
Abstract
In the 1980s, Orion Pharma, then a mid-ranking Nordic area pharmaceutical company, established a drug development programme on the inhibition of catechol O-methyltransferase (COMT). This enzyme, which plays an important role in the inactivation of catecholamine neurotransmitters and drugs with a catechol structure, thus came under consideration as a target in the innovative translational and clinical programme we describe in this historical review. The starting point was the conjecture that a peripherally acting COMT inhibitor might improve entry of levodopa into the brain. This had potentially significant implications for the medical treatment of Parkinson’s disease (PD). The rationale was that more efficient delivery of levodopa to the brain might allow the high therapeutic doses of levodopa to be reduced and the dose interval to be extended. Elucidation of structure–activity relations paved the way for the discovery and development of entacapone, a 5-nitrocatechol that was a potent and highly specific inhibitor of COMT. Experience in phase III clinical trials established that entacapone, used as an adjunct to regular or controlled-release levodopa preparations (also including a peripherally acting dopa-decarboxylase inhibitor), increased ON-time and reduced OFF-time and improved clinical condition in patients with PD experiencing wearing-off, often with a reduced daily levodopa dose. Several of these studies also identified that entacapone improved patients’ quality of life and was cost-effective. Subsequently, entacapone has been amalgamated into a triple-combination preparation (Stalevo®) with levodopa and carbidopa to create a flexible and convenient drug therapy for patients with PD who have end-of-dose motor fluctuations not stabilised on levodopa/dopa-decarboxylase inhibitor treatment. This review offers a historical perspective on a successful programme of drug development by researchers who played central roles in the progress from exploratory hypothesis to registered pharmaceutical product.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In the 1980s, inhibition of peripheral catechol O-methyltransferase (COMT) was identified as a potential avenue to refine the use of levodopa in Parkinson’s disease. |
An independent research and development programme of Orion Pharma led to the development of entacapone. |
In the course of that programme, extensive new understanding was gained into the properties and functions of COMT. |
Entacapone has now been in clinical use for more than two decades: currently it is most widely used in the Stalevo® formulation, combined with levodopa and carbidopa. |
Introduction
The catechol O-methyltransferase (COMT) inhibitor entacapone is among the first new chemical entities to have been developed by Orion Pharma as a treatment for Parkinson’s disease (PD). The first human trial was performed at the Helsinki University Hospital in 1992 [1], and—after a full clinical and regulatory development programme—entacapone entered clinical practice at the end of the 1990s and has since established itself as a levodopa-enhancing adjuvant therapy for the treatment of motor symptoms of PD [2]. The Committee for Medicinal Products for Human Use of the European Medicines Agency (EMA) has approved entacapone for use together with levodopa and a dopa-decarboxylase inhibitor (DDCI; usually benserazide or carbidopa) for patients with PD who are experiencing ‘fluctuations’ towards the end of the period between two doses of levodopa and who cannot be stabilised on those levodopa/DDCI combinations [3]. (The development of these fluctuations is linked with a reduction in the therapeutic effects of levodopa.) These phenomena usually emerge after a few years of levodopa/DDCI therapy; a corollary of this is that entacapone use is substantially confined to patients with established PD [4].
Bringing an original medicinal product from concept to marketed product was a significant accomplishment for a mid-size pharmaceutical company in the 1980s. Some of the human aspects of that journey are recalled in the Supplementary Material appended to this article. In this central part we consider some key considerations and events in the development of COMT inhibitors as clinical resources in PD, with specific emphasis on entacapone.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
This commentary draws on personal recollections and insights accrued during pioneering research into COMT inhibition and from within a drug development programme that had an explicitly commercial focus. Like any such memoirs they are therefore partial, even perhaps partisan, and certainly selective as a chronology or survey of the field. They are, nevertheless, faithfully representative of the research journey as we experienced it. Readers seeking a comprehensive account of the primary science and clinical trials’ data that support the positioning and use of entacapone in PD are referred to the various original studies referenced in this paper and to recent expert commentaries, such as those of Jenner et al. [2].
What Does COMT Do?
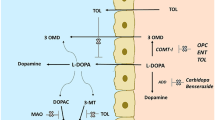
COMT mediates the O-methylation of a catechol substrate in positions 3 or 4, the former being the major route (Fig. 1a). Methylation takes place in the presence of S-adenosyl-methionine (a methyl-group donor) and ionic magnesium (Mg2+) as a cofactor and usually results in the complete loss of the pharmacological activity of the substrate molecule.

a The basic function of catechol O-methyltransferase (COMT) is the metabolism of catechol-containing molecules. The major route of metabolism is via O-methylation at position 3 of the catechol ring, with a minor pathway at position 4. S-Adenosyl-methionine (AdoMet) and ionic magnesium (Mg2+) are cofactors in the reaction. b The position of COMT in the pathways of peripheral levodopa (l-dopa), dopamine and noradrenaline metabolism. 3-OMD 3-ortho-methyl-dopa, AADC amino acid decarboxylase, D-β-H dopamine beta hydroxyl, MAO monoamine oxidase, HVA homovanillic acid, 3-MT 3-methyltyrosine, MHPG 3-methoxy-4-hydroxyphenylglycol, DHPG 3,4-dihydroxyphenylglycol, NMN normetanephrine, AdoHcy S-adenosyl-l-homocysteine, DOPAC 3,4-dihydroxyphenylacetic acid
COMT is indiscriminate in that it mediates the metabolism of any compounds containing a catechol moiety, including:
-
Catecholamines and levodopa (l-3,4-dihydroxyphenylalanine)
-
Dietary catechols, including flavonoids like quercetin and toxic polyphenols
-
Catechol-oestrogens (e.g. 2-hydroxyestrone)
-
Other drugs with a catechol structure, such as epinephrine and norepinephrine
It was understood from an early point that in this context “compounds containing a catechol moiety” included levodopa, though it was not initially anticipated that this would emerge as the principal target of our research. The ubiquitous position of COMT in the metabolic pathways of dopamine/levodopa/noradrenaline is summarised in Fig. 1b, though it must be stressed that for the most part COMT occupies a secondary position in those pathways. One anatomical exception to this is the medial prefrontal cortex of the brain, where COMT appears to be the pre-eminent catechol-metabolising enzyme. This creates a hypothetical potential for brain-specific COMT inhibition in aspects of cognition, memory and possibly schizophrenia; these subjects lie outside the scope of this discussion but published work gives a flavour of the possibilities in this area [5,6,7].
It should also be noted that COMT is central to the metabolism of catechol-oestrogens, raising the possibility of a role in hormonally influenced diseases, including breast cancer [8]. More generally, COMT inhibition may interfere with the metabolism of medicinal products containing a catechol group, implying that it may not be prudent to inhibit COMT activity completely.
COMT Inhibition and PD
COMT was first isolated and reported by Axelrod and Tomchick in 1958 [9], just months after Carlsson and colleagues [10, 11] had demonstrated the anti-PD effects of levodopa in rabbits, and thereby established the link between dopamine insufficiency and PD. Both of those discoveries may be seen as part of a wider decade-or-more-long exploration of catecholamine pharmacology and of PD, which culminated in the successful first use of levodopa to ameliorate symptoms of PD in human patients by Cotzias et al. in 1967.
As explained by Stanley Fahn [12] in his vigorous and exceptionally detailed account of those years, the seemingly (in retrospect) inevitable progress whereby levodopa became the core medical therapy for PD for more than 50 years was in fact anything but inevitable. Indeed, levodopa’s lengthy dominance seems surprising when one considers its limitations. These have been examined in detail in a recent expert review [13] and include prodrug status; poor oral absorption; short elimination half-life resulting in need for administration of several doses per day; poor penetration into the brain; and poor efficacy (with a resulting need for high doses) when given alone. Levodopa is also subject to extensive enzymic metabolism, mainly in the gut. It is thus no exaggeration to say that effective inhibition of that enzymic degradation has been central to levodopa’s success and its longevity in PD.
In particular, in the context of this review, it is instructive to note that several years before Cotzias et al. reported success with slow titration of levodopa to the high doses needed for therapeutic effect, McGeer and others in Vancouver [14] had tried a very similar approach with little or no success. In at least some of those unsuccessful attempts, levodopa had been co-administered with pyridoxine. As Fahn [12] notes, “pyridoxine would have speeded the conversion of the [levodopa]… to dopamine in the peripheral organs, with insufficient [levodopa] to get to high concentrations in the brain.” That sentence anticipates the rationale for COMT therapy in PD.
Figure 2 expands on that statement, illustrating that the delivery of levodopa in sufficient dosage to the dopamine-deficient brain of patients with PD is subject to disruption by two metabolic pathways, regulated respectively by dopamine decarboxylase (DDC) and COMT. In the context of PD, DDC inhibition was the first pathway to be addressed and DDC inhibition has been achieved to such an extent that levodopa plus a DDC inhibitor is now the standard starting point for levodopa therapy of PD. Used alone, however, DDC inhibition leads to increased formation, via COMT, of the slowly eliminated levodopa metabolite 3-ortho-methyl-dopa (3-OMD). Although this metabolite is considered inactive, its long half-life (> 10 h) and accumulation in response to extended levodopa/DDC inhibitor therapy may be undesirable [15]. (Aside from any inherent undesirability of 3-OMD accumulation, it may compete with levodopa for uptake into the brain [16].) Simultaneous COMT inhibition thus serves the twin purposes of further enhancing delivery of levodopa to the brain (beyond that achieved by DDC inhibition alone) and also preventing the accumulation of 3-OMD.

This figure first appeared in Trends in Pharmacological Sciences (TIPS) in 1989 as part of an article authored by Pekka Männistö and Seppo Kaakkola [16] making the case for the use of catechol O-methyltransferase (COMT) inhibition as adjunct therapy in Parkinson’s disease. The original figure legend stated: “The absorption, main metabolic routes and brain penetration of levodopa given orally alone (a), together with a peripheral dopa-decarboxylase inhibitor (DDI) (b), or with both a DDI and a COMT inhibitor (c). BBB, the blood–brain barrier.” DA dopamine, DDC-I dopa-decarboxylase inhibitor, L-dopa levodopa, 3OMD 3-ortho-methyl dopa
What We Learned About COMT
An important product of our research into COMT inhibition was the elucidation of the nature and location of COMT itself. COMT is not located in presynaptic neurones nor in the synaptic cleft (which is unfortunate, as it would be a more accessible target there). It is confined to postsynaptic neurones and affiliated glial cells, where it exists as two isoforms, both of which are strictly intracellular.
-
Soluble COMT (S-COMT) is a low-affinity, high-capacity isoform located in the cytoplasm and nucleus of cells in the intestines, liver and any tissue.
-
Membrane-bound COMT (MB-COMT) is a high-affinity, low-capacity isoform located throughout the body but also in the brain. It is bound only to the membranes of rough endoplasmic reticulum, not to external cell membranes.
All currently available COMT inhibitors inhibit both isoforms. It remains to be seen if selective inhibition of MB-COMT will open a new line of development for therapeutic agents in PD and other neurological or psychiatric conditions. (Or indeed if selective inhibition of S-COMT has clinical applications in other indications.) Progress in this area is hampered by the fact that the precise 3D molecular structure of MB-COMT has not been resolved. Some work towards identifying new COMT inhibitor candidates has been performed using the crystal structure of the S-COMT isoform but the potential limitations of this in the context of treatment of PD will be evident [17].
Development of Entacapone
Our initial experiments in COMT inhibition used the Upjohn compound U-0521 [18]. Effects were modest but sufficient as proof of concept to warrant further research. Taking advantage of Orion Pharma’s considerable strength in synthetic chemistry, an extensive range of candidate molecules was developed. Inter alia, this work established the importance of a nitrocatechol pharmacophore in an effective COMT inhibitor. Even the third marketed compound, opicapone, conforms to that requirement; in that sense, it is not accurate to characterise opicapone as a “third-generation” compound since it conforms to the chemical template of its predecessors. All nitrocatechols are tight-binding, slow-dissociating compounds, opicapone being extra-slow to dissociate. We took a scientific-strategy decision to publish the results of our research (after patenting) as a way of promoting external engagement and identifying commercial competition. Initial publications focused on candidate molecules such as nitecapone, the fate of which is discussed in our accompanying Supplementary Material [19,20,21].
Our foundational report on entacapone (originally known as OR-611) was published in 1992 and established the following features of the drug [22]:
-
Marked, profound and specific COMT inhibition in peripheral organs such as the duodenum and liver but limited inhibition in the striatum (Fig. 3). In this context, it must be noticed that entacapone is about 1000 times more potent than U-0521 as a COMT inhibitor (half-maximal inhibitory concentration [IC50] 0.01 μg vs. 6 μg. For other significant catecholamine-metabolising enzymes, including tyrosine hydroxylase, the IC50 of entacapone is at least 48 μg).
-
Clear-cut and dose-dependent reduction in 3-OMD levels.
-
Prolongation of plasma levels of levodopa above the threshold needed for therapeutic effect in PD but no augmentation of the levodopa effect and no inherent anti-parkinsonism activity.
-
No reduction in striatal levels of homovanillic acid, even at high dose (30 mg/kg in rats), signalling no inhibition of cerebral COMT.

This figure first appeared in Naunyn–Schmiedeberg Archives of Pharmacology in 1992 as part of an article authored by Erkki Nissinen, Inge-Britt Lindén, Eija Schultz and Pentti Pohto [22] reporting the biochemical and pharmacological properties of entacapone. The original figure legend stated: “Inhibition of catechol-O-methyltransferase activity in different rat tissues by entacapone 10 mg/kg p.o. Results shown are percentages (means ± SEM) of control values. ●, duodenum; ○, erythrocytes; ■, liver; ∇, striatum. The rats (n = 4–6) were killed at times up to 8 h after the treatment. Statistical significance: ***P < 0.001, **P < 0.01, *P < 0.05”. COMT catechol O-methyltransferase; p.o. per os, SEM standard error of the mean. Reproduced with permission
Additional preclinical investigations confirmed these core qualities of entacapone in PD [23], which were recorded at length and in detail in a portmanteau publication in 1999 [24] that continues to this day to be cited regularly in discussions of the field.
Current Research Landscape of Drug Discovery for PD
Research into PD therapies remains vigorous, with McFarthing et al. [25] reporting that new agents were being tested in 58 phase III trials in 2023. Most of the 139 trials identified are evaluating symptomatic treatments and 10 phase III studies are evaluating agents that either restore or replace dopamine or mimic its effects (mostly dopamine agonists [n = 6]). However, none of the phase III studies identified is evaluating a novel COMT inhibitor, even though the methods and resources now available to identify potential new candidate molecules are unrecognisably advanced from the era when our original research was conducted [26,27,28]. Whether this lack of evolution reflects the chilling effect of the tolcapone experience, a shift towards other targets in PD or the effect of conflicting priorities in drug development is hard to say with assurance. Certainly, preclinical research into COMT inhibition is continuing. At least one compound (ZIN27985035) [27] has emerged as a possible relatively selective inhibitor of MB-COMT, along with a novel chemical series (bicyclic hydropyridines) [29], the implied hope for which is that some of them may replicate or exceed the COMT inhibitory potency of tolcapone, without the associated hepatic toxicity. These and other ideas are, however, all at a very early stage of evaluation and may never be realised as viable pharmaceutical products.
Entacapone in 2024: How We Got Here
Given the paucity of the research pipeline described above, given also that the restrictions on tolcapone arising from its potential for hepatic toxicity relegate it to second-line status (notwithstanding its evident efficacy) and given that the depth and breadth of experience in clinical use substantially exceed those so far accrued with opicapone, it is our view that currently the foreseeable future of COMT inhibition in PD rests substantially with entacapone. For some time now, entacapone has overwhelmingly been used in the fixed-dose-ratio triple preparation of levodopa–carbidopa–entacapone branded as Stalevo®. This preparation, which presents levodopa in a dose range of 50–200 mg in a 4:1 ratio with carbidopa and a fixed dose of 200 mg of entacapone, is indicated on the basis of randomised controlled trials [30,31,32,33,34,35,36,37,38,39] for patients with idiopathic PD having end-of-dose motor fluctuations not stabilised on levodopa/DDC treatment to (i) substitute for immediate-release carbidopa/levodopa and entacapone previously administered as individual products or (ii) replace immediate-release carbidopa/levodopa therapy (without entacapone) in patients taking a total daily dose of levodopa of up to 800 mg and not experiencing dyskinesias.
Presentation in a fixed-dose combination ensures that entacapone is administered at the correct time point and at a dose optimal for the inhibition of COMT, regardless of the administered dose of levodopa. In addition to simplifying the dosing regimen, the pharmacokinetics of entacapone and levodopa are ‘time-locked’ such that maximal inhibition of COMT coincides with the levodopa plasma profile. Switching from dual therapy of levodopa plus a DDC inhibitor to Stalevo is advantageous for patients with wearing-off phenomena and meets with a high degree of patient acceptance [36, 37]. Use of Stalevo earlier in the progression of PD has been mooted but the available data are inconclusive (see Poewe [38] for a recent commentary on this possibility).
The pathway to the current status of Stalevo in PD is worth recording in more detail.
1998: Entacapone to Comtess®/Comtan® as a New Treatment for Patients with PD and Motor Fluctuations
Experience in phase I and phase II dose-finding studies identified 200 mg entacapone given concomitantly with levodopa–DDC inhibitor as the optimal dose to prolong the levodopa half-life significantly and thereby its therapeutic effect in PD, particularly reduced OFF-time. The 200-mg dose of entacapone (given concomitantly with each levodopa dose) was therefore used in two pivotal double-blind placebo-controlled efficacy studies. Clinical trial protocols for those studies were developed to follow the regulatory guidelines for the development of new therapies for PD published earlier by the US Food and Drug Administration (FDA). Professor Ariel Gordin played a critical role in the fulfilment of the clinical trials programme: it is not an exaggeration to say that, without his energy, extensive contacts with key experts and negotiating skills the whole enterprise might have foundered.
Results (based on patients’ home diaries) from both those studies (NOMECOMT in Nordic countries and SEESAW in the USA [30, 31]) confirmed that, compared to placebo, entacapone increased ON-time (i.e., the duration of levodopa’s clinical effect) to a statistically and clinically significant extent. Entacapone also reduced OFF-time, and relieved clinical disability (as assessed by the Unified Parkinson’s Disease Rating scale [UPDRS]), both in UPDRS-II (activities of daily living) and UPDRS-III (motor function). These improvements, together with positive global assessments both by physicians and patients themselves, indicated improvements in the quality of life achieved from the addition of entacapone.
The most frequently recorded adverse effect with entacapone in these studies was dyskinesia (25%, vs. 15% with placebo), a foreseeable consequence of the enhancement of levodopa effect that in current practice is usually manageable by reducing concomitant levodopa dose. Another side effect associated with entacapone was diarrhoea (10%, vs. 4%), though this was classified mostly as mild or moderate and rarely led to discontinuation in clinical studies.
Primarily on the basis of the outcomes of these pivotal studies, entacapone was approved to treat patients with PD and motor fluctuations first by the EMA in the European Union (1998), then by the FDA in 1999 and later in many other territories and jurisdictions. Entacapone was first launched under the trade name Comtess by Orion Pharma (in the Nordic countries and in some European countries, e.g. in Germany and the UK) and as Comtan (first in the USA and then in many other countries globally) by Novartis.
Other phase III and phase IIIb controlled studies were subsequently conducted using separate 200 mg entacapone tablets in patients with PD. Table 1, taken from a recent review of the trials programme [38], summarises the efficacy findings of pivotal controlled trials, while Table 2, from the same source, reminds us that there is never a free lunch in drug therapy and that entacapone, while regarded as a generally well-tolerated medication, is not free from adverse effects.
2003: Triple Combination (TC) to Stalevo
Given that well before the start of the twenty-first century it had become established good practice in many countries to present levodopa for PD in a combination tablet with a DDCI, it was a logical step in product development to combine entacapone with both those agents in a single-tablet TC. Carbidopa was selected as the DDCI for this purpose owing to its wide availability and use.
Guided by levodopa dosage data accrued from the pivotal studies mentioned above, three different strengths of TC were originally developed: 50/12.5/200, 100/25/200 and 150/37.5/200 (levodopa/carbidopa/entacapone; all in milligrams). Registration strategy and potential approvability based on bioequivalence were discussed with and endorsed by the EMA and FDA before clinical equivalence studies were conducted. These three strengths of TC were subsequently shown to be bioequivalent with corresponding doses of levodopa/carbidopa plus separate entacapone.
TC in all three strengths was duly approved by the EMA and FDA in 2003 and marketed under the trade name Stalevo by both Orion Pharma and Novartis. Further strengths of Stalevo were subsequently developed and approved (2006–9) to create the current seven-dose range. The four additional dose options are 75/18.75/200, 125/31.25/200, 175/43.75/200 and 200/50/200 (levodopa/carbidopa/entacapone; all in milligrams). The availability of Stalevo in this broad range of levodopa strengths offers flexibility for optimisation and tailoring of levodopa dosing schedules to the needs and circumstances of individual patients [39]. Entacapone (alone or in the Stalevo TC) has now been approved and introduced in clinical practice in over 120 countries worldwide.
A recent innovation in the use of entacapone is its incorporation into device-aided therapies for patients with advanced PD who can no longer be effectively controlled with optimised oral or transdermal medications. In particular, entacapone has been incorporated into levodopa–entacapone–carbidopa intestinal gel (LECIG) infusion. This is reported to be of equivalent therapeutic effectiveness to levodopa–carbidopa gel infusion (LCIG), with the attraction for patients that the external drug-delivery device is smaller for LECIG than for LCIG [40, 41]. Rates of early discontinuation of LECIG are higher than with LCIG for reasons that have not yet been elucidated. The published dataset is small, however, and there are no randomised controlled trials either of LECIG versus LCIG or of oral therapies, including COMT inhibitor, versus device-aided drug delivery. The safety of combined administration of entacapone and apomorphine in levodopa-treated patients with PD has been affirmed by Zijlmans and colleagues [42] in a multicentre, double-blind, placebo-controlled, cross-over study but comparisons of entacapone and apomorphine have not been reported and no ongoing trials in that area are currently registered at www.clinicaltrials.gov.
Importance of Preclinical Research
The current lack of new COMT inhibitors in clinical trials directs attention towards the position of preclinical evaluations in drug development. As befits a drug that was first in its class, entacapone was subjected to extensive investigations designed to answer the question “Is it safe to inhibit COMT effectively?” Given that COMT is a ubiquitous enzyme that metabolises all catecholamines, both dietary and medicinal, and given that endogenous catecholamines (e.g. dopamine, noradrenaline, adrenaline) are important both in periphery and centrally, there were questions around the potential of COMT inhibition to create risks for high blood pressure or arrhythmias; the possibility of an exaggerated tyramine (cheese) reaction; potential for interactions with monoamine oxidase inhibitors and other uptake inhibitors; and no information about any possible impact on catechol-oestrogens.
In the interests of brevity, and bearing in mind that entacapone is now in its third decade of clinical use, we summarise these matters by stating that all these anxieties and others were allayed [43,44,45,46,47]. Indeed, the published repertoire of preclinical investigations performed on entacapone provides the foundation for the COMT inhibitor class as a whole—so much so that claims for tolcapone and opicapone not infrequently refer to entacapone data. Assumptions of ‘class-effect’ pharmacology usually have limitations, however, and the history of tolcapone may be seen as an illustration of the potential pitfalls of over-extrapolation between drugs. We found no indication of liver toxicity in animal studies of entacapone. As part of our research into that matter, however, we identified that, whereas entacapone had no adverse impact on cardiac or hepatic mitochondrial energy production at clinically relevant doses, tolcapone certainly did [48, 49]. Mitochondrial damage was subsequently reported in the cases of tolcapone-induced fulminant hepatitis that led to that drug being withdrawn from the European Union in 1998 and restricted in other markets [50,51,52]. (Inter alia, and in the interests of completeness, we at this point very briefly acknowledge nebicapone, a long-acting inhibitor of peripheral COMT initially reckoned to be a very promising lead in this arena but one that eventually foundered in much the same way as tolcapone and for much the same reasons [53, 54]. [Opicapone is a later product of the same research programme.].)
Other preclinical data comparing entacapone and opicapone highlight differences that may be relevant to the treatment of PD. Specifically, administration of opicapone to rats treated with levodopa (12 mg/kg) and benserazide (3 mg/kg) produces a very marked spike in plasma levodopa levels 1–2 h after dosing but no substantial ‘plateau’ of moderately elevated levodopa (Fig. 4) [55]. Moreover, 7–12 h elapse before statistically significantly elevated levels of levodopa or dopamine are recorded in the brain, compared with an interval of 1–2 h for entacapone or tolcapone [55]. This profile of marked and rapid but short-lived elevation of plasma levodopa and long-delayed increase in brain levels of dopamine differs substantially from the prompt, moderate and sustained increase in levodopa and brain dopamine levels achieved with entacapone [56] and prima facie is not optimal for the treatment of PD motor symptoms. In addition, the effect of long-term suppression of a ubiquitous house-keeping enzyme such as COMT is not known. Possible effects on oestrogen metabolism [57] or glucose homeostasis [58] are among the matters to be considered.

This figure first appeared in the British Journal of Pharmacology in 2015 as part of an article authored by MJ Bonifácio and colleagues [55] reporting the pharmacological profile of opicapone. The original figure legend stated: “Levels of levodopa… in the bloodstream evaluated by means of microdialysis in the right jugular vein of rats administered 12 mg/kg levodopa and 3 mg/kg benserazide p.o., on 3 consecutive days. On day 1 (vehicle), rats received vehicle 2 h before levodopa/benserazide. On day 2 (opicapone), rats received opicapone 2 h prior to levodopa/benserazide. On day 3 (24 h post opicapone), rats that were treated with opicapone 24 h earlier, received vehicle 2 h before levodopa/benserazide. Dialysate samples were collected over 20 min periods starting at 150 min before levodopa/benserazide dosing until 310 min post-dose. Values are presented as mean ± SEM (n = 5–6) and were subtracted from their corresponding baseline values, calculated as the means of the samples collected before each levodopa/benserazide administration (from − 150 to − 10 min), to minimise inter day variability.” p.o. per os, SEM standard error of the mean. Reproduced with permission
In this context, it has to be noticed that observations from the open-label NEWSTA study in healthy volunteers illustrate how entacapone enables levodopa dosage to be refined in PD. Levodopa peak plasma concentration was not increased when the Stalevo dose was reduced by 25 mg after the first morning dose, but minimum levodopa levels and the area under the curve between 0 and 14 h were significantly higher than for levodopa–carbidopa dual therapy, and peak-to-trough fluctuations in levodopa levels were attenuated [56]. A pragmatic interpretation of these data is that patients’ first daily dose should be large enough to address early-day akinesia; lower doses may then be used through the rest of the day to mitigate the risks of dyskinesia.
The lessons from these observations are that COMT inhibitors are not the same or seamlessly interchangeable merely for being part of the same drug class and differences between drugs may have real-world implications. Inter-drug differences seen in preclinical evaluations may have consequences for clinical effectiveness and safety that may take some time to be fully appreciated and it is imprudent to assume that all such differences signify nothing important. Table 3 summarises some contrasts between entacapone and opicapone that illustrate this principle. A possibly important difference between the effects of opicapone and the other clinically available COMT inhibitors was signalled in a very recent report on the depressogenic effects of drugs through the World Health Organization pharmacovigilance database [59]. Whereas entacapone and tolcapone both registered a moderate value for the reporting odds ratio (ROR; 2.73 and 3.96, respectively), opicapone was at the top of the list of potentially depressogenic drugs, with a ROR value of 20.66 (95% confidence interval 15.62–27.33, with 56 cases reported). It remains to be demonstrated if this is due to a pharmacodynamic or pharmacokinetic characteristic of COMT inhibition by opicapone or whether it is a quality of the drug unrelated to COMT inhibition.
Conclusion
We started this account by saying that our position on COMT inhibitors is partial, even partisan, and perhaps that should be registered as a limitation of this article. The research programme that resulted in the discovery and development of entacapone and Stalevo was a major episode in the professional lives of several of us and set the stage for careers in pharmaceutical and biomedical research. It also helped to pay our living costs—so we have skin in the game. We trust, nevertheless, that in our recollections we have been faithful to the facts and that we have identified even-handedly some of the achievements and disappointments that attended this era of research.
Bringing a molecule such as entacapone through from discovery to regulatory approval and commercialisation is a very significant achievement for an organisation of any size, arguably even more so for one operating at the scale of Orion Pharma in the 1980s. Progress, as is so often the case in drug development, was far from linear and success by no means inevitable, as the fates of nitecapone, nebicapone and tolcapone, all summarised in this review, eloquently attest. But succeed entacapone certainly did and continues to do, nowadays most usually in the Stalevo formulation. In 2018, Orion Pharma resumed the sales and distribution rights to Stalevo for most of the countries in the European Union, a commercial arrangement that has helped to consolidate central nervous system disorders as one of the company’s central areas of expertise. In 2024, Orion Pharma will also take over sales in Japan, currently the most important individual national market for entacapone.
Clinically, the impact of entacapone on the management of PD has been very great. The ability to enhance, refine and augment the effects of levodopa will have delivered day-to-day benefits to many tens (and by now probably hundreds) of thousands of patients. The impact on individuals is of course impossible to assert with exactness but the aggregate effects demonstrated in controlled clinical trials are compelling. Can it be argued that successful implementation of COMT inhibition has hampered research into an effective alternative to levodopa? Almost certainly not. The limitations of levodopa were apparent even as (and arguably even before) it emerged as the central medical therapy for PD and there has been no lack of effort in looking for agents that might replace it. Nothing has so far emerged to challenge its pre-eminence. COMT inhibition has helped us to make the best of what we have and will continue to do so until something better than levodopa comes along: but we may be in for a long wait and while we wait the Stalevo formulation provides physicians and patients with an effective, versatile and easy-to-use medication to optimise control of PD symptoms.
We may also have a long wait to see a step-change successor to entacapone. There appear at the moment to be no decisive new advances in COMT inhibitor development. Given the time from bench to bedside is now routinely well in excess of a decade (and perhaps as long as two decades) and in the absence of any convincing alternative to levodopa, entacapone, already in its third decade of use, may be in service for the management of PD for many years yet. A recent network meta-analysis on the efficacy and safety of adjunctive drugs to levodopa for fluctuating PD showed how entacapone is among the most studied treatments, with an overall good efficacy and safety pattern when compared to the other adjunctive drugs [60].
The choice of a first-line COMT inhibitor rests at present substantially between entacapone and opicapone, and—in our opinion—the advantage is with the former. Firstly, weight of time and experience is firmly on the side of entacapone. Secondly, the suggestion that single daily dosing with opicapone offers improved convenience and simplicity takes no account of the availability of Stalevo in an extensive dose range. Moreover, the same pharmacokinetics that make once-daily opicapone dosing a practical proposition may have implications for the sustained inhibition of peripheral COMT that in our opinion warrant a continuing precautionary attitude. Thirdly, pricing in many territories makes a persuasive case for entacapone/Stalevo. The suggestion that “opicapone may be an option to consider when entacapone is not tolerated or is inadequate at controlling symptoms” [61] was made when opicapone first became available in the UK in 2017 but remains in our view a broadly correct appraisal of the relative position of these two drugs.
Data Availability
All data discussed in this review are in the public domain.
References
Kaakkola S, Teräväinen H, Ahtila S, Rita H, Gordin A. Effect of entacapone, a COMT inhibitor, on clinical disability and levodopa metabolism in parkinsonian patients. Neurology. 1994;44(1):77–80. https://doi.org/10.1212/wnl.44.1.77.
Jenner P, Poewe W, Reichmann H. The 20th anniversary of the first clinical use of Stalevo®. Eur J Neurol. 2023;30(Suppl 2):1–2. https://doi.org/10.1111/ene.15996.
Summary of the European public assessment report (EPAR) on Comtan (entacapone p.o.) https://www.ema.europa.eu/en/medicines/human/EPAR/comtan. Accessed 3 April 2024.
Marsili L, Marconi R, Colosimo C. Treatment strategies in early Parkinson’s disease. Int Rev Neurobiol. 2017;132:345–60. https://doi.org/10.1016/bs.irn.2017.01.002.
Louis CC, D’Esposito M, Moser JS. Investigating interactive effects of worry and the catechol-O-methyltransferase gene (COMT) on working memory performance. Cogn Affect Behav Neurosci. 2021;21(6):1153–63. https://doi.org/10.3758/s13415-021-00922-9.
Dauvilliers Y, Tafti M, Landolt HP. Catechol-O-methyltransferase, dopamine, and sleep-wake regulation. Sleep Med Rev. 2015;22:47–53. https://doi.org/10.1016/j.smrv.2014.10.006.
Nogueira NGHM, Bacelar MFB, Ferreira BP, Parma JO, Lage GM. Association between the catechol-O-methyltransferase (COMT) Val158Met polymorphism and motor behavior in healthy adults: a study review. Brain Res Bull. 2019;144:223–32. https://doi.org/10.1016/j.brainresbull.2018.11.002.
Tolba MF, Omar HA, Hersi F, Nunes ACF, Noreddin AM. The impact of catechol-O-methyl transferase knockdown on the cell proliferation of hormone-responsive cancers. Mol Cell Endocrinol. 2019;15(488):79–88. https://doi.org/10.1016/j.mce.2019.03.007.
Axelrod J, Tomchick R. Enzymatic O-methylation of epinephrine and other catechols. J Biol Chem. 1958;233(3):702–5.
Carlsson A. The occurrence, distribution and physiological role of catecholamines in the nervous system. Pharmacol Rev. 1959;11:490–3.
Carlsson A, Lindqvist M, Magnusson T. 3,4-Dihydroxyphenylalanine and 5-hydroxytryptophan as reserpine antagonists. Nature. 1957;180:1200.
Fahn S. The medical treatment of Parkinson disease from James Parkinson to George Cotzias. Mov Disord. 2015;30(1):4–18. https://doi.org/10.1002/mds.26102.
Jenner P. Stalevo®: a pioneering treatment for OFF periods in Parkinsons disease. Eur J Neurol. 2023;30(Suppl 2):3–8. https://doi.org/10.1111/ene.15994.
McGeer PL, Zeldowicz LR. Administration of dihydroxyphenylalanine to parkinsonian patients. Can Med Assoc J. 1964;90:463–6.
Da Prada M, Kettler R, Zürcher G, Schaffner R, Haefely WE. Inhibition of decarboxylase and levels of dopa and 3-O-methyldopa: a comparative study of benserazide versus carbidopa in rodents and of Madopar standard versus Madopar HBS in volunteers. Eur Neurol. 1987;27(Suppl 1):9–20. https://doi.org/10.1159/000116170.
Männistö PT, Kaakkola S. New selective COMT inhibitors: useful adjuncts for Parkinson’s disease? Trends Pharmacol Sci. 1989;10(2):54–6. https://doi.org/10.1016/0165-6147(89)90075-8.
Czarnota S, Johannissen LO, Baxter NJ, et al. Equatorial active site compaction and electrostatic reorganization in catechol-O-methyltransferase. ACS Catal. 2019;9:4394–401. https://doi.org/10.1021/acscatal.9b00174.
Nuutila J, Kaakkola S, Männistö PT. Potentiation of central effects of L-dopa by an inhibitor of catechol-O-methyltransferase. J Neural Transm. 1987;70(3–4):233–40. https://doi.org/10.1007/BF01253600.
Männistö PT, Kaakkola S, Nissinen E, Linden IB, Pohto P. Properties of novel effective and highly selective inhibitors of catechol-O-methyltransferase. Life Sci. 1988;43(18):1465–71. https://doi.org/10.1016/0024-3205(88)90258-5.
Nissinen E, Lindén IB, Schultz E, Kaakkola S, Männistö PT, Pohto P. Inhibition of catechol-O-methyltransferase activity by two novel disubstituted catechols in the rat. Eur J Pharmacol. 1988;153(2–3):263–9. https://doi.org/10.1016/0014-2999(88)90614-0. Erratum. In: Eur J Pharmacol. 1988 Nov 22;157(2–3):244
Lindén IB, Nissinen E, Etemadzadeh E, Kaakkola S, Männistö P, Pohto P. Favorable effect of catechol-O-methyltransferase inhibition by OR-462 in experimental models of Parkinson’s disease. J Pharmacol Exp Ther. 1988;247(1):289–93.
Nissinen E, Lindén IB, Schultz E, Pohto P. Biochemical and pharmacological properties of a peripherally acting catechol-O-methyltransferase inhibitor entacapone. Naunyn Schmiedebergs Arch Pharmacol. 1992;346(3):262–6. https://doi.org/10.1007/BF00173538.
Smith LA, Gordin A, Jenner P, Marsden CD. Entacapone enhances levodopa-induced reversal of motor disability in MPTP-treated common marmosets. Mov Disord. 1997;12(6):935–45. https://doi.org/10.1002/mds.870120616.
Männistö PT, Kaakkola S. Catechol-O-methyltransferase (COMT): biochemistry, molecular biology, pharmacology, and clinical efficacy of the new selective COMT inhibitors. Pharmacol Rev. 1999;51(4):593–628.
McFarthing K, Buff S, Rafaloff G, et al. Parkinson’s disease drug therapies in the clinical trial pipeline: 2023 update. J Parkinsons Dis. 2023;13(4):427–39. https://doi.org/10.3233/JPD-239901.
Roy RK, Patra N. Prediction of COMT inhibitors using machine learning and molecular dynamics methods. J Phys Chem B. 2022;126(19):3477–92. https://doi.org/10.1021/acs.jpcb.1c10278.
Subair TO, Akawa OB, Soremekun OS, Olotu FA, Soliman MES. Insight into the therapeutic potential of a bicyclic hydroxypyridone compound 2-[(2,4-dichlorophenyl)methyl]-7-hydroxy-1,2,3,4-tetrahydro-8H-pyrido[1,2-a]pyrazin-8-one as COMT inhibitor in the treatment of parkinson’s disease: a molecular dynamic simulation approach. Chem Biodivers. 2021;18(9):e2100204. https://doi.org/10.1002/cbdv.202100204.
Cruz-Vicente P, Passarinha LA, Silvestre S, Gallardo E. Recent developments in new therapeutic agents against Alzheimer and Parkinson diseases: in-silico approaches. Molecules. 2021;26(8):2193. https://doi.org/10.3390/molecules26082193.
Cruz-Vicente P, Gonçalves AM, Ferreira O, et al. Discovery of small molecules as membrane-bound catechol-O-methyltransferase inhibitors with interest in Parkinson’s disease: pharmacophore modeling, molecular docking and in vitro experimental validation studies. Pharmaceuticals (Basel). 2021;15(1):51. https://doi.org/10.3390/ph15010051.
Parkinson Study Group. Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Ann Neurol. 1997;42:747–55.
Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. NOMECOMT Study Group. Neurology. 1998;51:1309–14. https://doi.org/10.1212/wnl.51.5.1309.
Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M, CELOMEN Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with suboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (CELOMEN study). Acta Neurol Scand. 2002;105:245–55. https://doi.org/10.1034/j.1600-0404.2002.1o174.x.
Brooks DJ, Sagar H, UK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind, six month study. J Neurol Neurosurg Psychiatry. 2003;74:1071–9. https://doi.org/10.1136/jnnp.74.8.1071.
Myllylä VV, Kultalahti ER, Haapaniemi H, Leinonen M, FILOMEN Study Group. Twelve-month safety of entacapone in patients with Parkinson’s disease. Eur J Neurol. 2001;8:53–60. https://doi.org/10.1046/j.1468-1331.2001.00168.
Mizuno Y, Kanazawa I, Kuno S, Yanagisawa N, Yamamoto M, Kondo T. Placebo-controlled, double-blind, dose-finding study of entacapone in fluctuating parkinsonian patients. Mov Disord. 2007;22:75–80. https://doi.org/10.1002/mds.21218.
Eggert K, Skogar O, Amar K, et al. Direct switch from levodopa/benserazide or levodopa/carbidopa to levodopa/carbidopa/entacapone in Parkinson’s disease patients with wearing-off: efficacy, safety and feasibility—an open-label, 6-week study. J Neural Transm. 2010;117:333–42. https://doi.org/10.1007/s00702-009-0344-4.
Myllylä V, Haapaniemi T, Kaakkola S, et al. Patient satisfaction with switching to Stalevo: an open-label evaluation in PD patients. Acta Neurol Scand. 2006;114:181–6. https://doi.org/10.1111/j.1600-0404.2006.00703.x.
Poewe W. Catechol-O-methyltransferase inhibition with entacapone: evidence from controlled clinical trials in Parkinson’s disease. Eur J Neurol. 2023;30(Suppl 2):9–14. https://doi.org/10.1111/ene.15993.
Reichmann H. Real-world considerations regarding the use of the combination of levodopa, carbidopa, and entacapone (Stalevo®) in Parkinson’s disease. Eur J Neurol. 2023;30(Suppl 2):15–20. https://doi.org/10.1111/ene.15992.
Nyholm D, Jost WH. Levodopa-entacapone-carbidopa intestinal gel infusion in advanced Parkinson’s disease: real-world experience and practical guidance. Ther Adv Neurol Disord. 2022;26(15):17562864221108018. https://doi.org/10.1177/17562864221108018.
Viljaharju V, Mertsalmi T, Pauls KAM, et al. Levodopa-entacapone-carbidopa intestinal gel treatment in advanced parkinson’s disease: a single-center study of 30 patients. Mov Disord Clin Pract. 2024;11(2):159–65. https://doi.org/10.1002/mdc3.13926.
Zijlmans JC, Debilly B, Rascol O, Lees AJ, Durif F. Safety of entacapone and apomorphine coadministration in levodopa-treated Parkinson’s disease patients: pharmacokinetic and pharmacodynamic results of a multicenter, double-blind, placebo-controlled, cross-over study. Mov Disord. 2004;19(9):1006–11. https://doi.org/10.1002/mds.20188.
Illi A, Sundberg S, Koulu M, Scheinin M, Heinävaara S, Gordin A. COMT inhibition by high-dose entacapone does not affect hemodynamics but changes catecholamine metabolism in healthy volunteers at rest and during exercise. Int J Clin Pharmacol Ther. 1994;32(11):582–8.
Illi A, Sundberg S, Ojala-Karlsson P, Scheinin M, Gordin A. Simultaneous inhibition of catecholamine-O-methylation by entacapone and neuronal uptake by imipramine: lack of interactions. Eur J Clin Pharmacol. 1996;51(3–4):273–6. https://doi.org/10.1007/s002280050197.
Illi A, Sundberg S, Ojala-Karlsson P, Korhonen P, Scheinin M, Gordin A. The effect of entacapone on the disposition and hemodynamic effects of intravenous isoproterenol and epinephrine. Clin Pharmacol Ther. 1995;58(2):221–7. https://doi.org/10.1016/0009-9236(95)90200-7.
Illi A, Sundberg S, Ojala-Karlsson P, Scheinin M, Gordin A. Simultaneous inhibition of catechol-O-methyltransferase and monoamine oxidase A: effects on hemodynamics and catecholamine metabolism in healthy volunteers. Clin Pharmacol Ther. 1996;59(4):450–7. https://doi.org/10.1016/S0009-9236(96)90115-0.
Sundberg S, Scheinin M, Illi A, Akkila J, Gordin A, Keränen T. The effects of the COMT inhibitor entacapone on haemodynamics and peripheral catecholamine metabolism during exercise. Br J Clin Pharmacol. 1993;36(5):451–6. https://doi.org/10.1111/j.1365-2125.1993.tb00394.x.
Nissinen E, Kaheinen P, Penttilä KE, Kaivola J, Lindén IB. Entacapone, a novel catechol-O-methyltransferase inhibitor for Parkinson’s disease, does not impair mitochondrial energy production. Eur J Pharmacol. 1997;340(2–3):287–94. https://doi.org/10.1016/s0014-2999(97)01431-3.
Haasio K, Nissinen E, Sopanen L, Heinonen EH. Different toxicological profile of two COMT inhibitors in vivo: the role of uncoupling effects. J Neural Transm (Vienna). 2002;109(11):1391–401. https://doi.org/10.1007/s00702-002-0748-x.
Spahr L, Rubbia-Brandt L, Burkhard PR, Assal F, Hadengue A. Tolcapone-related fulminant hepatitis: electron microscopy shows mitochondrial alterations. Dig Dis Sci. 2000;45(9):1881–4. https://doi.org/10.1023/a:1005549304404.
Borges N. Tolcapone in Parkinson’s disease: liver toxicity and clinical efficacy. Expert Opin Drug Saf. 2005;4(1):69–73. https://doi.org/10.1517/14740338.4.1.69.
Olanow CW, Watkins PB. Tolcapone: an efficacy and safety review. Clin Neuropharmacol. 2007;30:287–94. https://doi.org/10.1097/wnf.0b013e318038d2b6.
Ferreira JJ, Rascol O, Poewe W, et al. A double-blind, randomized, placebo and active-controlled study of nebicapone for the treatment of motor fluctuations in Parkinson’s disease. CNS Neurosci Ther. 2010;16(6):337–47. https://doi.org/10.1111/j.1755-5949.2010.00145.x.
Kaakkola S. Problems with the present inhibitors and a relevance of new and improved COMT inhibitors in Parkinson’s disease. Int Rev Neurobiol. 2010;95:207–25. https://doi.org/10.1016/B978-0-12-381326-8.00009-0.
Bonifácio MJ, Torrão L, Loureiro AI, Palma PN, Wright LC, Soares-da-Silva P. Pharmacological profile of opicapone, a third-generation nitrocatechol catechol-O-methyl transferase inhibitor, in the rat. Br J Pharmacol. 2015;172(7):1739–52. https://doi.org/10.1111/bph.13020.
Ingman K, Naukkarinen T, Vahteristo M, Korpela I, Kuoppamäki M, Ellmén J. The effect of different dosing regimens of levodopa/carbidopa/entacapone on plasma levodopa concentrations. Eur J Clin Pharmacol. 2012;68(3):281–9. https://doi.org/10.1007/s00228-011-1121-5.
Zahid M, Saeed M, Lu F, Gaikwad N, Rogan E, Cavalieri E. Inhibition of catechol-O-methyltransferase increases estrogen-DNA adduct formation. Free Radic Biol Med. 2007;43(11):1534–40. https://doi.org/10.1016/j.freeradbiomed.2007.08.005.
Kanasaki M, Srivastava SP, Yang F, et al. Deficiency in catechol-O-methyltransferase is linked to a disruption of glucose homeostasis in mice. Sci Rep. 2017;7(1):7927. https://doi.org/10.1038/s41598-017-08513-w.
Li J, Zelmat Y, Storck W, et al. Drug-induced depressive symptoms: an update through the WHO pharmacovigilance database. J Affect Disord. 2024;350:452–67. https://doi.org/10.1016/j.jad.2024.01.119.
Sako W, Kogo Y, Koebis M, et al. Comparative efficacy and safety of adjunctive drugs to levodopa for fluctuating Parkinson’s disease - network meta-analysis. NPJ Parkinsons Dis. 2023;9(1):143. https://doi.org/10.1038/s41531-023-00589-8.
NICE Guidance. Parkinson’s disease with end-of-dose motor fluctuations: opicapone. Evidence summary [ES9], Published: 21 March 2017. https://www.nice.org.uk/advice/es9/chapter/estimated-impact-for-the-NHS#other-treatments. Accessed 24 Apr 2024.
Acknowledgements
Medical Writing and Editorial Assistance
The authors thank Peter Hughes (Hughes associates, Oxford, UK) for editorial assistance with the text, and Shrestha Roy (Orion Pharma, Espoo, Finland) for graphical assistance with the figures. No form of artificial intelligence was used in the preparation of the text. Medical writing and editorial assistance were funded by Orion Pharma, Espoo, Finland.
Funding
The Rapid Service Fee, Open Access fee, editorial assistance and other costs relating to the preparation and publication of this work were funded by Orion Pharma, Espoo, Finland.
Author information
Authors and Affiliations
Contributions
Pekka T. Mannisto, Tapani Keranen, Kari J. Reinikainen, Anna Hanttu and Piero Pollesello contributed equally to the development of the manuscript, including concept and design, drafting the manuscript and compiling figures and tables. Piero Pollesello was responsible for coordinating the contributions, finalising figures and tables and correspondence with the journal.
Corresponding author
Ethics declarations
Conflict of Interest
Pekka T. Mannisto, Tapani Keranen and Kari J. Reinikainen have been full- or part-time employees of Orion Pharma and worked on the discovery and development of entacapone and/or Stalevo. In the past 5 years all three have received honoraria from Orion Pharma for educational and promotional consultancies. Anna Hanttu and Piero Pollesello are current full-time employees of Orion Pharma. None of the co-authors have other financial interests in Orion Pharma.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Männistö, P.T., Keränen, T., Reinikainen, K.J. et al. The Catechol O-Methyltransferase Inhibitor Entacapone in the Treatment of Parkinson’s Disease: Personal Reflections on a First-in-Class Drug Development Programme 40 Years On. Neurol Ther (2024). https://doi.org/10.1007/s40120-024-00629-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40120-024-00629-2