, the safety monitoring committee recommended study termination due to safety. The 900-mg SAD cohort, the MAD arm, and the food study were not conducted.
Adverse Events
The overall summary of AEs by dose group is presented in Table 1. Among 48 subjects in the safety population (defined as subjects receiving a dose), 38 (79%) experienced at least one AE. Twenty subjects (42%) experienced at least one mild or worse treatment-related AE, six subjects (13%) experienced at least one moderate or worse treatment-related AE, and three (6%) subjects experienced at least one severe, treatment-related AE. All severe AEs occurred in the 600-mg dosing group. No deaths or serious adverse events were reported in the study, and no AEs led to early termination or withdrawal. Of the seven moderate or severe AEs related to treatment, participants experienced: moderate, intermittent systolic hypertension, moderate vomiting for 3 days, moderate acute inflammatory response for 3 days, moderate sinus bradycardia for 7 days, severe acute inflammatory response (3 individuals) lasting 5 days for two subjects and 10 days for one subject. The inflammatory responses in the 300-mg and 600-mg cohorts generally coincided with increased white blood cells on day 2 with transiently increased neutrophils, decreased lymphocytes and decreased platelets. The white blood cell and neutrophil counts then decreased by day 6 before approaching baseline on day 14. Furthermore, several subjects in the 600-mg dosage group experienced elevations in liver enzymes, most commonly a mild (n = 3) or moderate (n = 1) elevation in aspartate aminotransferase (AST) and/or alanine aminotransferase (ALT) on day 6. All elevations in liver enzymes in the 600-mg cohort were deemed related to study drug. All participants recovered from these events. Representative plots of the white blood cell count, AST and ALT results by dose group are noted in Supplemental Figs.1–3.
Across all dosages, the mean (SD) change from baseline for total leukocytes (109/l) was 1.58 (1.46) on day 2 and − 0.41 (1.43) on day 6. The mean (SD) change from baseline for lymphocytes (109/l) was 0.61 (0.62) on day 2 and − 0.07 (0.47) on day 6. The mean (SD) change from baseline for platelets (109/l) was − 6.7 (19.3) on day 2 and − 20.1 (28.6) on day 6.
Regarding electrocardiogram measurements, there was one AE for mild QTc prolongation in the 150-mg dose group that was deemed unrelated to the study drug. No subjects who had normal parameters at baseline developed clinically significant abnormalities post-baseline.
Exposure-Response Analysis
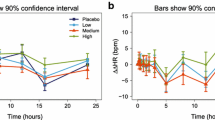
Figures 1, 2 and 3 characterize the relationship between drug exposure in whole blood (AUC) and total white blood count, lymphocyte count and platelet count. We observed a trend toward increased white blood cells on Day 2 and decreased white blood cells on Days 3 and six with whole-blood exposure. Similarly, there appeared to be a decrease in both lymphocytes and platelets with whole-blood exposure on Days 3 and 6. Although statistically significant, there was no apparent visual relationship between whole-blood AUC and AST or ALT.

Whole-blood AUC vs. leukocyte count. AUC area under the curve for the time-concentration profile

Whole-blood AUC vs. lymphocyte count. AUC area under the curve for the time-concentration profile

Whole-blood AUC vs. platelet count. AUC area under the curve for the time-concentration profile
PK Modeling and Simulation
Plasma samples from three participants who received 9 mg and one participant who received 150 mg had no measurable DM1157 concentration and were excluded. Additionally, whole-blood samples from one participant who received 9 mg and one who received 150 mg had no measurable concentration and were excluded. In total, 510 plasma samples from 32 subjects and 543 whole-blood samples from 34 participants were included in the PK analysis.
PK parameters in plasma from non-compartmental analysis are noted in Table 2, whereas PK parameters in whole blood are noted in Supplemental Table 1. Acceptance criteria for estimation of the terminal slope of the time-concentration profile (λz) were not met in any subject (insufficient number of half-lives observed after Tmax) with plasma concentrations and so the terminal phase PK parameters (half-life, CL/F, Vz/F, AUC0–inf) could not be analyzed. The median Tmax in plasma ranged from 0.5 to 2 h, and the geometric mean Cmax was 2.48–613.5 ng/ml. Both Cmax and AUC0–last appeared to increase in a non-linear (greater than dose proportional) fashion with dosage. Furthermore, exposure was higher for whole blood compared to plasma. For the 600-mg SAD cohort, the mean estimated half-life in whole blood was 29.53 h; CL/F was 53.23 l/h; Vz/F was 2268.06 l. The time-concentration profile in whole blood is depicted in Supplemental Fig. 4.
The final PopPK model was a two-compartment model with saturable protein binding, a delay in absorption and estimated between-subject variability for each parameter (exponential model). The prediction-corrected visual predictive checks show that the model can adequately predict the median trend and variability of the observed data (Supplemental Figs. 5–8).
Using the final PopPK model, dosing simulations were conducted for 1000 virtual subjects per dose, including intermediate doses of 222 mg and 450 mg that were not assessed in the trial. The simulations suggested that none of the virtual subjects met both the safety and efficacy criteria. The 9-mg and 27-mg dosages were predicted to meet safety criteria in 100% of simulations, 81 mg in 97% of simulations, 150 mg in 55% of simulations, 300 mg in 2% of simulations, and 600 mg was not predicted to meet the safety target in any simulations. From an efficacy standpoint, the primary systemic exposure efficacy target was reached 1% of the time at the 150-mg dosage, 36% of the time for the 300-mg dosage and 94% of the time for the 600-mg dosage. Results of the dosing simulations are noted in Fig. 4.

Dosing simulations. Model predicted AUC on the Y axis with doses on X axis. Box plots represent the mean (symbols), median (solid line), interquartile range (IQR; box) and min/max values within the 1.5*IQR boundary (whiskers). Left: Efficacy target. Right: Safety target. AUC area under the curve for the time-concentration profile
Discussion
In this first-in-human, phase 1 trial of DM1157 in healthy volunteers, we observed mild to moderate AEs in the 300-mg dosage group and more severe symptoms (e.g., headache, nausea, vomiting, fever, tachycardia, chills) in the 600-mg dosage group. In addition to this dose-adverse event relationship, all participants reported a comparable timeline for their symptoms, starting on the evening of day 2 post-dose, worsening on day 3 and then gradual resolution. There were no similar symptoms in study staff, symptoms did not occur in subjects dosed with placebo, and testing for viral illnesses was negative in selected participants. Overall, these findings suggest that the symptoms were study drug-related and not attributable to a communicable illness. Similar inflammatory responses have not been observed with chloroquine analogs, with typical adverse events being lightheadedness/dizziness, gastrointestinal distress, tinnitus, rash or vision changes [3]. However, agranulocytosis has been reported with the chloroquine analog amodiaquine [4]; reversible hepatotoxicity has been reported with administration of chloroquine in combination with proguanil [5] and very rarely with use of hydroxychloroquine in rheumatologic and dermatologic disease [6, 7]. Hepatotoxicity may reflect accumulation of intracellular chloroquine analogs within lysosomes, a process that interferes with the lysosomal acidification that is necessary for proteolysis, endocytosis, exosome release and phagolysosomal fusion [8, 9].
Population PK/PD models are powerful mathematical tools that relate drug concentration to effect and can be used to predict drug exposure at different dosages. The United States Food and Drug Administration routinely encourages the use of this modeling and simulation approach to guide drug development [10]. This study provides an important example of how modeling and simulation can be used to guide clinical trial design and study management. Specifically, we used a population PK model to quantitatively assess whether the DM1157 exposure needed for P. falciparum inhibition was achievable at safe doses. We found that there was no dose where both the safety and efficacy targets were simultaneously achieved; conversely, 27 mg was the highest dosage at which the model predicted patients would consistently maintain safe exposure. Accordingly, the combination of stopping rules and interim assessment with pharmacokinetic modeling resulted in the early termination of a study that could otherwise have resulted in significant harm to participants without any benefit to the clinical development of the therapeutic.
There are a few limitations of the study. First, there were limited in vivo experimental data to definitely determine the target therapeutic exposure of DM1157. Despite this potential limitation, several modeling exercises suggested that safe and effective dosages could not be simultaneously achieved, even if alternative exposure targets were used. Second, we could not quantify PK parameters for all subjects because of difficulty estimating the terminal elimination profile. However, the population PK model evaluation methods still suggested that the PK model fit the data well and therefore is unlikely to impact the study conclusions.
Conclusion
In this first-in-human, phase 1 trial of DM1157 in healthy volunteers, we observed dose-dependent adverse events consistent with an acute inflammatory response. By pre-defining dosage escalation stopping rules and conducting an interim PK/PD analysis, we were able to address the safety signals and determine that the study would be unable to safely achieve a dosage needed to observe an anti-malarial effect, thereby providing strong rationale to halt the study. This study provides an important example of the risks and challenges of conducting early phase research as well as the role of modeling and simulation to optimize participant safety.
References
World Health Organization (WHO). World Malaria Report 2019. WHO web site. https://www.who.int/publications/i/item/9789241565721. Accessed 3 Nov 2020.
Coronado LM, Nadovich CT, Spadafora C. Malarial hemozoin: from target to tool. Biochim Biophys Acta. 2014;1840:2032–41. https://doi.org/10.1016/j.bbagen.2014.02.009.
Mzayek F, Deng H, Mather FJ, et al. Randomized dose-ranging controlled trial of AQ-13, a candidate antimalarial, and chloroquine in healthy volunteers. PLoS Clin Trials. 2007;2(1):e6. https://doi.org/10.1371/journal.pctr.0020006.
Phillips-Howard PA, West LJ. Serious adverse drug reactions to pyrimethamine-sulphadoxine, pyrimethamine-dapsone and to amodiaquine in Britain. J R Soc Med. 1990;83(2):82–5. https://doi.org/10.1177/014107689008300208.
Wielgo-Polanin R, Lagarce L, Gautron E, et al. Hepatotoxicity associated with the use of a fixed combination of chloroquine and proguanil. Int J Antimicrob Agents. 2005;26(2):176–8. https://doi.org/10.1016/j.ijantimicag.2005.04.019.
Sunkara B, Roofeh D, Silver S, et al. The devil’s in the dosing: severe drug-induced liver injury in a hydroxychloroquine-naive patient with subacute cutaneous lupus erythematosus and porphyria cutanea tarda. Lupus. 2018;27(8):1383–6. https://doi.org/10.1177/0961203318768884.
Abdel Galil SM. Hydroxychloroquine-induced toxic hepatitis in a patient with systemic lupus erythematosus: a case report. Lupus. 2015;24(6):638–40. https://doi.org/10.1177/0961203314561667.
Kaufmann AM, Krise JP. Lysosomal sequestration of amine-containing drugs: analysis and therapeutic implications. J Pharm Sci. 2007;96(4):729–46. https://doi.org/10.1002/jps.20792.
Taveira-DaSilva AM, Moss J. Optimizing treatments for lymphangioleiomyomatosis. Expert Rev Resp Med. 2012;6(3):267–76. https://doi.org/10.1586/ers.12.26.
US Food and Drug Administration. End-of-Phase-2A Meetings. Guidance for Industry. FDA Web Site. https://www.fda.gov/media/72211/download. Published September 2009. Accessed 4 Nov 2020.
Acknowledgements
Funding
The phase I clinical trial was funded National Institutes of Health (HHSN272201500006I). The sponsor did not provide support for the journal’s Rapid Service Fee, which was funded by the authors.
Medical Writing and Editorial Assistance
We thank Erin Campbell, MS, for manuscript review. Ms. Campbell did not receive compensation for her contributions, apart from her employment at Duke University. We would also like to thank Walter Jones for contributions to the manuscript and Yixuan Zou for assistance with pharmacokinetic modeling.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Stephen J. Balevic: acquisition, analysis, or interpretation of data; manuscript drafting and revising it critically for important intellectual content. Shruti M. Raja: acquisition, analysis, or interpretation of data; manuscript drafting and revising it critically for important intellectual content. Rachel L. Randell: acquisition, analysis, or interpretation of data; manuscript drafting and revising it critically for important intellectual content. Gregory A. Deye: Conception or design of the work; manuscript drafting and revising it critically for important intellectual content. Thomas Conrad: acquisition, analysis, or interpretation of data; manuscript drafting and revising it critically for important intellectual content. Aya Nakamura: acquisition, analysis, or interpretation of data; manuscript drafting and revising it critically for important intellectual content. David H. Peyton: Conception or design of the work; manuscript drafting and revising it critically for important intellectual content. Sandra Shotwell: Conception or design of the work; manuscript drafting and revising it critically for important intellectual content. Katherine Liebman: Conception or design of the work; manuscript drafting and revising it critically for important intellectual content. Michael Cohen-Wolkowiez: Conception or design of the work; manuscript drafting and revising it critically for important intellectual content. Jeffrey T. Guptill: Conception or design of the work; manuscript drafting and revising it critically for important intellectual content.
Disclosures
Stephen J Balevic receives support from the National Institutes of Health, US Food and Drug Administration, Patient Centered Outcomes Institute, the Rheumatology Research Foundation’s Scientist Development Award, the Childhood Arthritis and Rheumatology Research Alliance, and consulting for UCB. Rachel L. Randell receives support from the National Institute of General Medical Sciences and the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD) of the National Institutes of Health (NIH) under Award Number T32GM086330. Rachel L. Randell's spouse has financial relationships with Merck & Co, and Biogen. Thomas Conrad is an employee of AstraZeneca. Michael Cohen-Wolkowiez receives support for research from the NIH [1U24-MD016258], National Institute of Allergy and Infectious Diseases [HHSN272201500006I, HHSN272201300017I, 1K24-AI143971], NICHD [HHSN275201000003I], US Food and Drug Administration [5U18-FD006298], and industry for drug development in adults and children (https://dcri.org/about-us/conflict-of-interest/). Jeffrey T. Guptill has been a consultant in past 12 months for Immunovant, Alexion, Momenta, Ra Pharma, Grifols, Jacobus, Becton Dickinson, Cabaletta, Regeneron, Argenx, Signant, UCB, Toleranzia and Piedmont Pharmaceuticals. He receives industry grant support from UCB pharma for a fellowship training grant. Full disclosure statement available at: https://dcri.org/about-us/conflict-of-interest/. He is site investigator for: Alexion, Momenta, Ra Pharma, Argenx and receives research support from NIH and the Myasthenia Gravis Foundation of America. Katherine Liebman and Sandra Shotwell were employees of DesignMedix at the time this work was performed.
Compliance with Ethics Guidelines
The study was approved by the Duke Institutional Review Board (Pro00091787), all subjects provided written consent, and the trial was conducted in accordance with the Declaration of Helsinki.
Data Availability
The datasets generated during and/or analyzed during the current study are not available as the sponsor does not have provisions to share data for phase 1 trials.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Balevic, S.J., Raja, S.M., Randell, R. et al. Adverse Reactions in a Phase 1 Trial of the Anti-Malarial DM1157: An Example of Pharmacokinetic Modeling and Simulation Guiding Clinical Trial Decisions. Infect Dis Ther 11, 841–852 (2022). https://doi.org/10.1007/s40121-022-00605-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-022-00605-z