
Abstract
Human immunodeficiency virus (HIV) therapy has evolved over the last 20 years from mono-drug therapy given five times daily to regimens consisting of three or four drugs combined in a single-tablet dosed once daily. To allow once-daily administration, several drugs require pharmacokinetic boosting by a concomitantly administered P-glycoprotein and cytochrome P450 inhibitor such as ritonavir. The availability of cobicistat provides an alternative to ritonavir to those who are intolerant to this drug, and the opportunity for co-formulated single-tablet regimens consisting of tenofovir/emtricitabine, cobicistat and elvitegravir, atazanavir or darunavir. The cobicistat/elvitegravir-based regimen is well tolerated and patients achieved high rates of HIV RNA suppression in clinical trials. Cobicistat inhibits renal tubular secretion of creatinine, resulting in increased serum creatinine concentrations and reduced estimated glomerular filtration rate, with a new set point reached after 4 weeks. Treatment limiting renal toxicity with cobicistat/elvitegravir and tenofovir disoproxil fumarate is infrequent and may be further reduced when cobicistat is co-formulated with tenofovir alafenamide fumarate, a novel formation of tenofovir currently undergoing clinical trials.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Since the introduction of zidovudine in 1987, human immunodeficiency virus (HIV) therapy has been revolutionised with the availability of over 30 agents across six drug classes. Current British HIV Association (BHIVA) guidelines recommend treatment with a nucleoside/nucleotide reverse transcriptase inhibitor (NRTI) backbone together with a non-nucleoside reverse transcriptase inhibitor (NNRTI), ritonavir-boosted protease inhibitor (PI), or integrase inhibitor (II) as a first-line therapy for the treatment-naïve HIV-positive individuals [1]. Whilst the majority of patients achieve an undetectable HIV RNA level on these treatments, high levels of adherence are required for successful treatment outcomes; recent clinical trials show significantly worse outcomes amongst sub-optimally adherent individuals (defined as adherence <95%) on NNRTI- and PI-based first-line regimens [2, 3]. Side effects remain the commonest reason for switching antiretroviral therapy [4, 5], and side effects are a common reason for late and missed doses [6]. Several agents [e.g. lamivudine, emtricitabine (FTC), efavirenz (EFV), nevirapine and raltegravir (RTG)] have a low genetic barrier to resistance and may be rendered ineffective by single nucleotide substitutions in the viral genome [7–9], while others [e.g. rilpivirine (RPV) and abacavir (ABC)] may have limited potency at high HIV viral load, are best avoided in patients with chronic kidney disease [e.g. tenofovir (TDF), atazanavir (ATV)], or in those at high risk of coronary heart disease (ABC), or should not be used in HLA B5701-positive patients (ABC) [1]. While many patients prefer a once-daily regimen consisting of a small number of tablets, some agents (e.g. RTG) require twice-daily dosing. As a result, antiretroviral therapy continuous to evolve as agents with favourable side-effect profiles, low pill burden, potency across viral loads, and limited cross resistance with existing antiretrovirals become available for use in clinical practice. Co-formulation of such drugs with the NRTI backbone into a single-tablet regimen is an attractive strategy to improve patient convenience, adherence, long-term outcomes and, in some countries, to lower prescription charges.
Cobicistat (COBI), a novel pharmacoenhancer, was recently licensed for the treatment of HIV infection when administered as Stribild® (Gilead Inc., Foster City, CA, USA), a single-tablet regimen containing COBI, elvitegravir (EVG), a novel II, and an NRTI backbone of TDF/FTC. Similar to many PI, EVG requires boosting in order to maintain therapeutic plasma concentrations. Co-administration of COBI maintains EVG plasma concentrations well above the protein-adjusted IC95 for wild-type HIV for more than 24 h, allowing once-daily administration [10]. COBI is also being developed as a pharmacoenhancer for HIV PI, with the potential to create fixed-dose combinations of COBI/ATV or COBI/darunavir (DRV). Finally, a novel formulation of tenofovir [tenofovir alafenamide fumarate (TAF)] is currently undergoing clinical trials which may lead to additional COBI-based combination tablets for HIV treatment [11]. In this review, we discuss the concept of pharmacoenhancing, the pharmacology of COBI, relevant clinical trial data and its potential role in clinical practice.
Methods
Clinical trials, pharmacokinetic and toxicity studies performed with COBI were reviewed for the purpose of this article. Relevant studies were identified by searching the published literature (PubMed) and conference abstracts from January 2008 up to July 2013 for “cobicistat”, “elvitegravir” and “Stribild”. The analysis in this article is based on previously conducted studies, and does not involve any new studies of human or animal subjects performed by any of the authors.
Ritonavir and the Concept of Boosting
Ritonavir (RTV) is an HIV PI which has been available since 1996. While its unfavourable side-effect profile at doses required to inhibit HIV replication limits its role as anti-HIV therapy, it has potent inhibitory effects on cytochrome P450 (CYP) and P-glycoprotein [12]. Inhibition of the efflux transporter P-glycoprotein results in increased drug absorption, and inhibition of CYP (especially 3A4) in reduced elimination of concomitantly administered medications. The pharmacokinetic profile of RTV has resulted in its widespread use as pharmacoenhancer of other PI, most commonly lopinavir, ATV and DRV. RTV prolongs the terminal elimination half-life of the co-administered PI and increases PI trough concentration, allowing once- or twice-daily administration of the “boosted” PI. This inhibitory effect on P-glycoprotein and CYP3A4 is achieved at low, sub-therapeutic doses (100–200 mg daily) that are generally better tolerated [12].
Drawbacks of Pharmacoenhancement
Inhibition of CYP3A4 (and other CYP iso-enzymes) will affect concurrently administered medications metabolised by this pathway. COBI interactions are less widely studied than RTV; while data are awaited it may be necessary to draw on the experience with RTV when predicting likely COBI interactions. Some drugs cannot be co-administered with CYP3A4 inhibitors due to significant increases in concentrations of the co-administered agent (e.g. fluticasone, simvastatin) while others require dose adjustment (e.g. rifabutin, for which interaction data with RTV and COBI is available, and clarithromycin, for which only the interaction with RTV has been studied—advice for COBI is extrapolated from this). In addition, neither RTV nor COBI is ‘clean’ in terms of CYP inhibition; the impact of both on hepatic enzymes is more complex than CYP3A4 inhibition alone (Table 1) [10], further increasing the potential for important drug–drug interactions. The low doses of ritonavir used for boosting may still be associated with tolerability and toxicity issues [13, 14]. There is a paucity of data regarding the tolerability of COBI as a single agent but when used to boost ATV, adverse events and tolerability were similar for COBI and RTV [15].
Pharmacoenhancers: Cobicistat Compared with Ritonavir
Similar to RTV, COBI is a potent inhibitor of CYP3A enzymes but has no antiviral activity against HIV. It was specifically developed as a pharmacoenhancer to be used alongside drugs that are metabolised through CYP, specifically EVG and the PI ATV and DRV. While COBI and RTV have similar inhibitory effects on CYP3A4 and 2B6, COBI has a weaker (2D6) or no (2C8 and 2C9) inhibitory effect on other CYP enzymes (Table 1) [10]. Additional pharmacokinetic studies of COBI revealed <twofold increased desipramine exposure (reflecting limited CYP2D6 inhibition), minimally reduced EFV exposure (suggesting no relevant interactions with CYP2B6 substrates) and small increases in digoxin exposure consistent with inhibition of intestinal P-glycoprotein [16]. Similar to RTV, cimetidine and trimethoprim, COBI is an inhibitor of the renal multidrug and toxin extrusion protein 1 (MATE1) [17]. As a consequence, serum creatinine levels are increased by approximately 10–15%, and creatinine-based estimates of creatinine clearance are reduced by approximately 10% (10–15 mL/min) with COBI exposure [18, 19], a somewhat more pronounced effect than observed with RTV.
COBI at a dose of 150 mg once daily increases EVG exposure to a similar degree as RTV 100 mg (Table 2A); the EVG Ctau with COBI was 11-fold above the protein binding-adjusted IC95 (44.5 ng/mL) of wild-type HIV [10]. COBI/ATV and RTV/ATV co-administration results in similar ATV pharmacokinetic profiles (Table 2B, C) [15, 20]. The ATV Ctau with COBI was well above the protein binding-adjusted IC90 of wild-type HIV (14 ng/mL) and in >90% of visits above the Department of Health and Human Sciences (DHHS) recommended target of 150 ng/mL [20]. COBI and RTV are also similar in their ability to boost DRV when given once or twice daily (Table 2D, E) [21]. The 30% lower mean Ctau with once-daily COBI/DRV administration is 18 times over the protein binding-adjusted EC50 of wild-type HIV and the recommended target for wild-type virus (55 ng/mL). Similar DRV concentrations were observed when COBI/DRV twice daily was co-administered with EVG or etravirine [22]. By contrast, tipranavir exposure was inadequately boosted by COBI 150 mg as compared to RTV 200 mg (both given twice daily) [22].
The pharmacokinetic parameters of COBI are similar when taken fasted or with light meals; high-calorie, high-fat meals reduce COBI AUCtau and C max by 18–24%. By contrast, COBI-boosted EVG exposure is increased when given with food, with AUCtau and C max increased by 22–36% with light meals and by 56–91% with high-calorie, high-fat meals. Although it is recommended that Stribild is administered with food [23], the fasted EVG C24h (250 ng/mL) was well over the protein-adjusted IC95 for wild-type HIV (44.5 ng/mL) [23], suggesting that Stribild should provide adequate EVG exposure in the vast majority of fasted patients. The pharmacokinetic parameters of COBI and EVG are not affected by co-administration of omeprazole, a proton pump inhibitor, or famotidine, an H2-receptor antagonist [24]. Neither COBI nor EVG requires dose modification in patients with severe renal impairment (creatinine clearance <30 mL/min) [25] or moderate liver disease (Child–Pugh–Turcotte class B) [26].
A pharmacokinetic study of 32 patients switched from Atripla® (Bristol Myers Squibb, New York, NY, USA & Gilead Inc, Foster City, CA, USA) (fixed-dose combination of EFV and TDF/FTC) to Stribild showed reduced EVG concentrations during the first week as a result of glucuronosyl transferase induction by EFV. However, the median EFV Ctau remained above the IC90 of wild-type HIV for at least 4 weeks and, by the end of the first week, the median EVG Ctau was threefold higher than the IC95, suggesting that EFV activity is maintained while EVG concentrations reach therapeutic concentrations [27]. A phase IIIb study is evaluating the safety of a regimen switch from Atripla to Stribild in terms of continued viral suppression.
Cobicistat and Drug–Drug Interactions
Due to its inhibition of CYP enzymes, it is anticipated that COBI exposure will result in drug–drug interactions similar to those seen with RTV (see above). However, few studies have examined the effects of COBI on the plasma concentrations of other drugs and until the results of such studies emerge, it would appear prudent to avoid COBI in patients who require drugs with a narrow therapeutic index (e.g. cancer chemotherapy, digoxin) or drugs that are contraindicated or require major dose adjustment in those on RTV. Further and up-to-date information is available on the HIV Drug Interactions webpage [28].
Cobicistat-Containing HIV Therapy: Results from the Phase III Clinical Trials Programme
The results of three studies have been presented to date; two studies investigated the efficacy and safety of Stribild [29–32], while the third study compared COBI with RTV, each co-administered with ATV and TDF/FTC [33].
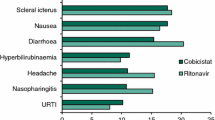
The GS-US-236-0102 and 0103 studies are ongoing phase III, double-blind, randomised, placebo-controlled trials of antiretroviral-naïve HIV-1-positive adults [31, 32]. Patients with a baseline HIV RNA measurement of >5,000 copies/mL were randomised 1:1 to Stribild or Atripla [0102 study], or to Stribild or TDF/FTC/ATV/RTV [0103 study]. To be eligible, patients were required to have a creatinine clearance (calculated by Cockcroft-Gault) of ≥70 mL/min. The primary endpoint was the proportion of patients with an undetectable HIV RNA level (<50 copies/mL) at 48 weeks in the intention to treat population using the Food and Drug Administration (FDA) snapshot analysis. In both studies, Stribild was non-inferior to the comparator and associated with high rates (84–87%) of HIV RNA suppression throughout 96 weeks, low rates (2–3%) of treatment-emergent NRTI/II resistance, and less dizziness or abnormal dreams (vs. EFV) and diarrhoea (vs. ATV/RTV) (Table 3). The GS-US-216-0114 study is an ongoing phase III, double-blind, randomised, placebo-controlled trial of antiretroviral-naïve HIV-1-positive adults (n = 692) with baseline HIV RNA measurements of >5,000 copies/mL and creatinine clearance ≥70 mL/min who were randomised 1:1 to COBI 150 mg or RTV 100 mg, each given together with ATV 300 mg and TDF/FTC once daily [33]. At 48 weeks, the COBI/ATV regimen was non-inferior to the RTV/ATV regimen, with 85% and 87% of patients achieving HIV RNA <50 copies/mL, respectively. Adverse events, including bilirubin elevations, jaundice, nausea and diarrhoea, and study drug discontinuations due to adverse events occurred with equal frequency in both arms [33]. Other ongoing studies investigate a switch from TDF/FTC plus an NNRTI to Stribild (ClinicalTrials.gov identifier: NCT01495702) or TDF/FTC plus a RTV-boosted PI to Stribild (ClinicalTrials.gov identifier: NCT01495702), and the use of Stribild or COBI in patients with impaired renal function (creatinine clearance 50–89 mL/min; ClinicalTrials.gov identifier: NCT01363011). A small single-arm study confirmed the safety of a switch from TDF/FTC plus RTG to Stribild [34].
Renal Safety
As described above, COBI inhibits the renal creatinine transporter MATE1. Although creatinine is freely filtered at the glomerulus, some 10–15% is actively secreted in the proximal tubule. Abrogation of tubular creatinine secretion results in mild increases in serum creatinine concentrations and mild reductions in estimated creatinine clearance. In healthy volunteers, COBI exposure resulted in reduced creatinine clearance (as measured with the Cockcroft-Gault formula) with minimal change in the actual (iohexol-measured) glomerular filtration rate (−9.9 vs. −2.7 mL/min in those with creatinine clearance ≥80 mL/min, and −11.9 vs. −3.6 mL/min in those with creatinine clearance 50–79 mL/min) [35]. Baseline creatinine clearance (range 50–140 mL/min) did not affect the magnitude of the reduction in creatinine clearance with COBI exposure [35]. In the 0102 and 0103 studies, serum creatinine levels in the Stribild arm increased by approximately 10–15% in the first 4 weeks, and creatinine clearance declined by 10–15 mL/min [29, 30]. However, at 4 weeks a new “set point” was reached, with minimal subsequent change up to week 96 (−2.6 vs. −1.0 mL/min for Stribild and Atripla in the 0102 study, −1.8 vs. −4.4 mL/min for Stribild and TDF/FTC/ATV/RTV in the 0103 study) [18, 19]. In the 0114 study, patients in the COBI arm experienced greater reductions in creatinine clearance (−13 vs. −9 mL/min) than in the RTV arm [33].
Five patients (1.4%) in the 0102 study, all in the Stribild arm, had renal events (reported as elevated serum creatinine in two, renal failure in two, Fanconi syndrome in one; a total of four patients had evidence of proximal tubulopathy that led to study drug discontinuation before week 48) [29]. Further two patients (0.6%) in the Stribild arm discontinued study drug between weeks 48 and 96, because of renal adverse events consisting of serum creatinine elevations not accompanied by proximal tubulopathy [31]. In the 0103 study, five patients (Stribild arm 3, ATV/RTV arm 2) discontinued study drug due to renal events before week 96; none had evidence of proximal tubulopathy [32]. In the 0114 study, 1.7% and 1.4% of patients discontinued study medication for renal events in the COBI and RTV arms, and 5 vs. 2 cases had proximal tubulopathy [33].
The low rate of renal discontinuations and renal tubular disease suggests an overall favourable renal safety profile of Stribild and COBI. Indeed, data from patients with creatinine clearance 50–89 mL/min who initiated Stribild or substituted RTV with COBI observed no increased rate of renal toxicity or renal discontinuations [36]. The increases in serum creatinine concentration and the reductions in estimates of creatinine clearance and glomerular filtration rate are unlikely to be of clinical importance. Some of the renal discontinuations were likely to be due to patients meeting pre-specified criteria for discontinuation rather than secondary to overt renal toxicity. Nonetheless, the population included in the clinical trials was at low risk of kidney injury and despite this a small number developed significant renal tubular disease requiring drug discontinuation. The risk factors for TDF-induced Fanconi syndrome and renal tubular disease remain poorly defined but may point to an interaction between COBI and tenofovir at renal tubular level, as previously suggested for RTV [37]. Although such an interaction is not predicted by in vitro studies (Fig. 1), clinicians will need to remain alert to the nephrotoxic potential of Stribild in clinical practice.

Effect of various drugs on tubular creatinine secretion [17]. Tubular secretion of creatinine and tenofovir is mediated through distinct membrane transporter molecules. Based on in vitro experiments, no interaction between cobicistat and tenofovir is predicted. MATE1–2 multidrug and toxin extrusion protein 1, MRP4 multidrug resistance protein 4, OAT1–3 organic anion transporter 1–3, OCT 2–3 organic cation transporter 2–3
Discussion
Cobicistat provides an alternative to ritonavir as a pharmacoenhancer for antiretroviral therapy and as a component of Stribild; it offers an effective, well-tolerated, integrase inhibitor-based single-tablet regimen for HIV treatment. In terms of PI, co-formulations of COBI/ATV and COBI/DRV are in development. The low incidence of neuro-psychiatric side effects with COBI/EVG compared with EFV, and the lower prevalence of diarrhoea with COBI/ATV compared with RTV/ATV, makes it a potentially attractive alternative to these commonly prescribed agents. The reduced pill burden and once-daily administration distinguish COBI/EVG from RTG, the only other II currently licensed. However, a single-tablet regimen based on the investigational integrase, dolutegravir, co-formulated with abacavir and lamivudine is expected to be licensed within the next 12 months and is currently under review by the FDA. Stribild’s lack of interaction with acid-reducing agents distinguishes it from ATV and RPV.
There remain several data gaps, and widespread uptake of Stribild and COBI may be hampered by these. The male predominance and high median CD4 cell count of the phase III trial participants limit data in women and patients with low CD4 cell counts, opportunistic infections, malignancy or other serious co-morbidities, although the WAVES study, comparing Stribild to Truvada® (Gilead Inc., Foster City, CA, USA) plus RTV/ATV in women, is currently recruiting.
COBI is associated with drug–drug interactions, few of which have been studied to date. Although virological failure with Stribild was uncommon, patients that did fail commonly did so with dual-class resistance, and it remains unclear whether these viral isolates remain susceptible to dolutegravir. Also, Stribild is only licensed for use in patients with creatinine clearance ≥70 mL/min thus is not suitable for patients with renal impairment. The inclusion of TDF in Stribild makes it a less attractive option for patients with, or at risk of, osteoporosis, although the renal and bone concerns are likely to be less if TAF becomes the preferred tenofovir formulation of COBI-based single-tablet regimens. Finally, in an increasingly cost-conscious environment, the relative benefits of Stribild and COBI will have to be weighed against any incremental cost relative to current proprietary medications as well as forthcoming generic formulations.
References
Williams I, Churchill D, Anderson J, Boffito M, Bower M, Cairns G, et al. British HIV Association guidelines for the treatment of HIV-1-positive adults with antiretroviral therapy 2012. HIV Med. 2012;13(Suppl 2):1–85.
Cohen CJ, Molina JM, Cahn P, Clotet B, Fourie J, Grinsztejn B, et al. Efficacy and safety of rilpivirine (TMC278) versus efavirenz at 48 weeks in treatment-naive HIV-1-infected patients: pooled results from the phase 3 double-blind randomized ECHO and THRIVE Trials. J Acquir Immune Defic Syndr. 2012;60(1):33–42.
Nelson M, Girard PM, Demasi R, Chen L, Smets E, Sekar V, et al. Suboptimal adherence to darunavir/ritonavir has minimal effect on efficacy compared with lopinavir/ritonavir in treatment-naive, HIV-infected patients: 96 week ARTEMIS data. J Antimicrob Chemother. 2010;65(7):1505–9.
Elzi L, Marzolini C, Furrer H, Ledergerber B, Cavassini M, Hirschel B, et al. Treatment modification in human immunodeficiency virus-infected individuals starting combination antiretroviral therapy between 2005 and 2008. Arch Intern Med. 2010;170(1):57–65.
Cooper V, Moyle GJ, Fisher M, Reilly G, Ewan J, Liu HC, et al. Beliefs about antiretroviral therapy, treatment adherence and quality of life in a 48-week randomised study of continuation of zidovudine/lamivudine or switch to tenofovir DF/emtricitabine, each with efavirenz. AIDS Care. 2011;23(6):705–13.
Duran S, Spire B, Raffi F, Walter V, Bouhour D, Journot V, et al. Self-reported symptoms after initiation of a protease inhibitor in HIV-infected patients and their impact on adherence to HAART. HIV Clin Trials. 2001;2(1):38–45.
Lennox JL, DeJesus E, Lazzarin A, Pollard RB, Madruga JV, Berger DS, et al. Safety and efficacy of raltegravir-based versus efavirenz-based combination. Lancet. 2009;374(9692):796–806.
Li JZ, Paredes R, Ribaudo HJ, Svarovskaia ES, Metzner KJ, Kozal MJ, et al. Low-frequency HIV-1 drug resistance mutations and risk of NNRTI-based. JAMA. 2011;305(13):1327–35.
Gianotti N, Tiberi S, Menzo S, Danise A, Boeri E, Galli L, et al. HIV-1 replication capacity and genotype changes in patients undergoing treatment. J Med Virol. 2008;80(2):201–8.
Mathias AA, Germa P, Lee M, et al. GS-9350: a pharmacoenhancer without anti-HIV activity. In: 16th Conference on Retroviruses and Opportunistic Infections. Montreal, Canada; February 8–11, 2009.
Zolopa A, Ortiz R, Sax P, et al. Comparative study of tenofovir alafenamide vs tenofovir disoproxil fumarate, each with elvitegravir, cobicistat, and emtricitabine, for HIV treatment. In: 20th Conference on Retroviruses and Opportunistic Infections. Atlanta, USA; March 3–6, 2013.
Hull MW, Montaner JS. Ritonavir-boosted protease inhibitors in HIV therapy. Ann Med. 2011;43(5):375–88.
Squires KE, Young B, DeJesus E, Bellos N, Murphy D, Ward D, et al. ARIES 144 week results: durable virologic suppression in HIV-infected patients simplified to unboosted atazanavir/abacavir/lamivudine. HIV Clin Trials. 2012;13(5):233–44.
Collot-Teixeira S, De Lorenzo F, Waters L, Fletcher C, Back D, Mandalia S, et al. Impact of different low-dose ritonavir regimens on lipids, CD36, and adipophilin expression. Clin Pharmacol Ther. 2009;85(4):375–8.
Ramanathan S, Warren D, Wei L, Kearney BP. Pharmacokinetic boosting of atazanavir with the pharmacoenhancer GS-9350 versus ritonavir. In: 49th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). San Francisco, USA; September 12–15, 2009.
German P, Mathias A, Wei L, Murray B, Warren D, Kearney B. The effect of cobicistat on cytochrome P450 2D6, 2B6 and P-glycoprotein using phenotypic probes. In: 12th International Workshop on Clinical Pharmacology of HIV Therapy. Miami, USA; April 13–15, 2011.
Lepist E-I, Murray B, Tong L, et al. Effect of cobicistat and ritonavir on proximal renal tubular cell uptake and efflux transporters. In: 61st Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). Chicago, USA; September 17–20, 2011.
Rockstroh JK, DeJesus E, Henry K, et al. Elvitegravir/cobicistat/emtricitabine/tenofovir DF (STB) has durable efficacy and differentiated safety compared to atazanavir boosted by ritonavir plus emtricitabine/tenofovir DF in treatment-naïve HIV-1 infected patients: week 96 results. In: 11th International Congress on Drug Therapy in HIV Infection. Glasgow, UK; November 11–16, 2012.
Zolopa A, Gallant J, Cohen C, et al. Elvitegravir/cobicistat/emtricitabine/tenofovir DF (STB) has durable efficacy and differentiated safety compared to efavirenz/emtricitabine/tenofovir DF (ATR) in treatment-naïve HIV-1 infected patients: week 96 results. In: 11th International Congress on Drug Therapy in HIV Infection. Glasgow, UK; November 11–15, 2012.
Elion R, Cohen C, Gathe J, Shalit P, Hawkins T, Liu HC, et al. Phase 2 study of cobicistat versus ritonavir each with once-daily atazanavir and fixed-dose emtricitabine/tenofovir df in the initial treatment of HIV infection. AIDS. 2011;25(15):1881–6.
Mathias A, Liu H, Warren D, Sekar V, Kearney BP. Relative bioavailability and pharmacokinetics of darunavir when boosted with the pharmacoenhancer GS-9350 versus ritonavir. In: 11th International Workshop on Clinical Pharmacology of HIV Therapy. Sorrento, Italy; April 7–9, 2010.
Ramanathan S, Wang H, Szwarcberg J, Kearney BP. Safety/tolerability, pharmacokinetics, and boosting of twice-daily cobicistat administered alone or in combination with darunavir or tipranavir. In: 13th International Workshop on Clinical Pharmacology of HIV Therapy. Barcelona, Spain; April 16–18, 2012.
German P, Warren D, Wei L, Zhong L, Hui J, Kearney BP. Effect of food on pharmacokinetics of elvitegravir, emtricitabine, tenofovir DF and the pharmacoenhancer GS-9350 as a fixed-dose combination tablet. In: 49th Interscience Conference on Antimicrobial Agents and Chemotherapy (ICAAC). San Francisco, USA; September 12–15, 2009.
Mathias A, Koziara J, Wei X, Warren D, Kearney BP. Effect of acid reducing agents on the relative bioavailability and pharmacokinetics of cobicistat-boosted elvitegravir. In: International Workshop on Clinical Pharmacology of HIV Therapy. Miami, USA; April 13–15, 2010.
German P, Wei X, Mizuno V, Cheng A, Kearney BP, Mathias A. Pharmacokinetics of elvitegravir and cobicistat in subjects with severe renal impairment. In: 13th International Workshop on Clinical Pharmacology of HIV Therapy. Barcelona, Spain; April 16–18, 2012.
Ramanathan S, Rhee M, Shen G, Custodio J, Kearney BP. Pharmacokinetics and safety of boosted-elvitegravir in subjects with hepatic impairment. In: 13th International Workshop on Clinical Pharmacology of HIV Therapy. Barcelona, Spain; April 16–18, 2012.
Ramanathan S, Wei X, Custodio J, Wang H, Dave A, Cheng A, Kearney BP. Pharmacokinetics of EVG/COBI/FTC/TDF single tablet regimen following treatment with EFV/FTC/TDF (Atripla®) in healthy subjects. In: 13th International Workshop on Clinical Pharmacology of HIV Therapy. Barcelona, Spain; April 16–18, 2012.
HIV Drug Interactions webpage. http://www.HIV-druginteractions.org. Last accessed 20 Aug 2013.
Sax PE, DeJesus E, Mills A, Zolopa A, Cohen C, Wohl D, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir versus co-formulated efavirenz, emtricitabine, and tenofovir for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3 trial, analysis of results after 48 weeks. Lancet. 2012;379(9835):2439–48.
DeJesus E, Rockstroh JK, Henry K, Molina JM, Gathe J, Ramanathan S, et al. Co-formulated elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir disoproxil fumarate for initial treatment of HIV-1 infection: a randomised, double-blind, phase 3, non-inferiority trial. Lancet. 2012;379(9835):2429–38.
Zolopa A, Sax PE, Dejesus E, Mills A, Cohen C, Wohl D, et al. A randomized double-blind comparison of coformulated elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate versus efavirenz/emtricitabine/tenofovir disoproxil fumarate for Initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;63(1):96–100.
Rockstroh JK, Dejesus E, Henry K, Molina JM, Gathe J, Ramanathan S, et al. A randomized, double-blind comparison of co-formulated elvitegravir/cobicistat/emtricitabine/tenofovir versus ritonavir-boosted atazanavir plus co-formulated emtricitabine and tenofovir DF for initial treatment of HIV-1 infection: analysis of week 96 results. J Acquir Immune Defic Syndr. 2013;62(5):483–6.
Gallant JE, Koenig E, Andrade-Villanueva J, Chetchotisakd P, Dejesus E, Antunes F, et al. Cobicistat versus ritonavir as a pharmacoenhancer of atazanavir plus emtricitabine/tenofovir disoproxil fumarate in treatment-naive HIV type 1-infected patients: week 48 results. J Infect Dis. 2013;208(1):32–9.
Mills A, Crofoot G, Ortiz R, Rashbaum B, Towner W, Ward D, et al. Safety and tolerability of switching from twice daily raltegravir plus truvada to stribild in virologically suppressed, HIV-1 infected subjects. Frontiers in Drug Development for Antiretroviral Therapies. San Diego, CA, USA; December 4–7, 2012.
German P, Liu HC, Szwarcberg J, Hepner M, Andrews J, Kearney BP, et al. Effect of cobicistat on glomerular filtration rate in subjects with normal and impaired renal function. J Acquir Immune Defic Syndr. 2012;61(1):32–40.
Post F, Winston J, Andrade-Villanueva J, Fisher M, Liu Y, Zhong L, et al. Elvitegravir/cobicistat/tenofovir DF/emtricitabine (STB) and cobicistat (COBI) in HIV infected patients with mild to moderate renal impairment. In: 7th IAS Conference on HIV Pathogenesis, Treatment, and Prevention. Kuala Lumpur, Malaysia; 30 June–03 July 2013.
Post FA, Holt SG. Recent developments in HIV and the kidney. Curr Opin Infect Dis. 2009;22(1):43–8.
Acknowledgments
No funding or sponsorship was received for this study or publication of this article. Frank A. Post is the guarantor for this article, and takes responsibility for the integrity of the work as a whole. Prior to peer review Gilead were offered the opportunity to review the article solely to ensure scientific accuracy of the details. Minor changes were made to the content as a result, at the discretion of the authors. No writing assistance or other editorial involvement was provided by the manufacturer.
Conflict of interest
Previously, LW has received funding to attend conferences or educational meetings, honoraria and/or funding for research from Gilead Sciences, Boehringer Ingelheim, Janssen-Cilag, ViiV healthcare and Merck, and FAP from Gilead Sciences, Bristol-Myers Squibb, Janssen-Cilag, GlaxoSmithKline, ViiV healthcare, Merck and Roche. BN and JB declare no conflict of interest.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Nathan, B., Bayley, J., Waters, L. et al. Cobicistat: a Novel Pharmacoenhancer for Co-Formulation with HIV Protease and Integrase Inhibitors. Infect Dis Ther 2, 111–122 (2013). https://doi.org/10.1007/s40121-013-0013-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40121-013-0013-7