
Abstract
Progressive supranuclear palsy (PSP) is a neurodegenerative disorder resulting from the deposition of misfolded and neurotoxic forms of tau protein in specific areas of the midbrain, basal ganglia, and cortex. It is one of the most representative forms of tauopathy. PSP presents in several different phenotypic variations and is often accompanied by the development of concurrent neurodegenerative disorders. PSP is universally fatal, and effective disease-modifying therapies for PSP have not yet been identified. Several tau-targeting treatment modalities, including vaccines, monoclonal antibodies, and microtubule-stabilizing agents, have been investigated and have had no efficacy. The need to treat PSP and other tauopathies is critical, and many clinical trials investigating tau-targeted treatments are underway. In this review, the PubMed database was queried to collect information about preclinical and clinical research on PSP treatment. Additionally, the US National Library of Medicine’s ClinicalTrials.gov website was queried to identify past and ongoing clinical trials relevant to PSP treatment. This narrative review summarizes our findings regarding these reports, which include potential disease-modifying drug trials, modifiable risk factor management, and symptom treatments.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Progressive supranuclear palsy (PSP) is a fatal tau-based neurodegenerative disease. |
The need for PSP therapy becomes more apparent as knowledge advances regarding its phenotypic variations. |
No tau-targeting therapies have been effective in modifying the course of PSP. |
Several tau-targeting therapies are in clinical trials. |
The discovery of effective tau-modifying pharmacotherapies could significantly impact the prognosis of patients with PSP and other neurodegenerative disorders such as Alzheimer’s and Parkinson’s diseases and corticobasal degeneration. |
Introduction
Progressive supranuclear palsy (PSP) is an adult-onset tauopathy and neurodegenerative disease that is growing in recognition and prevalence [1]. PSP is currently incurable [2]. Therefore, the development of PSP therapies is a highly relevant area of research, and this review summarizes the therapies that have been studied and those that are currently undergoing clinical trials for the treatment of PSP.
The prevalence of PSP has been studied over the years, and various levels of prevalence have been reported [1, 3, 4]. In a European age-adjusted study conducted between 2003 and 2012, the prevalence was 8.8 to 10.8 cases per 100,000 persons. One study in 1999 in Yonago, Japan, reported a prevalence of 5.8 cases per 100,000 persons [5]. A later study in the same city in 2010 reported a much-increased prevalence of 17 cases per 100,000 persons [1]. In 2001, estimates from the UK revealed a crude prevalence of 6.5 cases per 100,000 persons [6]. In a more recent study in Scotland in 2018, PSP had a regional crude point prevalence of 4.28 cases per 100,000 [7]. According to Zermansky and Ben-Shlomo, the prevalence of PSP varied from 0.97 to 6.54 cases per 100,000 persons in the eight studies they reviewed [8]. Although the exact numbers may vary, the increase in prevalence and recognition of this currently fatal disease makes further research into PSP therapies all the more pertinent.
Genetics and Biology of Tau/PSP
PSP is a neurodegenerative disease, and the initial degeneration process is unclear [2]. PSP has been identified as a tauopathy, a category including conditions such as corticobasal degeneration (CBD), Alzheimer’s disease (AD), and frontotemporal dementia (FTD) [2]. Tauopathies are a family of diseases that result from the aggregation of dysfunctional, misfolded tau proteins. These diseases often result in neuronal loss, gliosis, and deposition of this misfolded tau protein in affected brain areas [2]. Tau is a microtubule-associated protein that contributes to the stability of the cytoskeleton and is found predominantly in axons [1, 9]. It is the most abundant microtubule-associated protein in the brain, and its dysfunction can be detrimental [2]. As new tauopathies continue to be identified, the critical role of tau function in the brain becomes even clearer [2].
PSP is characterized by the aggregation of misfolded tau proteins into soluble and insoluble forms, which result in microtubule destabilization and neurofibrillary tangles. These neurofibrillary tangles can be neurotoxic and lead to cell death [2]. The pathological forms of tau are thought to result from an uneven ratio of tau isoforms [2]. The MAPT gene on chromosome 17q21 codes for the tau protein, and alternative splicing of this gene results in the production of six different isoforms of the tau protein [10]. These tau isoforms can form aggregates of 3-repeat (3R) or 4-repeat (4R) microtubule-binding domain repeats, which are determined by the presence or exclusion of exon 10 [10, 11]. In the healthy human brain, there are similar levels of 3R and 4R isoforms of tau protein [2]. In tauopathies such as PSP, uneven ratios of these isoforms can lead to tau aggregation into soluble and insoluble forms, which result in microtubule destabilization, the formation of neurofibrillary tangles, and the subsequent neuronal degeneration seen in these conditions [2]. PSP and CBD are 4R tauopathies in which the filaments are arranged in straight, tubular formations [1]. CBD and PSP differ in certain aspects, including the biochemical fingerprints of tau fragments in each condition, clinical features, and neuropathological features [12]. In AD, the 4R tau protein often aggregates in paired helical filaments [2]. In contrast, multiple systems atrophy (MSA) is an atypical parkinsonian disorder that involves accumulation and aggregation of α-synuclein, mostly in the cytoplasm of oligodendrocytes. MSA is characterized by classic parkinsonian motor symptoms, dysautonomia, and cerebellar ataxia, with rapid progression and low life expectancy [13, 14].
As more research is conducted regarding PSP genetics, the importance of the MAPT gene has become more apparent. A recent study reported that differential splicing of the MAPT exons between tau-based diseases such as AD and PSP, as well as differential expression of MAPT isoforms that result from various combinations of exon 2 and 10 splicing, might contribute to the course of these neurodegenerative diseases [10]. Additionally, the presence of splicing factors, such as RSC1, affects exon 2 and 10 splicing, and these factors are differentially expressed in PSP and AD. The MAPT gene is highly significant in the pathogenesis of PSP and other neurodegenerative tau-based diseases [10].
Fifteen different mutations in MAPT are noted in PSP cases, and patients with PSP with MAPT mutations have an earlier age of onset than those without mutations [15]. Additionally, patients with MAPT mutations often have a positive family history of PSP [15]. Mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are also being studied, with variable results [15]. LRRK2 mutations are some of the most common genetic causes of Parkinson’s disease (PD), and a recent study showed that common variation at the LRRK2 locus is indicative of PSP severity, suggesting that LRRK2 inhibition might serve as a therapeutic approach in patients with tauopathies [15, 16]. Patients with the LRRK2 mutation had a much older age of onset than those with MAPT mutations, with a primary clinical presentation of parkinsonism [15].
The first large genome-wide association study in patients with PSP also identified several new potential genetic associations [17, 18]. Three new genetic risk factors were identified, namely STX6, EIF2AK3, and MOBP, which are suspected to be involved in tau production, processing, and trafficking [15]. A more recent genome-wide association study identified several more possible risk loci, including SLCO1A2, DUSP10, and RUNX2 [2, 15, 19].
It is postulated that posttranslational modifications of tau protein can increase the likelihood of protein aggregation or interaction with microtubules, leading to these tauopathies [1]. In PSP, the pathological tau protein is often phosphorylated [1, 20, 21], acetylated [1, 22], and thioflavin-S positive [1, 23]. The phosphorylation of tau protein has been thoroughly studied [24]. Patients with these tauopathies have higher levels of phosphorylated tau in their brains, and the specific phosphorylated tau profiles differ among specific tauopathies [25]. Additionally, the potential disease-inducing effects of other posttranslational modifications are under study, including acetylation, OGlcNacylation, and N-glycosylation, as well as other mechanisms [2, 24].
Tau has been found to have prion-like properties, which enable it to propagate from cell to cell through transsynaptic mechanisms [26]. It is postulated that tau, like prions, propagates through exosomes, thus causing widespread tau aggregation and neuronal cell death in tau-dependent neurodegenerative diseases [27,28,29].
In addition to direct tau neurotoxicity, it is hypothesized that tau contributes to neurodegeneration through oxidative stress [30]. Oxidative stress is implicated in the development of multiple neurodegenerative diseases, including AD [30, 31]. Several studies have suggested that hyperphosphorylation of tau and subsequent microtubule dysfunction could produce reactive oxygen species and resultant oxidative stress [30, 32]. However, other data have also shown that the aggregation of tau and microtubule dysfunction could result from the initial accumulation of reactive oxygen species [30, 33].
Inflammation may also contribute to the development of PSP and has been associated with the enzyme 5-lipoxygenase (5-LO), which induces inflammation via leukotriene production [13, 34]. 5-LO is upregulated in brain samples of patients with tauopathies. Additionally, inhibition of 5-LO in mouse models has resulted in significant improvements in memory and synaptic function and a reduction in tau phosphorylation, likely through a method that reduces cyclin-dependent kinase 5 (CDK5) activity [35].
Activation of microglia, the most important immune modulator in the human nervous system, is also postulated as contributing to the development of PSP [13, 36]. It is thought that microglial activation drives tau pathology and contributes to the spread of tau in the brain [17]. In a 2010 study, microglial neuroinflammation was found to promote tau phosphorylation and aggregation, which may lead to neurodegenerative conditions such as PSP [37].
Neuropathology of PSP
Tau deposit accumulation in neurons and glial cells causes neurotoxicity and neuronal cell death [38]. The brains of patients with PSP have tufted astrocytes [1, 15] and other neuropathological findings, including globose neurofibrillary tangles in the gray matter and coiled tau bodies in oligodendrocytes in the white matter [1, 39]. Further neuronal findings in PSP include tau threads and pretangles [2]. The area of tau deposition depends on the specific phenotype of PSP, but the most common brain areas affected are the brainstem, globus pallidus, subthalamic nucleus, substantia nigra, and dentate nucleus of the cerebellum [1, 2, 40]. Tau sometimes deposits in the dentate nucleus and frontal, temporal, and parietal cortices [1]. The exact pattern of tau deposition varies with the disease phenotype [2]. For instance, the total tau load is generally less in the cortex in PSP-parkinsonism (PSP-P) and PSP-progressive gait freezing, while it is more prominent in the PSP-speech language subtype [41]. Significant differences in the tau burden and cytopathology are present among the various clinical phenotypes of PSP. Tau accumulation usually begins in the subcortical regions and proceeds to the neocortical regions and the cerebellum [41].
Biomarkers
Identifying disease-progression biomarkers in the pathophysiology of PSP continues to be a significant area of PSP research into diagnosis and therapy [2]. Previous studies have aimed to identify cerebrospinal fluid (CSF) biomarkers that might differentiate PSP from other similarly presenting neurodegenerative diseases, but none of these biomarkers were able to be confirmed at autopsy [17, 42]. However, data revealed that patients with PSP had normal or low concentrations of phosphorylated tau and total tau in the CSF, whereas patients with AD had higher total tau levels than normal [17, 43]. Another notable biomarker used to treat PSP and other atypical parkinsonian syndromes is a neurofilament light chain (Nfl), an intermediate filament and nonspecific biomarker indicative of neuronal injury [1, 44]. In patients with PSP, Nfl concentrations in CSF were two to five times higher than those of healthy controls, patients with PD, PD dementia, and dementia with Lewy bodies [17, 42, 45]. However, these differences were not identified when CSF from patients with PSP was compared to CSF from patients with corticobasal syndrome (CBS) and MSA [17, 42, 45]. Therefore, while some preliminary data suggest some PSP-identifying biomarkers, further research is required to determine those biomarkers that can aid in the accurate differentiation of PSP from other neurodegenerative disorders. Biomarkers may also be useful in the detection and prediction of PSP disease progression. When CSF analyses were performed over time in a multicenter PSP clinical trial, the only CSF biomarker that changed with time was Nfl concentrations [17, 46]. Baseline plasma Nfl concentrations were predictive of PSP progression over the course of 1 year [17, 47]. Additionally, serum Nfl levels have been found to correlate closely with CSF levels of Nfl, and higher baseline levels of Nfl have been associated with poorer clinical and radiological outcomes in patients with PSP [1, 47]. Finally, another biomarker called RT-QulC is under study and shows potential for aiding in the diagnosis of alpha-synuclein-based diseases (e.g., PD, dementia with Lewy bodies, and MSA), as well as in the diagnosis of tauopathies such as PSP [1].
Radiological methods can also be used in the identification of neurodegenerative disorders such as PSP [1]. Magnetic resonance imaging (MRI) in PSP often shows various types of midbrain atrophy, which are described as the “hummingbird” [48], “morning glory” [49], and “Mickey Mouse” signs [50]. The magnetic resonance parkinsonism index is useful in the diagnosis of PSP, with greater than 80% sensitivity and specificity in differentiating patients with PSP from those without [1, 51,52,53].
Positron emission tomography (PET) imaging has also been utilized in differentiating PSP from other neurodegenerative diseases [1]. Currently, several tracers that bind to tau proteins have been studied, but various limitations have been identified. 18F-AV-1451, also known as flortaucipir, is a specific PET radiotracer used to identify and image tau in the brain [54]. 18F-FDG-PET is a radiotracer of metabolic activity and does not specifically identify tau pathology. Other radiotracers are possibly beneficial but inaccessible due to economic concerns in daily practice [54].
Single-photon emission computed tomography (SPECT) has been studied in the diagnosis of PSP, but it is nonspecific. In studies using the radiotracer 99mTc-HMPAO in patients with tauopathic atypical parkinsonism, patients with PSP were found to have thalamic hypoperfusion, which did not help differentiate between patients with PSP, PSP-P, and CBS [54,55,56]. In a recent study evaluating frontal assessment battery (FAB) results alongside SPECT findings in patients with PSP-Richardson’s syndrome (PSP-RS), PSP-P, and CBS, impairment in executive functions was identified as a possible key differentiating factor in patients with variants of PSP [54]. Of note, executive functioning seemed to generally be more significantly impaired in patients with PSP-RS than in patients with PSP-P when using the FAB. Additionally, using SPECT, patients with PSP-P were found to have the least deteriorated perfusion compared to patients with PSP-RS and CBS. Overall, this study identified that when using FAB and SPECT perfusion assessments of the frontal lobes, there was slightly more deterioration in patients with CBS. Overall, it was determined that FAB and SPECT were useful in differentiating between these conditions when each was used alone but that the combined approach was not useful [54].
Clinical Features
PSP can present with a variety of phenotypic presentations. These differing presentations can result in PSP being easily confused with other neurodegenerative disorders, such as FTD and PD [17]. Guidelines such as those put out by the National Institute of Neurological Disorders and Stroke (NINDS) can assist in differentiating these neurodegenerative diseases. For instance, according to NINDS, early falls due to postural instability concurrent with supranuclear gaze palsy or slowed vertical saccades were the most helpful factors in distinguishing PSP from PD [1, 57].
The development of PSP is divided into stages, beginning with the presymptomatic stage. This stage is defined as individuals who are asymptomatic but are at high risk of developing symptoms of PSP in the future. Currently, this stage can only be identified by postmortem examination [1, 17]. The following early symptomatic stage of PSP is known as “suggestive of PSP stage” [17]. A few clinical signs and symptoms are present in this stage, but they are not sufficient for diagnosing PSP [17]. It is then possible for the patient to progress to any of the following PSP subtypes, including PSP-RS, PSP-P, or PSP with mixed pathology, but the specific subtype cannot be identified at this point in the disease process [17]. In the future, diagnostic biomarkers for PSP may help identify potential disease progression [17]. Further research into this period of disease progression is relevant, as greater insight into disease recognition in the presymptomatic stage may be helpful in preventive care.
The classic disease subtype of PSP is known as Richardson’s syndrome (PSP-RS) [17]. The early signs of PSP-RS include slowing of vertical saccades, as well as other nonspecific ocular symptoms, including dry, red, and sore eyes; photophobia, blurred vision, and difficulty focusing, as well as spontaneous involuntary eyelid closure or apraxia of eyelid opening, and impaired spontaneous blink rate [57]. Early indicators also include slow, slurred, or growling speech and difficulties swallowing [57]. Later in the disease course, a supranuclear gaze palsy can result, which may cause difficulty eating. The late presentation of this symptom can delay diagnosis in many cases [57]. The PSP-RS subtype also includes initial postural instability, leading to falls early in the disease course [2]. Overactivity of the frontalis, procerus, and corrugator muscles may result in eyelid retraction and a staring gaze [57]. Motor changes in PSP-RS have classically been described as a lurching gait or “that of a drunken sailor or dancing bear,” with unexplained backward falls without loss of consciousness [57]. Other common symptoms include bradykinesia and slow, ataxic, spastic, and hypophonic speech. In PSP-RS, half of the patients also develop personality changes, such as apathy, disinhibition, and cognitive slowing, within 2 years of diagnosis [17, 57].
Later in the disease, patients often develop unintelligible speech and recurrent choking, which may lead to the need for a percutaneous gastrostomy [57]. Patients also become wheelchair-dependent as a result of recurrent falls and fractures and often become dependent on others 3 to 4 years into the disease [57].
The median age of survival in the original series of studies regarding PSP-RS was approximately 5 years from disease onset. In more recent studies, the reported duration from onset of disease to death had increased to a range of 5 to 8 years [57]. The most common causes of death due to PSP-RS included aspiration pneumonia, primary neurogenic respiratory failure, and pulmonary embolism [57]. PSP-RS can be differentiated and identified as a clinical syndrome with associated pathological findings of PSP [57].
While PSP-RS is the classic presentation of PSP, more phenotypic presentations have been discovered. PSP-P (PSP-parkinsonism) is characterized by early disease features that include axial rigidity, speech impairment, gait difficulties, limb rigidity, bradykinesia, and a positive levodopa response, with a later onset of PSP-specific symptoms [17, 57]. PSP-RS and PSP-P are the most common among the variants of PSP. PSP-RS makes up about 60% of PSP cases, and PSP-P makes up approximately 30% [54].
PSP with progressive gait freezing is defined as the development of an isolated gait disorder years before the development of any other PSP-RS features [17, 58]. This isolated gait disorder may include symptoms such as gait disturbance with start hesitation and subsequent freezing of gait, as well as difficulties in initiating or completing speech and writing [17, 58].
PSP-corticobasal syndrome is the best-recognized presentation of CBD, which is defined as the accumulation of 4R tau aggregates [17, 59]. Phenotypically, this PSP subtype presents very similarly to PSP-RS [17]. However, these subtypes differ in their biochemical features, morphology, and anatomical distribution [17]. Symptoms include progressive limb rigidity, apraxia, cortical sensory loss, alien limb, and bradykinesia, all of which are unresponsive to levodopa in contrast to PSP-P [17].
PSP-speech language is a subtype of PSP that presents as a nonfluent or agrammatic variant of primary progressive aphasia, a syndrome in which there is agrammatism in language production or effortful, halting speech with inconsistent speech sounding errors and distortions [17, 60]. In this subtype, the speech and language symptoms are predominant in early disease stages, with motor symptoms of PSP-RS presenting later in the disease course [17].
PSP with frontal presentation is defined as exhibiting behavioral changes characteristic of FTD years before the onset of motor symptoms of PSP-RS. Patients present with a clinical syndrome of behavioral variant FTD, which includes early progressive deterioration of personality and social comportment; behavior changes such as apathy, rigidity, disinhibition, and hyperorality; and cognitive decline [17].
PSP with predominant cerebellar ataxia presents with cerebellar ataxia as the principal symptom, with motor symptoms of PSP-RS presenting later in the disease course [17]. Finally, PSP with mixed pathology is defined as the presence of PSP with another concurrent neuropathology, such as AD, PD, deposition of transactive response DNA binding protein 43 (TDP-43), argyrophilic grain disease, or other cerebrovascular diseases [17].
As noted above, diagnosis of PSP can be challenging because of the late onset of some defining symptoms as well as its similar presentation to many other neurodegenerative diseases. However, some key characteristics can be utilized to increase the likelihood of identifying PSP. PSP is clinically probable when a patient presents with both vertical supranuclear palsy and postural instability [1, 61]. PSP is clinically possible in cases of isolated vertical gaze palsy or slowed saccades with postural instability [1, 61]. At this time, a postmortem pathology confirmation showing “cardinal neuropathologic” features is still required for a definitive diagnosis of PSP [1].
Risk Factors, Associated Conditions
The role of associated conditions and modifiable risk factors in the progression and management of PSP has also been studied. One case–control study identified a modest yet significant association between hypertension and the development of PSP [62]. Another case–control study found that two potentially modifiable clinical features associated with prediagnostic PSP were cerebrovascular disease and diabetes mellitus [63]. Type 2 diabetes mellitus has previously been studied in its association with brain atrophy and neurodegeneration. The type 2 diabetes mellitus features, including glucose dysregulation, insulin resistance, and impaired insulin signaling, have been found to lead to the accumulation of amyloid beta and hyperphosphorylation of tau, alongside other inflammatory and oxidative stress-related processes [63, 64]. Additionally, the accumulation of amyloid beta and hyperphosphorylated tau may also contribute to pancreatic beta cell dysfunction, which could serve to cause or worsen type 2 diabetes mellitus [63, 64]. Another study evaluated the effects of glycemic variability below and above a hemoglobin A1C of 5.7% on cerebral perfusion. Patients with PSP or CBS were evaluated with A1C and SPECT, and patients with higher glycemic variability were found to have reduced cerebral perfusion to regions involved in these neurodegenerative diseases, notably the hippocampus, pons, left thalamus, and right insula [65]. In a prior review discussing the treatment of atypical parkinsonian disorders, insulin resistance targeting was also evaluated in the management of MSA. In studies in mouse models and patients with MSA, insulin resistance was reported in oligodendrocytes and neurons of the putamen, with impairment of insulin and insulin-like growth factor 1 signaling. This insulin resistance was thought to potentially affect oligodendrocyte functioning and lead to neurodegeneration of the putamen [13, 66].
Cerebrovascular disease has also been identified as a potential modifiable risk factor for PSP, and studies have identified vascular pathology involved with the development of some cases of PSP and CBS [65, 67, 68]. Overall, further study is needed regarding the prevention of PSP via management of risk factors, especially since PSP itself is currently untreatable once it develops.
PSP is currently incurable and is only treatable with therapies that alleviate symptoms [2]. Therefore, the identification of disease-modifying PSP therapies is a critical area of research. Current strategies of PSP treatment include targeting tau protein aggregation, microtubule dysfunction, and tau posttranslational modification by mechanisms that include immunotherapy, gene therapy, and vaccines [1]. Other unique approaches could include transcriptional disruption of the tau gene using repurposed small molecules [69].
Methods
In this review, the PubMed (National Library of Medicine) database was queried using the key search words of “progressive supranuclear palsy,” “tau,” “monoclonal antibodies,” “treatment,” “clinical trials,” and “progressive supranuclear palsy and diabetes/hypertension.” Past and ongoing clinical trials were also identified by searching for “progressive supranuclear palsy” on the US National Library of Medicine’s ClinicalTrials.gov website. Publication dates of the articles reviewed were from 1977 through 2023. An internet search using Google Search (Alphabet Inc.) was also done to identify articles and unpublished updates, including preliminary results from incomplete clinical trials. The last date these databases were searched was December 13, 2023. For most of the sources, the inclusion criteria required a peer-reviewed journal article documenting information from a preclinical or clinical trial that had the potential to affect treatment for patients with PSP. In selected cases, online articles and forum publications of preliminary results from unfinished clinical trials were included for thoroughness.
Additionally, articles discussing conditions similar to PSP, such as AD, MSA, PD, and CBS, were included because of potential overlap in the efficacy of treatment options. Studies were first grouped by symptomatic and disease-modifying treatments, which were then categorized by the mechanism of treatment action. Outcomes reported included primary outcome measures for most studies and some secondary outcome measures when there were significant and relevant results. Outcome effect measures were reported as they were presented in the primary articles.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Treatment
Non-Disease-Modifying Treatment
Despite numerous drug trials conducted in the past decades, PSP remains a universally fatal disease [17]. In some cases, patients with the PSP-P or PSP-RS phenotypic subtypes showed initial benefits in response to levodopa therapy [17, 70], especially among those with bradykinesia and rigidity. However, this benefit was often transient without effects on the overall disease trajectory [17, 70]. Currently, treatment remains mainly symptomatic, and several non-disease-modifying treatments are in clinical trials (Table 1).
Transcranial and Deep Brain Stimulation
Several clinical trials have evaluated the efficacy of transcranial and deep brain stimulation on PSP progression. One clinical trial (NCT01174771) that evaluated the safety and efficacy of noninvasive cortical stimulation for the treatment of PSP and CBD was completed in 2014, but no results have been reported. Another clinical trial (NCT02734485) sought to evaluate the efficacy of deep transcranial magnetic stimulation over the left Broca area and dorsolateral prefrontal cortex for promoting recovery of motor and cognitive function in patients with PSP. The last update of this study was reported in 2016, and no results have been reported. A more recent clinical trial (NCT04655079) is testing the efficacy and safety of anodal transcranial direct current stimulation (a-tDCS) over the left dorsolateral prefrontal cortex to treat PSP. a-tDCS has previously been shown to be effective for the treatment of psychiatric disorders. The study was expected to be completed in August 2022, but results have not yet been published. Trials for other nonpharmacological therapies, such as noninvasive repetitive cerebellar transcranial magnetic stimulation (NCT04468932) and spinal cord stimulation (NCT04367116), have been completed or are still ongoing for safety and efficacy for treating motor dysfunction in PSP. Still, these studies have not yet published results.
Another clinical trial, completed in 2019, evaluated the efficacy of cerebellar theta burst repetitive transcranial magnetic stimulation (rTMS) (NCT04222218). In this study, 20 patients with PSP underwent a series of sham or real cerebellar rTMS in a crossover study design. Outcome measures included static balance tests, timed up-and-go tests, and a short physical performance battery. Cerebellar rTMS was found to have a significant effect on stability in patients with PSP [71]. A follow-up study (NCT04237948) that evaluated the efficacy of combining transcranial direct magnetic stimulation and physical therapy for the treatment of PSP was performed, with estimated completion in December 2019. However, no results have been reported yet. Deep brain stimulation of the pedunculopontine region has been attempted in patients with advanced PSP-RS; however, no clear benefits were noted [72]. On the contrary, harmful adverse effects occurred, including perioperative hemorrhages in two patients of the eight study patients [17, 72].
Physical Therapy
Nonpharmacological interventions, such as physical therapy, improve symptomatic and functional manifestations of PSP [17, 73]. Several more studies regarding the efficacy of specific physical therapy programs on PSP progression are underway or completed. One interventional therapy study (NCT05139342) is currently evaluating the efficacy of a 2-week expiratory muscle strength training regimen to treat patients with either PD, MSA, or PSP; 75 patients are expected to be enrolled. A separate clinical trial (NCT04608604) of 180 individuals with PSP-RS, MSA, or PD is currently evaluating the efficacy of two different physiotherapy programs on the motor symptoms and gait parameters of these neurological diseases. Another study evaluated the efficacy of 4 weeks of multidisciplinary intensive rehabilitation treatment (MIRT), previously used for PD, with or without a driven gait orthosis therapy protocol for treating patients with PSP (NCT02109393). Twenty-four patients with PSP were enrolled. Twelve participants underwent MIRT with a treadmill-plus therapy regimen, while the other 12 used a driven gait orthosis. Overall, the driven gait orthosis did not offer additional benefit compared to the treadmill-plus regimen, but the efficacy of aerobic, multidisciplinary, intensive rehabilitation for patients with PSP was confirmed [73]. Robot-assisted gait training has also been studied and was shown to be feasible and safe for patients with PSP [74].
Other Treatment Approaches
Rivastigmine, an acetylcholinesterase and butyrylcholinesterase inhibitor used in the treatment of AD and PD, is currently undergoing a phase III clinical trial (NCT02839642) to evaluate its efficacy in the treatment of the motor, cognitive, and behavioral impairment in PSP. Previous findings regarding the treatment of PSP with acetylcholinesterase inhibitors are inconsistent. In a case series of five patients with PSP, treatment with rivastigmine for 3 to 6 months improved cognitive function [75]. However, in a randomized controlled trial of 21 patients with PSP treated with donepezil, an acetylcholinesterase inhibitor, the results showed mildly improved cognitive function with worsening motor function [76]. The current phase III trial of rivastigmine is a randomized, double-blind, placebo-controlled study with 106 participants with probable PSP-RS. Participants in the treatment arm receive an oral treatment of rivastigmine twice daily for 24 months, and patients in the control arm receive a matched dose of placebo. Primary outcomes include changes from baseline in the number of falls and near falls, and secondary outcomes include safety, changes in the Progressive Supranuclear Palsy Rating Scale (PSPRS) score from baseline, and patient and caregiver quality of life. The study was reportedly completed in November 2022, but no results have been released yet.
Additionally, a phase IV clinical trial (NCT04014387) is currently underway to evaluate the efficacy and safety of zolpidem, a benzodiazepine receptor agonist, and suvorexant, a dual orexin receptor antagonist, in the treatment of sleep quality in patients with PSP. Another phase IV trial (NCT03924414) is testing the efficacy of a single intravenous infusion of zoledronic acid for the prevention of fractures in patients with PD and Parkinson-like disorders, such as PSP.
A more specific therapy for PSP symptoms is pretarsal botulinum treatment, which has been shown to improve apraxia of the eyelid [17]. Speech therapy, occupational therapy, social work, and palliative care are also critical in caring for patients with PSP, emphasizing the importance of multidisciplinary treatment paradigms for this disease [1, 73].
Risk Factor Modification/Management of Associated Conditions
Studies are currently underway to evaluate whether the management of modifiable risk factors aids in the treatment of patients with PSP [62, 63]. A recent review discussing treatment options for parkinsonian disorders, including PSP, MSA, and CBD, noted studies that evaluated insulin for the treatment of MSA and PD [13]. This study evaluated the effects of intranasal insulin injection on the motor and cognitive functioning of patients with MSA and PD. Patients who received insulin had better cognitive and motor functioning than patients in the placebo group [13, 77]. A separate study noted that glucagon-like peptide 1 (GLP-1) analogues are being studied for the treatment of MSA and that preclinical studies of GLP-1 analogue injections showed favorable results for lowering insulin resistance and reducing neuronal loss in the putamen [13, 66]. A phase II trial is underway to evaluate the efficacy of GLP-1 analogues in the treatment of early-stage MSA. The findings in MSA treatment suggest possibilities for the utility of insulin resistance management in patients with PSP.
Disease-Modifying Treatments
Several clinical trials are currently underway to identify a disease-modifying treatment for PSP. These trials target tau aggregation, microtubule dysfunction, and tau posttranslational modifications [17]. In determining treatment for tauopathies, it is critical to consider the cause of tau-dependent neurodegeneration. There are two types of tau dysfunction that are thought to lead to neurodegeneration: toxic tau gain-of-function or loss-of-function; both lead to tau aggregation and neurotoxicity [17, 78]. Studies have shown that toxic tau aggregation in one brain region can spread through transsynaptic methods and propagate to other brain areas [26]. Below, we present the main therapeutic interventions currently explored in clinical trials on PSP.
Tau Loss-of-Function Therapies
Microtubule-Stabilizing Agents
The first set of tau-based treatments targets tau loss-of-function, which can lead to microtubule destabilization. In healthy neurons, tau is enriched in axons and aids in providing strength and stability to microtubules [79,80,81]. When tau is dysfunctional and forms insoluble aggregates, microtubules may become unstable, leading to disruption of fast axonal transport [79, 82]. Therefore, treatment with microtubule-stabilizing agents may compensate for tau dysfunction in these neurodegenerative diseases [79].
Davunetide is a microtubule-stabilizing octapeptide and is derived from glial cells in response to vasoactive intestinal peptide exposure. Davunetide has been found to function as a neuroprotective agent and to prevent microtubule disruption in cell culture models [46]. Davunetide has also shown this benefit in transgenic mouse models of tauopathy [83]. However, a phase II/III clinical trial conducted on patients with PSP-RS showed no efficacy of davunetide (NCT01110720) [46]. In this study, 313 individuals with PSP-RS were randomly assigned to davunetide (n = 157) or placebo (n = 156). Baseline characteristics did not differ between the groups, and medication compliance was similar between the groups. After monitoring patients for 52 weeks, no significant differences in the primary efficacy endpoints were found. The primary efficacy endpoints included the PSPRS and the Schwab and England Activities of Daily Living scale [46]. No significant differences in the number or timing of deaths between the two groups were found, and all MRI measurements evaluated in the study showed progressive atrophy over the year of monitoring [46]. Both treatment groups were found to have similar rates of serious adverse events that were attributable to disease progression [46]. However, more patients in the davunetide arm than the placebo arm had adverse events, mainly epistaxis and nasal congestion, resulting in study discontinuation [46]. Overall, davunetide was ineffective for treating PSP [46].
TPI-287 is a new blood–brain-barrier-permeable taxane that also acts as a microtubule stabilizer [17, 84]. TPI-287 was found to accumulate in higher concentrations in the brain than in the blood; it reduced hyperphosphorylated tau levels in the brain and improved cognitive performance in tau-transgenic mice [84]. Two phase I randomized controlled trials tested the safety and efficacy of this drug in 29 patients with AD (NCT01966666) and in 44 patients with the 4R tauopathies of PSP and CBD (NCT02133846) [84]. Patients in both studies received either 20, 6.3, or 2 mg/m2 of TPI-287 or a matched placebo once every 3 weeks for 9 weeks. In the AD trial, all three instances of serious adverse effects were anaphylactoid reactions that occurred in the treatment arm, and the study was terminated early for safety reasons [84]. In the 4R tauopathy trial, there were no severe adverse reactions. However, more patients in the treatment arm experienced falls than in the placebo arm [84]. Patients in the treatment arms of both trials experienced higher incidences of headaches, dizziness, constipation, diarrhea, and nausea [84]. In addition, patients in the 4R tauopathy treatment arm had dose-related worsening of global cognitive function compared to patients in the placebo group [84]. Although TPI-287 was ineffective in the treatment of 4R tauopathies, this study revealed critical differences in the safety and tolerability of neurodegenerative disease treatments based on the disease process [84].
Epothilone D is another blood–brain-barrier-permeable microtubule-stabilizing agent. When administered to tau transgenic mice with no initial tau abnormality, it was found to prevent axonal microtubule loss and dystrophy, as well as improve cognitive performance [1, 79, 85]. A phase I trial (NCT01492374) evaluated the safety and pharmacodynamic effects of epothilone D (BMS-241027) in the treatment of participants with mild AD. Forty participants were randomized to receive either intravenous 0.003 mg/kg, 0.01 mg/kg, or 0.03 mg/kg of epothilone D or dose-matched placebo for 9 weeks. The primary outcomes included safety and CSF levels of N-terminal tau fragments. The last update on this study was in 2014, and no results have been published yet.
Tau Gain-of-Function Therapies
In contrast to tau dysfunction, it is proposed that toxic tau gain-of-function promotes aggregation and prion-like spreading of pathogenic tau between cells [2, 26]. There is evidence to suggest that tau fragments can be secreted and “seed” from one cell to the other and for abnormal tau to induce dysfunction and aggregation of normal tau [86, 87]. As a result, anti-tau antibodies are being investigated [2]. In transgenic mouse models, it has been shown that passive immunization with anti-tau monoclonal antibodies suppresses tau pathology and improves cognitive and motor function [17, 88, 89].
Monoclonal Antibodies
The monoclonal antibody bepranemab (UCB0107) recently underwent a phase Ib study (NCT04185415) to evaluate its safety, tolerability, and pharmacokinetics in the treatment of patients with PSP. According to Vaswani and Olsen [86], bepranemab is a humanized murine monoclonal antibody that targets an epitope of tau adjacent to the microtubule-binding repeats. In an in vitro model of tau aggregation, monoclonal antibodies that included a central tau epitope prevented tau seeding and propagation more strongly than antibodies with N-terminal epitopes [86]. In a previously conducted phase I trial (NCT03464227), in which results were released in abstract form, 52 healthy male adult participants were intravenously administered bepranemab or placebo [90]. Adverse effects occurred in 47% of the participants, but the adverse effects were not severe and were unrelated to the treatment [86, 90]. In the more recent phase Ib study of bepranemab (NCT04185415), 25 individuals were enrolled. All participants had a possible or probable diagnosis of PSP, and the trial was a double-blind, placebo-controlled study. Participants received bepranemab or intravenously administered saline, and their responses were monitored from baseline until the final follow-up visit at week 68. That trial was completed on November 17, 2021, and no results have been posted. An open-label extension stage (NCT04658199) began in November 2020 and is estimated to be completed in November 2026. This study was designed to evaluate the long-term safety and tolerability of bepranemab for the treatment of PSP. Eligible patients included those with a possible or probable diagnosis of PSP-RS. Nineteen patients are estimated to have been enrolled. Patients receive a predefined intravenous dose of bepranemab, and primary outcomes compare the safety and tolerability from baseline to the end of the study at 60 months.
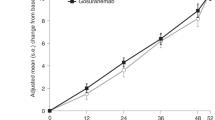
Two N-terminal tau-targeting monoclonal antibodies, gosuranemab (BMS-986168, BIIB092) and tilavonemab (ABBV-8E12), have entered phase II clinical trials for PSP [17, 91]. Although tau is primarily an intracellular protein, there are a variety of tau fragments that can be found extracellularly [91]. These tau fragments lack the microtubule-binding region and C-terminus sequences of intracellular tau and are abundant in CSF [91]. Previous studies have identified that this extracellular N-terminal tau could drive neuron-to-neuron pathologies such as PSP [91]. Gosuranemab is a monoclonal antibody directed against extracellular N-terminal tau that has a high affinity for the fibrillar tau form identified in patients with AD and PSP [91]. When studied in transgenic mouse models, gosuranemab was found to reduce levels of unbound tau in CSF and interstitial fluid [91]. A phase I study (NCT02294851) conducted with gosuranemab demonstrated a marked decrease in this extracellular CSF N-terminal tau without any deaths or serious adverse outcomes [91, 92]. A separate phase Ib randomized controlled trial (NCT02460094) also demonstrated significant decreases, by approximately 90% at all doses, of CSF unbound N-terminal tau with gosuranemab treatment [91, 93]. The purpose of the phase II clinical trial (NCT03068468) was to identify whether decreases in CSF extracellular N-terminal tau correlated with improved prognosis of PSP [91]. The study included 486 participants with a diagnosis of possible or probable PSP [91]. Other eligibility criteria included 41 to 86 years of age, a score of at least 20 on the Mini-Mental State Examination, and no other notable neurological or psychiatric disorders [91]. Participants were randomized and assigned to either gosuranemab (n = 321) or placebo (n = 165). Patients in the treatment arm received 50 mg/mL gosuranemab intravenously once every 4 weeks for 48 weeks, and patients in the control group received matched dosing of a placebo intravenous infusion. The patients were evaluated at 52 weeks by comparing baseline and posttreatment PSPRS scores, which was the primary efficacy endpoint. In this study, although the levels of CSF extracellular N-terminal tau did decrease in gosuranemab groups compared to the control group, there was no significant effect on other disease markers, such as Nfl and midbrain atrophy [91]. In addition, there were no significant differences between gosuranemab and placebo groups in changes from baseline using the PSPRS score [91]. Ultimately, gosuranemab treatment for 52 weeks did not significantly impact PSP disease progression as compared with control groups [91].
A second monoclonal antibody, tilavonemab (ABBV-8E12), was evaluated in a phase II trial for safety and efficacy (NCT02985879) [94]. Tilavonemab is a monoclonal antibody that targets the N-terminus of human tau [94]. In mouse models, tilavonemab reduced the loss of brain volume, slowed the progression of tau pathology, and increased cognitive performance [89, 94]. This most recent study was a phase II, multicenter, randomized, placebo-controlled, double-blind study at 66 hospitals and clinics in Australia, Canada, France, Germany, Italy, Japan, Spain, and the USA [94]. Study participants included 377 people 40 years of age or younger diagnosed with possible or probable PSP who were symptomatic for less than 5 years. The participants were randomly assigned to tilavonemab 2000 mg (n = 126), tilavonemab 4000 mg (n = 125), or matching placebo (n = 126) administered intravenously on days 1, 15, and 29, and then every 28 days through the end of the 52-week treatment period [94]. The primary efficacy outcome was the change in total PSPRS score from baseline to week 52 [94]. Baseline PSPRS total scores did not differ between the two treatment groups and the placebo group [94]. In addition, the most common adverse events reported during the study were consistent with complications of PSP, including falls, contusions, and skin lacerations [94]. Overall, the results showed that tilavonemab lacked efficacy in the treatment of PSP in humans [94].
Further evidence of the effects of treatment with tilavonemab was evaluated in two autopsies of patients with PSP after treatment with tilavonemab [95]. One patient had been enrolled in the phase II trial mentioned above [94, 95], and the other was in a phase I trial with the same monoclonal antibody [95, 96]. The patients were confirmed to have had PSP via autopsy [95]. The tau burden in these two patients was compared to that of 10 other patients with autopsy-confirmed PSP, and there was no significant difference in tau density or distribution [95]. Ultimately, there were no other findings to indicate a response to tilavonemab [95].
In another autopsy report from January 2023, a patient was reported to have been enrolled in a phase II trial and had received tilavonemab [97]. On autopsy, the patient was found to have neuronal loss, gliosis, and widespread deposits of phosphorylated tau in neurofibrillary tangles, tufted astrocytes, coiled bodies, and threads [97]. These findings were identified in the inferior olivary nucleus, dentate nucleus of the cerebellum, substantia nigra, midbrain tegmentum, subthalamic nuclei, globus pallidus, putamen, and precentral gyrus [97]. The location and characteristics of these findings confirmed a diagnosis of PSP [97]. However, there were no findings, such as slowed loss of brain volume or decreased tau burden, to suggest that treatment with tilavonemab had been efficacious [94]. These findings further suggested the inefficacy of tilavonemab in the treatment of PSP [97].
Vaccinations: AADvac1, ACI-35, JACl-35
Active vaccinations against pathological tau epitopes have been investigated, and three potential tau vaccines have begun human clinical trials: AADvac1, ACI-35, and JACl-35 [17, 98, 99].
AADvac1 is an active immunotherapy that produces antibodies that target conformational epitopes in the microtubule-binding region of tau, which aims to prevent tau aggregation and promote tau clearance [98, 99]. When tested in transgenic rat and mouse models of Alzheimer’s tauopathy, administration of AADvac1 resulted in decreased neurofibrillary tangles and insoluble tau buildup as well as in improvement of the neurobehavioral impairment of the models [98]. AADvac1 underwent two phase I clinical trials (NCT01850238 and the follow-up study NCT02031198). The first phase I trial was a 12-week, randomized, double-blind, placebo-controlled trial with a 12-week open-label extension in patients with mild-to-moderate AD [100]. Twenty-four patients were randomized to the treatment arm and six to the placebo arm. Patients in the treatment arm received three subcutaneous doses of AADvac1 at monthly intervals, which was then followed by the open-label stage, in which all patients from the treatment and control arms were allocated to AADvac1 and received three more monthly subcutaneous doses [100]. The primary endpoint was adverse events. AADvac1 was found to have a favorable safety profile with excellent immunogenicity in this first phase 1 trial [100]. In the follow-up phase I study, the patients received two booster doses of AADvac1 at 24-week intervals and were followed for a total of 72 weeks. The primary outcome was the long-term safety of AADvac1. The vaccine was found to have a benign safety profile, and patients with high immunoglobulin G (IgG) titers had slower rates of atrophy in MRI evaluations and less of a decline in cognitive assessment (NCT02031198) [101]. AADvac1 moved on to a phase II trial in the treatment of mild to moderate AD (NCT02579252) [102]. This trial was a 24-month randomized controlled trial that included 208 participants randomized into control and treatment groups. Participants in the treatment arm received six doses of the vaccine at 4-week intervals and five individual booster doses at 3-month intervals. Participants in the placebo group received a matched dose of placebo. The primary outcome was safety, and secondary outcomes included dementia progression, cognitive function, immunogenicity, and ability to perform activities of daily living. Initial results of this phase II study were released in 2019. The AADvac1 vaccine was highly immunogenic (98.2% of participants generated anti-tau antibodies), safe, and well tolerated. AADvac1 was also found to cause a statistically significant change in CSF and blood biomarkers, which indicated that the vaccine slows the progression of the neurodegenerative process to that more commonly seen in healthy individuals [102, 103]. If this treatment proves helpful in patients with AD, its tau-targeting capabilities may also aid in treating patients with PSP.
ACI-35 is an active vaccine that is undergoing a phase Ib/IIa clinical trial (NCT04445831) for the treatment of early AD. ACI-35 is a liposome-based vaccine that targets epitopes of tau, which are often phosphorylated in pathological states, most notably serine residues [104]. JACI-35 is a protein conjugate of the same tau protein in the ACI-35 vaccine. However, at the Clinical Trials on Alzheimer’s Disease conference on December 2022, JACI-35 was reported to be less effective than ACI-35, and the trial moving forward will be with ACI-35 [105].
In a 3-month regimen of subcutaneous injections of ACI-35 in mice, the mice produced a rapid immune response, with the production of high IgG antibody levels specifically targeting phosphorylated tau. This regimen also resulted in a mild reduction of pathological hyperphosphorylated tau in these mouse models, which may indicate the future utility of this active vaccine in treating other tauopathies [99, 104]. In this current phase Ib/IIa trial, a total of 57 participants were initially randomized to receive either low, high, or medium doses of the ACI-35 vaccine, either low or medium doses of the JACI-35 vaccine or placebo administration at specified time points over 48 weeks. At the Clinical Trials on Alzheimer’s Disease conference in December 2022, interim results were released, reporting that the vaccine was overall safe and well tolerated. Additionally, the vaccine was found to have a “rapid and durable antibody response” specific to phosphorylated tau and paired helical filaments in people with AD [105]. The last study update was submitted in October 2023.
Inhibiting Tau Aggregation
Other pharmacological agents have been developed to target tau aggregation or posttranslational modifications [17]. For instance, methylene blue (MB) derivatives have been studied for the treatment of AD and frontotemporal degeneration. MB is an approved treatment for methemoglobinemia that inhibits tau aggregation in vitro and in transgenic mouse models. It has also been found to dissolve paired helical filaments (PHF), which form neurofibrillary tangles and correlate with pyramidal cell destruction and the development of dementia in AD brain tissue [106,107,108]. This was found to occur as a result of MB reversing the proteolytic stability of protease-resistant PHFs by inhibiting tau-tau binding at repeat domains, which serves to enforce the stability of PHFs [109].
In a phase II clinical trial (NCT00515333), MB (TRx0014) was found to be a safe and potentially effective monotherapy in the treatment of AD, with clinical improvement after 50 weeks of 138 mg of MB per day in patients with mild to moderate AD, as measured by the Alzheimer’s Disease Assessment Scale-cognitive subscale [108, 110].
Leuco-methylthioninium (bis)hydromethanesulfonate (LMTM), also known as hydromethylthionine, is a stable and reduced form of MB with superior pharmaceutical properties, which has been shown to maintain tau aggregation inhibitor properties in vivo and in vitro [108, 110]. In two phase III trials through TauRx Therapeutics Ltd, LMTM was found to have a concentration-dependent response at steady state plasma levels in the range of 0.3–0.8 ng/mL at an 8 mg/day dose, with a plateau at higher doses, in slowing disease progression in patients with AD both as monotherapy and add-on therapy, but with maximum effect reduced by half in add-on therapy (NCT01689233, TRx-237-005, and NCT01689246, TRx-237-015) [111]. Currently, a phase III randomized controlled trial (NCT03446001) to test the efficacy and safety of 8-mg and 16-mg doses of LMTM (TRx-237-039) for the treatment of AD is underway; it was estimated to be completed in April 2023, and no results have been released. An open-label, delayed-start stage of the study is now ongoing as an expanded access program for patients who had previously taken TRx0237 in a clinical trial or compassionate use program. Further study is required in this area, but given the positive findings in AD studies, the tau aggregation-inhibitor effects of LMTM may be potentially beneficial in treating tauopathies such as PSP as well.
Inhibiting Tau Hyperphosphorylation
Another drug target is CDK5 (cyclin-dependent kinase 5), which contributes to tauopathy pathogenesis by phosphorylating tau [1]. CDK5 is activated in postmitotic neurons via the neuron-specific activator p35, and CDK-p35 plays a vital role in brain development and physiological synaptic activity [112]. In diseased brains, CDK5 is thought to be hyperactivated by p25, which is the N-terminal truncated form of p35 [112]. This hyperactivation is suspected to abnormally activate CDK5 in tauopathies such as AD, in which CDK5 phosphorylates tau at serine/threonine residues in serine/threonine-proline sequences [112]. In addition, increased levels of CDK5 have been identified in the tissue and neurons of patients with PSP. Immunohistochemistry of CDK5 displayed greater immunoreactivity within neurons containing tau aggregates [113]. Tolfenamic acid (TA), an NSAID currently used to treat migraines in Europe, has been shown to decrease CDK5 levels in amyloid precursor protein knock-in transgenic mouse models [69, 114, 115]. TA is another emerging treatment of tauopathies, and its therapeutic potential in modifying disease processes has also been explored in a transgenic animal model that carries the human tau gene (hTau) [69]. In a more recent study evaluating the effects of TA on hTau, behavioral tests demonstrated the efficacy of TA in improving spatial learning deficits and memory impairments in young and aged hTau mice, which only occurred in the presence of the hTau gene [69]. Western blot analysis of the hTau protein revealed a reduction in total tau as well as in site-specific hyperphosphorylation of tau in response to TA administration [69]. Immunohistochemical analysis for phosphorylated tau protein revealed a reduction in staining in the frontal cortex, hippocampus, and striatum in animals treated with TA. TA thus holds the potential to be a disease-modifying agent for the treatment of tauopathies [69]. TA underwent a 12-week phase IIa randomized controlled trial (NCT04253132) for the treatment of individuals with PSP. A total of 24 participants with PSP were randomized to receive 50, 300, or 600 mg daily of either TA or placebo. Primary outcomes include safety and tolerance of TA in patients with PSP. This trial was estimated to end on December 31, 2022, and results have not yet been published.
Further treatment methods are designed to inhibit the hyperphosphorylation of tau, which is postulated to lead to tau aggregation [17]. Glycogen synthase kinase 3 (GSK-3) is believed to hyperphosphorylate tau in pathological states such as PSP [116]. Inhibition of GSK-3, a serine/threonine kinase, has been shown to reduce tau phosphorylation and may be an effective drug target [17, 116]. Tideglusib (NP031112), a small-molecule inhibitor of GSK-3-beta, was studied in a phase II trial (NCT01049399) [116]. All enrolled participants had a possible or probable diagnosis of PSP and were randomized to receive either 800 mg of tideglusib, 600 mg of tideglusib, or placebo once daily for 52 weeks. Primary outcomes included changes from baseline in PSPRS scores. Although the drug was found to be safe, it was not significantly clinically efficacious in treating patients with mild-to-moderate PSP [116]. Lithium was also found to inhibit GSK-3, but a clinical trial of lithium was stopped for intolerability [17].
Sodium valproate (VPA), an antiepileptic drug, also inhibits GSK-3 at clinically feasible concentrations [117]. A double-blind, randomized, placebo-controlled trial (NCT00385710) through Nantes University Hospital in France evaluated the efficacy of treatment with VPA on the disease progression of patients with possible or probable diagnoses of PSP [117]. Twenty-eight patients were enrolled and received 1500 mg/day of VPA or matching placebo for 24 months [117]. The primary endpoint was the change from baseline in the patients’ PSPRS scores at 12 and 24 months [117]. Secondary endpoints included the effects of VPA on cognitive and behavioral status, the tolerability of treatment, and patient compliance [117]. Ultimately, no differences were found between treatment with VPA versus placebo in clinical rating scores after 24 months [117]. PSPRS score was significantly higher in the VPA group at 12 months, but there was no significant difference in the scores at 24 months [117]. Furthermore, no differences were identified in secondary endpoints between the VPA and placebo groups. Ultimately, VPA was determined not to be an effective disease-modifying treatment for PSP [117].
Inhibiting Tau Acetylation
It is proposed that the acetylation of soluble tau species precedes hyperphosphorylation and contributes to the pathogenesis of tauopathies [17]. Therefore, inhibition of this acetylation process may be a promising drug target [17, 118]. Salsalate is a commercially available NSAID used in the treatment of rheumatoid arthritis, osteoarthritis, and other rheumatological disorders [119]. Salsalate is proposed as a possible treatment for PSP and other tauopathies, as preclinical studies have indicated that salsalate inhibits tau acetylation [119]. Furthermore, levels of acetylated tau are elevated in patients with tauopathies [119, 120], and studies have shown that treatment of tau transgenic mice with salsalate rescues tau-induced memory deficits and prevents hippocampal atrophy [118, 119]. Salsalate was studied in a phase I futility study (NCT02422485) alongside another phase I trial (NCT02460731) that evaluated the efficacy of treating patients with PSP with young, healthy male donor plasma transfusions [119]. The salsalate trial included 10 patients, and the young plasma trial included five. Although both treatments were safe and well tolerated overall, there was no significant effect on disease progression after 6 months of treatment [119].
Inhibiting Pathologic Tau Processing
Yet another emerging therapeutic approach is the addition of O-linked β-N-acetylglucosamine (O-GlcNAc) residues to proteins, which has been shown to offer protection against pathogenic processing of amyloid precursor protein as well as tau [1, 121]. By modifying tau with even a single O-GlcNAc residue, the toxic self-assembly and misfolding of tau is inhibited, which is postulated to result from O-GlcNAcylation competing with tau phosphorylation [121]. O-GlcNAcylation may have implications for treating neurodegenerative diseases such as PSP, AD, PD, amyotrophic lateral sclerosis (ALS), and Huntington’s disease [121]. Inhibitors of O-GlcNAcase (OGA) have been shown to protect against neurodegeneration in tau transgenic mice, and an OGA inhibitor (LY3372689) has entered a phase II clinical trial (NCT05063539). This study includes 330 participants with AD, randomized into high-dose treatment, low-dose treatment, or placebo groups. The primary outcome includes a change from baseline in AD progression when evaluated at 76 and 124 weeks. This study is estimated to be completed in June 2024 [17, 122].
Antisense Oligonucleotides
Other mechanisms under development to manage tau aggregation include antisense oligonucleotides and splicing modulators that target hairpin RNA structures [17]. Tau is coded for by the MAPT gene on chromosome 17, and alternative splicing of exon 10 causes the production of the pathological 4R tau isoform [123]. Therefore, it is suspected that excluding exon 10 may prove useful in decreasing pathological tau production [123]. Exon 10 splicing of tau protein is regulated by hairpin RNA structures that are destabilized by pathological MAPT point mutations [17, 124]. A study involving MAPT antisense oligonucleotides, which bind to and stabilize the hairpin RNA of exon 10, resulted in reduced human tau protein concentrations both in vitro and in vivo using mouse models [17, 124]. Therefore, using antisense oligonucleotides or splicing modulators to normalize the 3R/4R tau ratios purported to cause disease in tauopathies may prove to be a viable therapeutic approach [17, 124]. This mechanism has been previously investigated in the treatment of ALS [17, 124]. Treatment with antisense oligonucleotides was beneficial in rat models of ALS [124, 125]. Similar treatment approaches were studied in animal models of tauopathies, with positive results. In mouse models, treatment with antisense oligonucleotides reduced human tau mRNA and protein in mice expressing mutant human tau, which resulted in the development of fewer tau inclusions, the reversal of pre-existing phosphorylated tau, and the prevention of neuronal death, ultimately resulting in extended survival of these treated mice. In addition, when nonhuman primates were studied using this treatment, tau antisense oligonucleotides were distributed throughout the brain and spinal cord, resulting in decreased tau mRNA and protein levels in the brain, spinal cord, and CSF. The use of MAPT antisense oligonucleotides has not yet been tested in humans, but these findings suggest that the use of this treatment in humans with pre-existing or early tau-based neurodegeneration may prolong life and reduce disease burden [124].
One tau antisense oligonucleotide currently in clinical trials is NIO752 (NCT04539041). It is being studied in a phase I, multicenter, double-blind, placebo-controlled, multiple-dose escalation study to evaluate the safety, tolerability, and pharmacokinetics of this antisense oligonucleotide in patients with PSP. In this study, approximately 58 patients with PSP were randomized in a 3:1 ratio to receive NIO752 or a placebo. NIO752 was administered intrathecally multiple times over 3 months, with the participants evaluated over a 9-month follow-up period, or it was administered multiple times over 9 months with participants evaluated for a 3-month follow-up period. Primary outcomes include the number of adverse events and outcomes, changes in severity scores for the Columbia-Suicide Severity Rating Scale, and levels of infection indicators in the blood. The study is estimated to be completed in November 2024.
Anti-Inflammatory Therapies
Given the suspected association between microglial activation and inflammatory cytokines in the development of PSP, a case–control study regarding the efficacy of NSAID use in the management of PSP was conducted. Information was collected from 276 patients and 278 controls, and the results showed no association between NSAID exposure and risk of PSP or clinical expression [126].
AZP2006, a small molecule and lead compound of the piperazine family, is another potential treatment that is being studied for its efficacy in treating AD and other related disorders, including PSP. In a study published in 2021, AZP2006 increased levels of the protein progranulin (PGRN) [127]. PGRN is a glycoprotein that is secreted or transported to lysosomes in neurons and the immune system [127]. It is associated with regulating neuroinflammation, neurite branching and outgrowth, lysosome function, and neuron survival [127]. High levels of PGRN, broken down into granulin, are associated with pathological conditions, while low circulating levels are associated with neurodegenerative diseases such as FTD [127]. In animal models of FTD, PD, and AD, increasing levels of PGRN were found to have neuroprotective effects and reduce both the pathological and clinical features of the disease [127,128,129,130]. A study investigating the effect of PGRN levels on rat cortical neurons and microglia exposed to amyloid beta showed that AZP2006 increased PGRN release and exerted an anti-inflammatory effect on the neurons. The effects of AZP2006 were also tested in vivo in a mouse model of AD as well as a mouse model of senescence [127]. In both models, treatment with AZP2006 reduced cognitive impairment, prevented loss of central synapses and neurons, and reduced tau hyperphosphorylation [127]. Currently, AZP2006 is in a phase II clinical trial to evaluate its tolerability, safety, pharmacokinetics, and effect on CSF biomarkers in patients with PSP. Thirty-six participants will be enrolled and randomized into treatment and control groups. Treatment groups received either 60 mg/day of oral AZP2006 for 84 days or 80 mg/day for 10 days, followed by 50 mg/day for 74 days. The placebo group received a daily oral dose of a placebo solution for 84 days. Primary outcomes included adverse events and pharmacokinetics of AZP2006 as evaluated by CSF biomarkers (NCT04008355). The latest update on this study was in February 2023, and the results have not yet been published.
Enhancing Tau Degradation
Other treatment options being studied are those targeting pathologic tau degradation pathways. In a study of Drosophila melanogaster models of human tau pathology, mammalian neuroblastoma cells, and a mouse model of tau overexpression, microRNAs that enhanced tau degradation were identified. In the Drosophila model, the miR-9 family of microRNAs was found to enhance tau degradation. The miR-9 target gene CG11070 and its mammalian orthologue UBE4B, an E3/E4 ubiquitin ligase, were found to reduce neurodegeneration and pathologic phenotypes in Drosophila models of human tau overexpression [131]. Total and phosphorylated tau levels were also decreased with overexpression of CG11070 and UBE4B. In the mammalian neuroblastoma cells, overexpression of UBE4B and STUB1, which encodes the E3 ligase CHIP (C-terminus of Hsc70 interacting protein), was found to increase ubiquitination and degradation of tau [131]. In addition, in a mouse model of tau overexpression, overexpression of UBE4B and STUB1 was found to increase oligomeric tau degradation [131]. Overall, UBE4B and STUB1 may serve as potential treatment targets for tau-based pathologies but have yet to be tested in humans [131].
Inhibition of Microglial Activation
Another treatment approach is via the inhibition of microglial activation. Current animal studies are investigating strategies to target microglia-specific fractalkine receptors (e.g., CX3CR1), as well as interleukin (IL)-1β, which may assist in slowing or arresting tau spread [17, 37]. Fractalkine (CX3CL1) is a chemokine that regulates inflammation via interaction with neuron–microglia communication [132]. Neurons secrete CX3CL1 in both soluble and membrane-bound forms [132,133,134]. Plasma levels of CX3CL1 were increased in the early stages of AD and decreased in severe AD [132, 135]. In mouse models lacking CX3CR1, tau hyperphosphorylation was enhanced, and this hyperphosphorylation was dependent on functional toll-like receptor (TLR) 4 and IL-1 receptors [37]. In a separate study, overexpression of fractalkine significantly reduced tau pathology in a mouse model of tau deposition [136]. Further research is necessary to elucidate the relationship between microglial activity, fractalkine, IL-1, and TLR signaling pathways, and tau production and aggregation in the pathogenesis of tauopathies.
Other Treatment Approaches
In Progress/Incomplete
Aside from the approaches noted above, other treatment methods have been attempted and are currently being tested.
TPN-101 is another drug currently undergoing clinical trials for the treatment of PSP. A phase IIa trial (NCT04993768) is evaluating the safety of administering TPN-101 versus placebo. TPN-101 is a derivative of the nucleotide reverse transcriptase inhibitor stavudine, which has traditionally been used in the treatment of human immunodeficiency virus. Antiretroviral drugs have previously been studied for treating ALS and FTD. In these two neurodegenerative disorders, loss of the nuclear RNA binding protein transactive response DNA binding protein 43 (TDP-43) into cytoplasmic aggregates is correlated with the development of neurodegeneration in ALS and FTD [137]. When studied in postmortem human brain tissue from patients with FTD or ALS, nuclear TDP-43 loss was associated with gene expression changes that affected RNA processing, nucleocytoplasmic transport, histone processing, and DNA damage [137]. The loss of TDP-43 was also associated with chromatin decondensation around long interspersed nuclear elements (LINEs), as well as increased LINE1 DNA content [137]. Loss of TDP-43 in cultured human neuronal cells was associated with increased retrotransposition, and this increased accumulation of RNA products resulted in an immune response, which is thought to drive the neuroinflammation and neurodegeneration in patients with ALS or FTD [137, 138]. When studied in Drosophila models of ALS and FTD, the use of TPN-101 was found to prolong lifespan and suppress dendritic collapse [139, 140]. The ongoing phase IIa trial is a multicenter, double-blind, randomized, placebo-controlled parallel group four-arm treatment study with an open-label treatment stage in patients with PSP. Patients undergo 24 weeks of treatment with either 100, 200, or 400 mg daily of TPN-101 or placebo. Subsequently, all patients from the treatment and control arms will receive 4 weeks of open-label treatment with 400 mg/day of TPN-101. Patients undergo evaluation 13 times within 52 weeks. Primary treatment outcomes include the number of treatment-related adverse events compared to placebo and the pharmacokinetics and pharmacodynamics of TPN-101. Clinical outcomes, as assessed by the PSPRS, are also being evaluated. The study was expected to enroll 40 participants and is anticipated to be completed in December 2023.
The Rho-dependent protein kinases ROCK1 and ROCK2 have also been identified as potential drivers of disease progression in PSP [141]. Levels of ROCK1 and ROCK2, as well as p70 S6 kinase and mammalian target of rapamycin, are all increased in the brains of humans with PSP and CBD [141]. Previous studies have shown that pharmacologic inhibition of Rho kinases in neurons reduced levels of detergent-soluble and insoluble tau via stimulation of autophagy, leading to protein aggregate degradation and suppression of tau mRNA production [141]. Fasudil, a clinically approved ROCK inhibitor, was also found to mitigate pathogenic 4R tau levels by inducing autophagic pathways in Drosophila models of tauopathy [141]. Fasudil is currently undergoing a phase IIa open-label clinical trial (NCT04734379) to evaluate its safety, tolerability, and effect on biomarkers of neurodegeneration in patients with PSP and CBD. Fifteen patients with MRIs consistent with PSP-RS or CBD will be enrolled, and monitoring of adverse events is the primary outcome. Secondary outcomes include phosphorylated tau levels. Biomarkers of neurodegeneration, including Nfl and total tau fragment levels, and imaging biomarkers of neurodegeneration, including changes in brain volume and white matter integrity, are also under evaluation. Patients received 180 mg/day of orally administered fasudil, and treatment continued for 48 weeks. Patients were evaluated at week 1 and weeks 12, 24, 36, and 48; they will also be evaluated at week 52 posttreatment. The study was estimated to be completed in November 2023, but no results have been released yet.
NBMI (N1,N3-bis(2-mercaptoethyl)isophthalamide) is a metal chelator drug and antioxidant that has also undergone clinical trials for the treatment of neurodegenerative diseases [13, 142]. The efficacy and safety of using NBMI in the treatment of PSP and MSA were evaluated in a phase IIa trial (NCT04184063). In this study, 20 patients diagnosed with PSP or MSA were treated for 28 days with either orally administered NBMI or placebo, and primary outcomes included changes in disease progression regarding motor and nonmotor symptoms of PSP and MSA. This study was completed in June 2021, but results have not yet been reported.
Alpha-lipoic acid and acetyl l-carnitine, which act as antioxidants, have also been studied as potential treatments for PSP [143]. In a phase I/II human trial of 11 participants with PSP (NCT01537549), each participant received 600 mg of alpha-lipoic acid and 1.5 g of acetyl l-carnitine daily for 6 months. The most common adverse effects included restlessness, dizziness, insomnia, and seizures. Information regarding the effects on cerebral oxidative stress markers has yet to be published.
A clinical pilot trial initiated in 2004 tested the efficacy of dietary supplements pyruvate, creatinine, and niacinamide, which act as antioxidants, in slowing disease progression in 20 participants with PSP (NCT00605930) [143]. This trial was last updated in 2017, and results have not yet been released.
Coenzyme Q10 (CoQ10), which is also thought to serve as an antioxidant, has also been studied in a phase II double-blind, randomized, placebo-controlled clinical trial (NCT00382824) to evaluate its efficacy, tolerability, and safety in treating PSP [143]. CoQ10 has previously been identified as a potentially beneficial treatment in PD, and this study evaluated whether that treatment benefit could also apply to patients with PSP [144]. The study enrolled 61 participants. Patients in the treatment arm received 2400 mg/day of CoQ10, while patients in the control group received a matching dose of placebo. Primary outcomes after 12 months of treatment included changes from baseline in PSPRS scores and Unified Parkinson’s Disease Rating Scale scores. Secondary outcome measurements included activities of daily living and Mini Mental State Examination scores, which were monitored at baseline and 3, 6, 9, and 12 months [145]. The treatment was safe and well tolerated, but there were no statistically significant differences from baseline in primary or secondary outcomes between placebo and treatment groups [145]. CoQ10 did not significantly slow disease progression or improve symptoms in patients with PSP [145]. Another phase II trial (NCT00328874) was conducted to evaluate the effect of CoQ10 treatment on brain energy metabolites in patients with PSP as quantified by MRI, as well as on clinical symptoms of PSP. The results of this study have not yet been published.
Treatment of neurological conditions such as PSP with stem cells is also currently in clinical trials. In an ongoing clinical trial (NCT02795052), 500 participants with neurological disorders, including PSP, will undergo an autologous bone marrow aspiration and separation of bone marrow-derived stem cell fraction, which will then be administered intravenously and intranasally in the lower one-third of the nasal passages. Primary outcome includes changes in neurological function, which will be assessed before treatment and 1, 3, 6, and 12 months after treatment. This study is estimated to be completed in July 2025.
A phase II clinical trial (NCT00005903) sponsored by the National Institute of Neurological Disorders and Stroke sought to evaluate the safety and initial efficacy of continuously infused recombinant methionyl human glial-derived neurotrophic factor (GDNF) in the treatment of patients with PSP. Neurotrophic factors are proteins that activate cell signaling pathways that regulate neuronal survival, differentiation, growth, and regeneration [146]. However, these neurotrophic factors have been challenging to study, given their limited penetration of the blood–brain barrier [146]. In animal models of PD, continuous GDNF infusion or administration of a viral vector into the cerebral ventricles or directly into the denervated putamen had neuroprotective and neuroregenerative effects [146].
Previous studies indicated that infusion of GDNF into the brains of patients with PD via an intracerebroventricular catheter showed no benefit and instead led to significant side effects, including nausea, vomiting, anorexia, weight loss, paresthesia, and hyponatremia [146].
In the most recent study on patients with PSP, all patients had two catheters, one on each side of the brain, which delivered saline or GDNF directly into the brain for 6 months through an infusion pump placed in the abdomen. Half of the patients received a placebo in the right side of the brain and GDNF in the left; the other half received GDNF in the right side of the brain and a placebo in the left. Efficacy was evaluated by neurologic function as well as by biochemical and radiographic methods. The last update on this study was reported in 2008, and no results have been published yet. The utility of BDNF and GDNF has also been highlighted in a prior atypical parkinsonian disorder treatment review as a potential treatment for MSA [13]. In mouse models of MSA, GDNF infusion reduced neuropathological progression and improved motor dysfunction [13, 147].
Additionally, increased levels of BDNF and GDNF in transgenic mouse brains via administration of fluoxetine, a selective serotonin reuptake inhibitor, improved motor skills, which further suggests the relevance of exploring these treatment modalities in other neurodegenerative diseases such as PSP [13, 148].
Complete
Riluzole is a benzothiazole drug that functions as a glutamatergic signaling modulator and was tested in the phase III trial Neuroprotection and Natural History in Parkinson Plus Syndromes (NNIPPS) (NCT00211224) for the management of patients with PSP or MSA. Riluzole has been found to have multiple effects that aid in neuroprotection in preclinical studies, including anti-excitotoxic activity, blocking of voltage-dependent sodium channels, scavenging of free radicals, anti-apoptotic effects, neurotrophic effects, and inhibition of protein aggregation [149,150,151,152,153,154]. Riluzole has previously been identified to prolong survival in patients with ALS [149, 155]. The primary outcome of this phase III trial was survival. In this study, 760 patients with PSP or MSA were randomized to receive riluzole or placebo daily with follow-up at 36 months. However, riluzole did not affect the survival or disease progression of patients with Parkinson-plus syndromes [91, 149].
RT001, a stabilized fatty acid compound, is being investigated as a treatment for PSP. RT001 is designed to prevent lipid peroxidation [1, 156]. Lipid peroxidation is selectively involved in the pathogenesis of PSP, along with the accumulation and aggregation of pathological tau [156, 157]. On autopsy evaluation of the brains of individuals with PSP, lipid peroxidation byproducts such as toxic aldehydes were found in brain areas selectively involved in PSP [157]. In addition, previous studies in patients with PSP have identified defects in the oxidative phosphorylation in their muscle mitochondria [156, 158]. RT001 is a deuterated isotope of linoleic acid designed to make membrane polyunsaturated fatty acids resistant to lipid peroxidation [156]. In this most recent study published in 2021, the effects of RT001 on various oxidative and biogenetic parameters were tested in the mesenchymal stem cells derived from patients with PSP (NCT01824121) [156]. In the mesenchymal stem cells of the participants, in vitro incubation with RT001 was found to reduce lipid peroxidation and mitochondrial reactive oxygen species production, as well as improve other measures of mitochondrial function [156]. The study also reported the results of expanded-access use of RT001 in three patients with PSP; each was treated with the medication for over 27 months [156]. Oral use of RT001 in these patients was well tolerated, and the treatment stabilized the rate of decline in the patient’s functional rating scales over time [156]. As a result of the possibility of a placebo effect, as well as the small sample size and lack of a control group, further study of RT001 using randomized controlled trials is warranted [156].
A phase I clinical trial (NCT01824121) was conducted to test the efficacy of autologous mesenchymal stem cells to treat PSP by administration into the internal carotid artery. This study included five patients who were evaluated 1 year after infusion. All patients had survived except for one, who had died from an accidental fall not related to cell administration or disease progression. After 1 year, the treated patients had stable motor function rating scale scores for at least 6 months during the 1-year follow-up [159]. In a follow-up study, two more patients were treated with mesenchymal stem cell therapy [160]. Six of the seven total patients were living 1 year after cell infusion, and asymptomatic spotty lesions were observed by brain MRI in six of the seven patients [160]. The last patient in the preliminary cohort exhibited transient symptomatic ischemic alterations [160]. No severe adverse events were noted in the last two treated patients [160]. One patient had an increase in motor function 3 months after cell infusion [160]. Another patient had a deterioration in motor function during the 6-month follow-up period [160]. Finally, another patient had a decline in the PSPRS score at the 6-month follow-up, which persisted at the 1-year follow-up [160]. As noted by a prior review discussing the treatment of atypical parkinsonian disorders, mesenchymal stem cell therapy has been studied in patients with MSA and was safe and well tolerated in those patients. In addition, intrathecal administration of adipose-obtained mesenchymal stem cells was associated with slower disease progression in patients with MSA [13, 161]. Overall, although these studies provided valuable initial information on the treatment of rare diseases with mesenchymal stem cells, further investigation into methods to increase safety and efficacy in treating PSP is pertinent [160].
Conclusion
Over the years, progress has been made in improving diagnostic guidelines for PSP, as well as exploring treatment options by approaching different steps in the pathology and development of PSP. However, there is still significantly more ground to be covered in the diagnosis and treatment of PSP. Although many of the prior clinical trials had robust features such as high patient numbers in some trials, the evaluation of treatment effects by multiple measures, and the comparison of treatment effects in different disease processes, they also had limitations related to PSP. One of these limitations is diagnostic uncertainty in PSP, leading to difficulty confirming diagnostic accuracy. Another limitation is the small number of patients in many trials. Additionally, given that PSP often presents late in the disease course, many of these interventions were initiated very late in the disease process. This latency has made it challenging to assess the utility of the treatment, as treatments could have been initiated so late in disease progression that they would no longer make a difference. Therefore, prioritizing earlier and more accurate methods for diagnosing PSP will be pertinent as trials continue to be performed.
Many current trials have been discussed above, and there is much more work to be done in the treatment of these types of neurodegenerative disorders. As discussed above, there are many theories for underlying causes of PSP, many of which have developing therapies underway. With the recognition of some potentially modifiable risk factors in the development of PSP, future treatments may target prevention by managing these underlying risk factors, given the challenges of producing a disease-modifying therapy. Additionally, with recognition of the different phenotypic variations of PSP, further research into their pathological variances may provide avenues for more individualized patient treatment. Finally, with the improvement of diagnostic criteria and earlier diagnosis, treatment options may be more effective and feasible in managing PSP pathology before it becomes irreversible.
PSP is a progressive neurodegenerative disease that is universally fatal; there are no disease-modifying treatments. Currently, treatment includes symptomatic therapy with symptom-directed medication, physical therapy, and occupational therapy. Many drug trials, most targeting tau dysfunction, aggregation, and posttranslational modifications, are ineffective at this point. However, several novel treatments are currently undergoing clinical trials and may hold promise as disease-modifying therapies for individuals with tauopathies in the future. As a result, the continued pursuit of effective tau-targeted therapies to treat neurodegenerative diseases such as PSP is highly relevant. With the advent of medications targeting tau, the prognosis of patients living with PSP could be drastically improved, as well as the lives of other patients living with tauopathies such as CBD and AD.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
References
Coughlin DG, Litvan I. Progressive supranuclear palsy: advances in diagnosis and management. Parkinsonism Relat Disord. 2020;73:105–16.
Giagkou N, Hoglinger GU, Stamelou M. Progressive supranuclear palsy. Int Rev Neurobiol. 2019;149:49–86.
Coyle-Gilchrist IT, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86(18):1736–43.
Fleury V, Brindel P, Nicastro N, Burkhard PR. Descriptive epidemiology of parkinsonism in the Canton of Geneva, Switzerland. Parkinsonism Relat Disord. 2018;54:30–9.
Kawashima M, Miyake M, Kusumi M, Adachi Y, Nakashima K. Prevalence of progressive supranuclear palsy in Yonago, Japan. Mov Disord. 2004;19(10):1239–40.
Nath U, Ben-Shlomo Y, Thomson RG, et al. The prevalence of progressive supranuclear palsy (Steele-Richardson-Olszewski syndrome) in the UK. Brain. 2001;124(Pt 7):1438–49.
Swallow DMA, Counsell CE. Prevalence of progressive supranuclear palsy and corticobasal syndrome in Scotland. Neuroepidemiology. 2022;56(4):291–7.
Zermansky A, Ben-Shlomo Y. Epidemiology of progressive supranuclear palsy and multiple system atrophy. In: Litvan I, editor. Atypical parkinsonian disorders. Totowa: Humana; 2005. p. 23–31.
Binder LI, Frankfurter A, Rebhun LI. The distribution of tau in the mammalian central nervous system. J Cell Biol. 1985;101(4):1371–8.
Bowles KR, Pugh DA, Oja LM, et al. Dysregulated coordination of MAPT exon 2 and exon 10 splicing underlies different tau pathologies in PSP and AD. Acta Neuropathol. 2022;143(2):225–43.
Goedert M, Spillantini MG, Potier MC, Ulrich J, Crowther RA. Cloning and sequencing of the cDNA encoding an isoform of microtubule-associated protein tau containing four tandem repeats: differential expression of tau protein mRNAs in human brain. EMBO J. 1989;8(2):393–9.
Ling H, Macerollo A. Is it useful to classify PSP and CBD as different disorders? Yes. Mov Disord Clin Pract. 2018;5(2):145–8.
Przewodowska D, Marzec W, Madetko N. Novel therapies for parkinsonian syndromes-recent progress and future perspectives. Front Mol Neurosci. 2021;14:720220.
Monzio Compagnoni G, Di Fonzo A. Understanding the pathogenesis of multiple system atrophy: state of the art and future perspectives. Acta Neuropathol Commun. 2019;7(1):113.
Wen Y, Zhou Y, Jiao B, Shen L. Genetics of progressive supranuclear palsy: a review. J Parkinsons Dis. 2021;11(1):93–105.
Jabbari E, Koga S, Valentino RR, et al. Genetic determinants of survival in progressive supranuclear palsy: a genome-wide association study. Lancet Neurol. 2021;20(2):107–16.
Boxer AL, Yu JT, Golbe LI, Litvan I, Lang AE, Hoglinger GU. Advances in progressive supranuclear palsy: new diagnostic criteria, biomarkers, and therapeutic approaches. Lancet Neurol. 2017;16(7):552–63.
Hoglinger GU, Melhem NM, Dickson DW, et al. Identification of common variants influencing risk of the tauopathy progressive supranuclear palsy. Nat Genet. 2011;43(7):699–705.
Sanchez-Contreras MY, Kouri N, Cook CN, et al. Replication of progressive supranuclear palsy genome-wide association study identifies SLCO1A2 and DUSP10 as new susceptibility loci. Mol Neurodegener. 2018;13(1):37.
Hanger DP, Anderton BH, Noble W. Tau phosphorylation: the therapeutic challenge for neurodegenerative disease. Trends Mol Med. 2009;15(3):112–9.
Wray S, Saxton M, Anderton BH, Hanger DP. Direct analysis of tau from PSP brain identifies new phosphorylation sites and a major fragment of N-terminally cleaved tau containing four microtubule-binding repeats. J Neurochem. 2008;105(6):2343–52.
Irwin DJ, Cohen TJ, Grossman M, et al. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain. 2012;135(Pt 3):807–18.
Schmidt ML, Schuck T, Sheridan S, et al. The fluorescent Congo red derivative, (trans, trans)-1-bromo-2,5-bis-(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (BSB), labels diverse beta-pleated sheet structures in postmortem human neurodegenerative disease brains. Am J Pathol. 2001;159(3):937–43.
Stamelou M, Boxer AL. Disease-modifying treatments for progressive supranuclear palsy. Mov Disord Clin Pract. 2015;2(1):3–5.
Samimi N, Sharma G, Kimura T, et al. Distinct phosphorylation profiles of tau in brains of patients with different tauopathies. Neurobiol Aging. 2021;108:72–9.
Liu L, Drouet V, Wu JW, et al. Trans-synaptic spread of tau pathology in vivo. PLoS One. 2012;7(2):e31302.
Jackson NA, Guerrero-Munoz MJ, Castillo-Carranza DL. The prion-like transmission of tau oligomers via exosomes. Front Aging Neurosci. 2022;14:974414.
Polanco JC, Scicluna BJ, Hill AF, Gotz J. Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J Biol Chem. 2016;291(24):12445–66.
Fevrier B, Vilette D, Archer F, et al. Cells release prions in association with exosomes. Proc Natl Acad Sci USA. 2004;101(26):9683–8.
Alavi Naini SM, Soussi-Yanicostas N. Tau hyperphosphorylation and oxidative stress, a critical vicious circle in neurodegenerative tauopathies? Oxid Med Cell Longev. 2015;2015:151979.
Borza LR. A review on the cause-effect relationship between oxidative stress and toxic proteins in the pathogenesis of neurodegenerative diseases. Rev Med Chir Soc Med Nat Iasi. 2014;118(1):19–27.
Stamer K, Vogel R, Thies E, Mandelkow E, Mandelkow EM. Tau blocks traffic of organelles, neurofilaments, and APP vesicles in neurons and enhances oxidative stress. J Cell Biol. 2002;156(6):1051–63.
Dias-Santagata D, Fulga TA, Duttaroy A, Feany MB. Oxidative stress mediates tau-induced neurodegeneration in Drosophila. J Clin Investig. 2007;117(1):236–45.
Kim SY, Kim TB, Moon KA, et al. Regulation of pro-inflammatory responses by lipoxygenases via intracellular reactive oxygen species in vitro and in vivo. Exp Mol Med. 2008;40(4):461–76.
Giannopoulos PF, Chu J, Sperow M, et al. Pharmacologic inhibition of 5-lipoxygenase improves memory, rescues synaptic dysfunction, and ameliorates tau pathology in a transgenic model of tauopathy. Biol Psychiatry. 2015;78(10):693–701.
Maphis N, Xu G, Kokiko-Cochran ON, et al. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain. 2015;138(Pt 6):1738–55.
Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68(1):19–31.
Usenovic M, Niroomand S, Drolet RE, et al. Internalized tau oligomers cause neurodegeneration by inducing accumulation of pathogenic tau in human neurons derived from induced pluripotent stem cells. J Neurosci. 2015;35(42):14234–50.
Dickson DW, Kouri N, Murray ME, Josephs KA. Neuropathology of frontotemporal lobar degeneration-tau (FTLD-tau). J Mol Neurosci. 2011;45(3):384–9.
Hauw JJ, Daniel SE, Dickson D, et al. Preliminary NINDS neuropathologic criteria for Steele–Richardson–Olszewski syndrome (progressive supranuclear palsy). Neurology. 1994;44(11):2015–9.
Kovacs GG, Lukic MJ, Irwin DJ, et al. Distribution patterns of tau pathology in progressive supranuclear palsy. Acta Neuropathol. 2020;140(2):99–119.
Magdalinou NK, Paterson RW, Schott JM, et al. A panel of nine cerebrospinal fluid biomarkers may identify patients with atypical parkinsonian syndromes. J Neurol Neurosurg Psychiatry. 2015;86(11):1240–7.
Wagshal D, Sankaranarayanan S, Guss V, et al. Divergent CSF tau alterations in two common tauopathies: Alzheimer’s disease and progressive supranuclear palsy. J Neurol Neurosurg Psychiatry. 2015;86(3):244–50.
Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577–89.
Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75(1):116–26.
Boxer AL, Lang AE, Grossman M, et al. Davunetide in patients with progressive supranuclear palsy: a randomised, double-blind, placebo-controlled phase 2/3 trial. Lancet Neurol. 2014;13(7):676–85.
Rojas JC, Karydas A, Bang J, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. 2016;3(3):216–25.
Kato N, Arai K, Hattori T. Study of the rostral midbrain atrophy in progressive supranuclear palsy. J Neurol Sci. 2003;210(1–2):57–60.
Adachi M, Kawanami T, Ohshima H, Sugai Y, Hosoya T. Morning glory sign: a particular MR finding in progressive supranuclear palsy. Magn Reson Med Sci. 2004;3(3):125–32.
Massey LA, Micallef C, Paviour DC, et al. Conventional magnetic resonance imaging in confirmed progressive supranuclear palsy and multiple system atrophy. Mov Disord. 2012;27(14):1754–62.
Quattrone A, Nicoletti G, Messina D, et al. MR imaging index for differentiation of progressive supranuclear palsy from Parkinson disease and the Parkinson variant of multiple system atrophy. Radiology. 2008;246(1):214–21.
Quattrone A, Morelli M, Williams DR, et al. MR parkinsonism index predicts vertical supranuclear gaze palsy in patients with PSP-parkinsonism. Neurology. 2016;87(12):1266–73.
Nigro S, Arabia G, Antonini A, et al. Magnetic Resonance Parkinsonism Index: diagnostic accuracy of a fully automated algorithm in comparison with the manual measurement in a large Italian multicentre study in patients with progressive supranuclear palsy. Eur Radiol. 2017;27(6):2665–75.
Alster P, Migda B, Madetko N, et al. The role of frontal assessment battery and frontal lobe single-photon emission computed tomography in the differential diagnosis of progressive supranuclear palsy variants and corticobasal syndrome—a pilot study. Front Neurol. 2021;12: 630153.
Alster P, Nieciecki M, Koziorowski D, et al. Is brain perfusion a differentiating feature in the comparison of progressive supranuclear palsy syndrome (PSPS) and corticobasal syndrome (CBS)? J Clin Neurosci. 2020;77:123–7.
Alster P, Nieciecki M, Koziorowski DM, et al. Thalamic and cerebellar hypoperfusion in single photon emission computed tomography may differentiate multiple system atrophy and progressive supranuclear palsy. Medicine (Baltimore). 2019;98(30):e16603.
Williams DR, Lees AJ. Progressive supranuclear palsy: clinicopathological concepts and diagnostic challenges. Lancet Neurol. 2009;8(3):270–9.
Owens E, Josephs KA, Savica R, et al. The clinical spectrum and natural history of pure akinesia with gait freezing. J Neurol. 2016;263(12):2419–23.
Constantinides VC, Paraskevas GP, Paraskevas PG, Stefanis L, Kapaki E. Corticobasal degeneration and corticobasal syndrome: a review. Clin Parkinson Relat Disord. 2019;1:66–71.
Rusina R, Bajtosova R, Csefalvay Z, et al. Comorbid neurodegeneration in primary progressive aphasia: clinicopathological correlations in a single-center study. Behav Neurol. 2022;2022:6075511.
Litvan I, Agid Y, Calne D, et al. Clinical research criteria for the diagnosis of progressive supranuclear palsy (Steele–Richardson–Olszewski syndrome): report of the NINDS-SPSP international workshop. Neurology. 1996;47(1):1–9.
Rabadia SV, Litvan I, Juncos J, et al. Hypertension and progressive supranuclear palsy. Parkinsonism Relat Disord. 2019;66:166–70.
Kwasny MJ, Oleske DM, Zamudio J, Diegidio R, Hoglinger GU. Clinical features observed in general practice associated with the subsequent diagnosis of progressive supranuclear palsy. Front Neurol. 2021;12: 637176.
Bharadwaj P, Wijesekara N, Liyanapathirana M, et al. The link between type 2 diabetes and neurodegeneration: roles for amyloid-β, amylin, and tau proteins. J Alzheimers Dis. 2017;59(2):421–32.
Alster P, Dunalska A, Migda B, Madetko N, Krolicki L. The rate of decrease in brain perfusion in progressive supranuclear palsy and corticobasal syndrome may be impacted by glycemic variability—a pilot study. Front Neurol. 2021;12: 767480.
Bassil F, Canron MH, Vital A, et al. Insulin resistance and exendin-4 treatment for multiple system atrophy. Brain. 2017;140(5):1420–36.
Josephs KA, Ishizawa T, Tsuboi Y, Cookson N, Dickson DW. A clinicopathological study of vascular progressive supranuclear palsy: a multi-infarct disorder presenting as progressive supranuclear palsy. Arch Neurol. 2002;59(10):1597–601.
Koga S, Roemer SF, Kasanuki K, Dickson DW. Cerebrovascular pathology presenting as corticobasal syndrome: an autopsy case series of “vascular CBS.” Parkinsonism Relat Disord. 2019;68:79–84.
Chang JK, Leso A, Subaiea GM, et al. Tolfenamic acid: a modifier of the tau protein and its role in cognition and tauopathy. Curr Alzheimer Res. 2018;15(7):655–63.
Lamb R, Rohrer JD, Lees AJ, Morris HR. Progressive supranuclear palsy and corticobasal degeneration: pathophysiology and treatment options. Curr Treat Options Neurol. 2016;18(9):42.
Pilotto A, Rizzetti MC, Lombardi A, et al. Cerebellar rTMS in PSP: a double-blind sham-controlled study using mobile health technology. Cerebellum. 2021;20(4):662–6.
Scelzo E, Lozano AM, Hamani C, et al. Peduncolopontine nucleus stimulation in progressive supranuclear palsy: a randomised trial. J Neurol Neurosurg Psychiatry. 2017;88(7):613–6.
Clerici I, Ferrazzoli D, Maestri R, et al. Rehabilitation in progressive supranuclear palsy: effectiveness of two multidisciplinary treatments. PLoS One. 2017;12(2):e0170927.
Sale P, Stocchi F, Galafate D, et al. Effects of robot assisted gait training in progressive supranuclear palsy (PSP): a preliminary report. Front Hum Neurosci. 2014;8:207.
Liepelt I, Gaenslen A, Godau J, et al. Rivastigmine for the treatment of dementia in patients with progressive supranuclear palsy: clinical observations as a basis for power calculations and safety analysis. Alzheimers Dement. 2010;6(1):70–4.
Litvan I, Phipps M, Pharr VL, Hallett M, Grafman J, Salazar A. Randomized placebo-controlled trial of donepezil in patients with progressive supranuclear palsy. Neurology. 2001;57(3):467–73.
Novak P, Pimentel Maldonado DA, Novak V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: a double-blinded placebo-controlled pilot study. PLoS One. 2019;14(4):e0214364.
Khanna MR, Kovalevich J, Lee VM, Trojanowski JQ, Brunden KR. Therapeutic strategies for the treatment of tauopathies: hopes and challenges. Alzheimers Dement. 2016;12(10):1051–65.
Zhang B, Carroll J, Trojanowski JQ, et al. The microtubule-stabilizing agent, epothilone D, reduces axonal dysfunction, neurotoxicity, cognitive deficits, and Alzheimer-like pathology in an interventional study with aged tau transgenic mice. J Neurosci. 2012;32(11):3601–11.
Cleveland DW, Connolly JA, Kalnins VI, Spiegelman BM, Kirschner MW. Physical properties and cellular localization of tau, a microtubule-associated protein which induces assembly of purified tubulin. J Cell Biol. 1977;75:A283.
Cleveland DW, Hwo SY, Kirschner MW. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J Mol Biol. 1977;116(2):227–47.
Lee VM, Daughenbaugh R, Trojanowski JQ. Microtubule stabilizing drugs for the treatment of Alzheimer’s disease. Neurobiol Aging. 1994;15(Suppl 2):S87–9.
Magen I, Ostritsky R, Richter F, et al. Intranasal NAP (davunetide) decreases tau hyperphosphorylation and moderately improves behavioral deficits in mice overexpressing alpha-synuclein. Pharmacol Res Perspect. 2014;2(5):e00065.
Tsai RM, Miller Z, Koestler M, et al. Reactions to multiple ascending doses of the microtubule stabilizer TPI-287 in patients with Alzheimer disease, progressive supranuclear palsy, and corticobasal syndrome: a randomized clinical trial. JAMA Neurol. 2020;77(2):215–24.
Brunden KR, Yao Y, Potuzak JS, et al. The characterization of microtubule-stabilizing drugs as possible therapeutic agents for Alzheimer’s disease and related tauopathies. Pharmacol Res. 2011;63(4):341–51.
Vaswani PA, Olsen AL. Immunotherapy in progressive supranuclear palsy. Curr Opin Neurol. 2020;33(4):527–33.
Mudher A, Colin M, Dujardin S, et al. What is the evidence that tau pathology spreads through prion-like propagation? Act Neuropathol Commun. 2017;5(1):99.
Kondo A, Shahpasand K, Mannix R, et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature. 2015;523(7561):431–6.
Yanamandra K, Kfoury N, Jiang H, et al. Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron. 2013;80(2):402–14.
Buchanan T, De Bruyn S, Fadini T, Watanabe S, Germani M, Mesa A, editors. A randomised, placebo-controlled, first-in-human study with a central tau epitope antibody–UCB0107. Int Congr Park Dis Mov Disord Nice; 2019 Sept 22–26, 2019.
Dam T, Boxer AL, Golbe LI, et al. Safety and efficacy of anti-tau monoclonal antibody gosuranemab in progressive supranuclear palsy: a phase 2, randomized, placebo-controlled trial. Nat Med. 2021;27(8):1451–7.
Qureshi IA, Tirucherai G, Ahlijanian MK, Kolaitis G, Bechtold C, Grundman M. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement (N Y). 2018;4:746–55.
Boxer AL, Qureshi I, Ahlijanian M, et al. Safety of the tau-directed monoclonal antibody BIIB092 in progressive supranuclear palsy: a randomised, placebo-controlled, multiple ascending dose phase 1b trial. Lancet Neurol. 2019;18(6):549–58.
Hoglinger GU, Litvan I, Mendonca N, et al. Safety and efficacy of tilavonemab in progressive supranuclear palsy: a phase 2, randomised, placebo-controlled trial. Lancet Neurol. 2021;20(3):182–92.
Koga S, Dickson DW, Wszolek ZK. Neuropathology of progressive supranuclear palsy after treatment with tilavonemab. Lancet Neurol. 2021;20(10):786–7.
West T, Hu Y, Verghese PB, et al. Preclinical and clinical development of ABBV-8E12, a humanized anti-tau antibody, for treatment of Alzheimer’s disease and other tauopathies. J Prev Alzheimers Dis. 2017;4(4):236–41.
Beck G, Yamashita R, Kido K, et al. An autopsy case of progressive supranuclear palsy treated with monoclonal antibody against tau. Neuropathology. 2023;43(4):326–32.
Novak P, Zilka N, Zilkova M, et al. AADvac1, an active immunotherapy for Alzheimer’s disease and non Alzheimer tauopathies: an overview of preclinical and clinical development. J Prev Alzheimers Dis. 2019;6(1):63–9.
Panza F, Solfrizzi V, Seripa D, et al. Tau-based therapeutics for Alzheimer’s disease: active and passive immunotherapy. Immunotherapy. 2016;8(9):1119–34.
Novak P, Schmidt R, Kontsekova E, et al. Safety and immunogenicity of the tau vaccine AADvac1 in patients with Alzheimer’s disease: a randomised, double-blind, placebo-controlled, phase 1 trial. Lancet Neurol. 2017;16(2):123–34.
Novak P, Schmidt R, Kontsekova E, et al. FUNDAMANT: an interventional 72-week phase 1 follow-up study of AADvac1, an active immunotherapy against tau protein pathology in Alzheimer’s disease. Alzheimers Res Ther. 2018;10:108.
Vaz M, Silvestre S. Alzheimer’s disease: recent treatment strategies. Eur J Pharmacol. 2020;887:173554.
Novak P, Kovacech B, Katina S. et al. ADAMANT: a placebo-controlled randomized phase 2 study of AADvac1, an active immunotherapy against pathological tau in Alzheimer's disease. Nat Aging. 2021;1(6):521–534. https://doi.org/10.1038/s43587-021-00070-2.
Theunis C, Crespo-Biel N, Gafner V, et al. Efficacy and safety of a liposome-based vaccine against protein Tau, assessed in tau.P301L mice that model tauopathy. PLoS One. 2013;8(8):e72301.
Shugart J. Two new stabs at vaccinating people against pathologic tau 2022. https://www.alzforum.org/news/conference-coverage/two-new-stabs-vaccinating-people-against-pathologic-tau. Accessed Mar 5, 2024.
Harrington CR, Storey JM, Clunas S, et al. Cellular models of aggregation-dependent template-directed proteolysis to characterize tau aggregation inhibitors for treatment of Alzheimer disease. J Biol Chem. 2015;290(17):10862–75.
Melis V, Magbagbeolu M, Rickard JE, et al. Effects of oxidized and reduced forms of methylthioninium in two transgenic mouse tauopathy models. Behav Pharmacol. 2015;26(4):353–68.
Wischik CM, Staff RT, Wischik DJ, et al. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer’s disease. J Alzheimers Dis. 2015;44(2):705–20.
Wischik CM, Edwards PC, Lai RY, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by phenothiazines. Proc Natl Acad Sci USA. 1996;93(20):11213–8.
Gauthier S, Feldman HH, Schneider LS, et al. Efficacy and safety of tau-aggregation inhibitor therapy in patients with mild or moderate Alzheimer’s disease: a randomised, controlled, double-blind, parallel-arm, phase 3 trial. Lancet. 2016;388(10062):2873–84.
Schelter BO, Shiells H, Baddeley TC, et al. Concentration-dependent activity of hydromethylthionine on cognitive decline and brain atrophy in mild to moderate Alzheimer’s disease. J Alzheimers Dis. 2019;72(3):931–46.
Kimura T, Ishiguro K, Hisanaga S. Physiological and pathological phosphorylation of tau by Cdk5. Front Mol Neurosci. 2014;7:65.
Borghi R, Giliberto L, Assini A, et al. Increase of cdk5 is related to neurofibrillary pathology in progressive supranuclear palsy. Neurology. 2002;58(4):589–92.
Adwan L, Subaiea GM, Zawia NH. Tolfenamic acid downregulates BACE1 and protects against lead-induced upregulation of Alzheimer’s disease related biomarkers. Neuropharmacology. 2014;79:596–602.
Adwan L, Subaiea GM, Basha R, Zawia NH. Tolfenamic acid reduces tau and CDK5 levels: implications for dementia and tauopathies. J Neurochem. 2015;133(2):266–72.
Tolosa E, Litvan I, Hoglinger GU, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014;29(4):470–8.
Leclair-Visonneau L, Rouaud T, Debilly B, et al. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin Neurol Neurosurg. 2016;146:35–9.
Min SW, Chen X, Tracy TE, et al. Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits. Nat Med. 2015;21(10):1154–62.
VandeVrede L, Dale ML, Fields S, et al. Open-label phase 1 futility studies of salsalate and young plasma in progressive supranuclear palsy. Mov Disord Clin Pract. 2020;7(4):440–7.
Min SW, Cho SH, Zhou Y, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67(6):953–66.
Ryan P, Xu M, Davey AK, et al. O-GlcNAc modification protects against protein misfolding and aggregation in neurodegenerative disease. ACS Chem Neurosci. 2019;10(5):2209–21.
Bartolome-Nebreda JM, Trabanco AA, Velter AI, Buijnsters P. O-GlcNAcase inhibitors as potential therapeutics for the treatment of Alzheimer’s disease and related tauopathies: analysis of the patent literature. Expert Opin Ther Pat. 2021;31(12):1117–54.
Schoch KM, DeVos SL, Miller RL, et al. Increased 4R-tau induces pathological changes in a human-tau mouse model. Neuron. 2016;90(5):941–7.
DeVos SL, Miller RL, Schoch KM, et al. Tau reduction prevents neuronal loss and reverses pathological tau deposition and seeding in mice with tauopathy. Sci Transl Med. 2017;9(374):eaag0481.
Smith RA, Miller TM, Yamanaka K, et al. Antisense oligonucleotide therapy for neurodegenerative disease. J Clin Investig. 2006;116(8):2290–6.
Marras C, Cunningham CR, Hou J, et al. Anti-inflammatory drug use and progressive supranuclear palsy. Parkinsonism Relat Disord. 2018;48:89–92.
Callizot N, Estrella C, Burlet S, et al. AZP2006, a new promising treatment for Alzheimer’s and related diseases. Sci Rep. 2021;11(1): 16806.
Arrant AE, Filiano AJ, Unger DE, Young AH, Roberson ED. Restoring neuronal progranulin reverses deficits in a mouse model of frontotemporal dementia. Brain. 2017;140(5):1447–65.
Mateo I, Gonzalez-Aramburu I, Pozueta A, et al. Reduced serum progranulin level might be associated with Parkinson’s disease risk. Eur J Neurol. 2013;20(12):1571–3.
Minami SS, Min SW, Krabbe G, et al. Progranulin protects against amyloid beta deposition and toxicity in Alzheimer’s disease mouse models. Nat Med. 2014;20(10):1157–64.
Subramanian M, Hyeon SJ, Das T, et al. UBE4B, a microRNA-9 target gene, promotes autophagy-mediated Tau degradation. Nat Commun. 2021;12(1):3291.
Hebron ML, Algarzae NK, Lonskaya I, Moussa C. Fractalkine signaling and Tau hyper-phosphorylation are associated with autophagic alterations in lentiviral Tau and Abeta1-42 gene transfer models. Exp Neurol. 2014;251:127–38.
Harrison JK, Jiang Y, Chen S, et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci USA. 1998;95(18):10896–901.
Hatori K, Nagai A, Heisel R, Ryu JK, Kim SU. Fractalkine and fractalkine receptors in human neurons and glial cells. J Neurosci Res. 2002;69(3):418–26.
Kim TS, Lim HK, Lee JY, et al. Changes in the levels of plasma soluble fractalkine in patients with mild cognitive impairment and Alzheimer’s disease. Neurosci Lett. 2008;436(2):196–200.
Nash KR, Lee DC, Hunt JB Jr, et al. Fractalkine overexpression suppresses tau pathology in a mouse model of tauopathy. Neurobiol Aging. 2013;34(6):1540–8.
Liu EY, Russ J, Cali CP, Phan JM, Amlie-Wolf A, Lee EB. Loss of nuclear TDP-43 is associated with decondensation of LINE retrotransposons. Cell Rep. 2019;27(5):1409–1421.e6.
Rodriguez S, Sahin A, Schrank BR, et al. Genome-encoded cytoplasmic double-stranded RNAs, found in C9ORF72 ALS-FTD brain, propagate neuronal loss. Sci Transl Med. 2021;13(601):eaaz4699. https://doi.org/10.1126/scitranslmed.aaz4699.
Fort-Aznar L, Ugbode C, Sweeney ST. Retrovirus reactivation in CHMP2BIntron5 models of frontotemporal dementia. Hum Mol Genet. 2020;29(16):2637–46.
Krug L, Chatterjee N, Borges-Monroy R, et al. Retrotransposon activation contributes to neurodegeneration in a Drosophila TDP-43 model of ALS. PLoS Genet. 2017;13(3):e1006635.
Gentry EG, Henderson BW, Arrant AE, et al. Rho kinase inhibition as a therapeutic for progressive supranuclear palsy and corticobasal degeneration. J Neurosci. 2016;36(4):1316–23.
Secor JD, Kotha SR, Gurney TO, et al. Novel lipid-soluble thiol-redox antioxidant and heavy metal chelator, N,N′-bis(2-mercaptoethyl)isophthalamide (NBMI) and phospholipase D-specific inhibitor, 5-fluoro-2-indolyl des-chlorohalopemide (FIPI) attenuate mercury-induced lipid signaling leading to protection against cytotoxicity in aortic endothelial cells. Int J Toxicol. 2011;30(6):619–38.
VandeVrede L, Ljubenkov PA, Rojas JC, Welch AE, Boxer AL. Four-repeat tauopathies: current management and future treatments. Neurotherapeutics. 2020;17(4):1563–81.
Muller T, Buttner T, Gholipour AF, Kuhn W. Coenzyme Q10 supplementation provides mild symptomatic benefit in patients with Parkinson’s disease. Neurosci Lett. 2003;341(3):201–4.
Apetauerova D, Scala SA, Hamill RW, et al. CoQ10 in progressive supranuclear palsy: a randomized, placebo-controlled, double-blind trial. Neurol Neuroimmunol Neuroinflamm. 2016;3(5):e266.
Patel NK, Gill SS. GDNF delivery for Parkinson’s disease. Acta Neurochir Suppl. 2007;97(Pt 2):135–54.
Ubhi K, Rockenstein E, Mante M, et al. Neurodegeneration in a transgenic mouse model of multiple system atrophy is associated with altered expression of oligodendroglial-derived neurotrophic factors. J Neurosci. 2010;30(18):6236–46.
Ubhi K, Inglis C, Mante M, et al. Fluoxetine ameliorates behavioral and neuropathological deficits in a transgenic model mouse of alpha-synucleinopathy. Exp Neurol. 2012;234(2):405–16.
Bensimon G, Ludolph A, Agid Y, et al. Riluzole treatment, survival and diagnostic criteria in Parkinson plus disorders: the NNIPPS study. Brain. 2009;132(Pt 1):156–71.
Doble A. The role of excitotoxicity in neurodegenerative disease: implications for therapy. Pharmacol Ther. 1999;81(3):163–221.
Heiser V, Engemann S, Brocker W, et al. Identification of benzothiazoles as potential polyglutamine aggregation inhibitors of Huntington’s disease by using an automated filter retardation assay. Proc Natl Acad Sci USA. 2002;99(Suppl 4):16400–6.
Yoo MH, Hyun HJ, Koh JY, Yoon YH. Riluzole inhibits VEGF-induced endothelial cell proliferation in vitro and hyperoxia-induced abnormal vessel formation in vivo. Investig Ophthalmol Vis Sci. 2005;46(12):4780–7.
Caumont AS, Octave JN, Hermans E. Specific regulation of rat glial cell line-derived neurotrophic factor gene expression by riluzole in C6 glioma cells. J Neurochem. 2006;97(1):128–39.
Shortland PJ, Leinster VH, White W, Robson LG. Riluzole promotes cell survival and neurite outgrowth in rat sensory neurones in vitro. Eur J Neurosci. 2006;24(12):3343–53.
Miller RG, Mitchell JD, Lyon M, Moore DH. Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). Cochrane Database Syst Rev. 2002;2:CD001447.
Angelova PR, Andruska KM, Midei MG, et al. RT001 in progressive supranuclear palsy-clinical and in-vitro observations. Antioxidants (Basel). 2021;10(7):1021. https://doi.org/10.3390/antiox10071021.
Odetti P, Garibaldi S, Norese R, et al. Lipoperoxidation is selectively involved in progressive supranuclear palsy. J Neuropathol Exp Neurol. 2000;59(5):393–7.
Di Monte DA, Harati Y, Jankovic J, Sandy MS, Jewell SA, Langston JW. Muscle mitochondrial ATP production in progressive supranuclear palsy. J Neurochem. 1994;62(4):1631–4.
Canesi M, Giordano R, Lazzari L, et al. Finding a new therapeutic approach for no-option Parkinsonisms: mesenchymal stromal cells for progressive supranuclear palsy. J Transl Med. 2016;14(1):127.
Giordano R, Canesi M, Isalberti M, et al. Safety and effectiveness of cell therapy in neurodegenerative diseases: take-home messages from a pilot feasibility phase I study of progressive supranuclear palsy. Front Neurosci. 2021;15: 723227.
Singer W, Dietz AB, Zeller AD, et al. Intrathecal administration of autologous mesenchymal stem cells in multiple system atrophy. Neurology. 2019;93(1):e77–87.
Acknowledgements
Medical Writing/Editorial Assistance.
We thank the staff of Neuroscience Publications at Barrow Neurological Institute for assistance with manuscript preparation.
Funding
Supported by National Institutes of Health grants R01AG059008, R01AG073212, P30AG072980, and the Barrow Neurological Foundation. The Rapid Service Fee will be paid by the authors.
Author information
Authors and Affiliations
Contributions
Marwan N. Sabbagh conceptualized the project and provided critical review and edits to the manuscript. Elise E. Dunning drafted manuscript. Boris Decourt, Holly A. Shill, and Nasser H. Zawia all provided critical review and edits to the manuscript. Marwan N. Sabbagh approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Elise E. Dunning and Boris Decourt have nothing to disclose. Nasser H. Zawia discloses ownership of NeuroTau. Holly A. Shill has received research support from Intra-cellular Therapeutics, Transposon, Parkinson Study Group/UCB, Parkinson’s Foundation, NINDS, Supernus/US World Meds, MJFF, Jazz Pharmaceuticals, Barrow Neurological Foundation and Cerevel Therapeutics. She has served as a consultant for the Parkinson Study Group/Nq, Biogen, Abbvie, Sage/Biogen, Praxis, KeifeRx, and Fasikl and Jazz Pharmaceuticals. Marwan N. Sabbagh discloses ownership interest (stock or stock options) in NeuroTau, uMethod Health, Lighthouse Pharmaceuticals, and Athira; he consults for Alzheon, Biogen, Roche-Genentech, T3D, Eisai, Lilly, and KeifeRx.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Dunning, E.E., Decourt, B., Zawia, N.H. et al. Pharmacotherapies for the Treatment of Progressive Supranuclear Palsy: A Narrative Review. Neurol Ther 13, 975–1013 (2024). https://doi.org/10.1007/s40120-024-00614-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-024-00614-9