
Abstract
Paroxysmal sympathetic hyperactivity (PSH) mainly occurs after acquired brain injury (ABI) and often presents with high fever, hypertension, tachycardia, tachypnea, sweating, and dystonia (increased muscle tone or spasticity). The pathophysiological mechanisms of PSH are not fully understood. Currently, there are several views: (1) disconnection theory, (2) excitatory/inhibitory ratio, (3) neuroendocrine function, and (4) neutrophil extracellular traps. Early diagnosis of PSH remains difficult, given the low specificity of its diagnostic tools and unclear pathogenesis. According to updated case analyses in recent years, PSH is now more commonly observed in patients with stroke, with tachycardia and hypertension as the main clinical manifestations, which is not fully consistent with previous data. To date, the PSH Assessment Measure tool is optimal for the early identification of PSH and stratification of symptom severity. Clinical strategies for the management of PSH are divided into three main points: (1) reduction of stimulation, (2) reduction of sympathetic excitatory afferents, and (3) inhibition of the effects of sympathetic hyperactivity on target organs. However, use of drugs and standards have not yet been harmonized. Further investigation on the relationship between PSH severity and long-term neurological prognosis in patients with ABI is required. This review aimed to determine the diagnostic and management challenges encountered in PSH after ABI.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Paroxysmal sympathetic hyperactivity (PSH) is more commonly observed in patients with stroke with tachycardia and hypertension as the main clinical manifestations |
The PSH-Assessment Measure tool is optimal for the early identification of PSH and stratification of symptom severity and has been increasingly recognized by clinicians |
Although the pathophysiological mechanisms of PSH remain unclear, the excitatory/inhibitory ratio model is still generally accepted, and endocrine-related theories are receiving more attention |
Early recognition of the location and severity of ABIs is critical for their clinical management |
Further investigation to determine the relationship between PSH severity and long-term neurological prognosis in patients with acquired brain injury is required |
Introduction
Acquired brain injury (ABI) is a leading cause of morbidity and mortality worldwide, and its clinical presentation is complex and variable, with paroxysmal sympathetic hyperactivity (PSH) being one of the clinical presentations. A clear definition of PSH syndrome is lacking, and progress in understanding its pathophysiology has been slow. Wilder Penfield first described the clinical features of PSH after traumatic brain injury and named it “midbrain epilepsy” [1]. This was followed by a series of terms based on the clinical symptoms, site of injury, and presumed mechanism. The term “paroxysmal sympathetic hyperactivity” was coined by Alejandro Rabinstein in 2007 [2]. PSH was first recommended as a standard term in 2010 [3]. In 2014, an evaluation team proposed a clear definition and diagnostic criteria through a review of 349 cases, recommending that the term “PSH” replace previous terms to describe the “syndrome of paroxysmal transient increases in sympathetic activity (heart rate, blood pressure, respiratory rate, body temperature, sweating) and motor (posturing) activity after ABI” [4]. The clinical management of PSH has also gained increasing attention in recent years, but several discrepancies and insufficient uniformity in current treatment protocols remain. This review aimed to determine the diagnostic and management challenges encountered in PSH after ABI.
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Retrieval Strategy
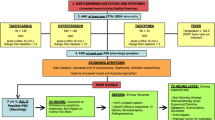
The retrieval strategy and criteria for selecting references for this review (Fig. 1) were determined by searching the online databases PubMed, Web of Science, and Google Scholar from 2015 to 2022 using the keyword “paroxysmal sympathetic hyperactivity” and identifying case reports or series that were complete and met the following inclusion criteria: (1) diagnosis of PSH; (2) diagnosis of ABI; (3) published from January 1, 2015, to December 31, 2022; (4) published in English.

Flowchart for inclusion and exclusion
Epidemiology of Paroxysmal Sympathetic Hyperactivity (PSH)
A review of 349 case reports on PSH published before 2010 found that approximately 80% of PSH cases occurred after traumatic brain injury (TBI), 10% after hypoxic brain injury, and 5% after stroke, and the remaining 5% were associated with hydrocephalus, tumors, hypoglycemia, infections, or unknown causes [3]. PSH is more likely to occur in pediatric patients following hypoxic-ischemic injury and encephalitis. In patients with severe TBI, the prevalence of PSH ranges from 8 to 33%, and the risk of developing PSH after TBI may be higher in adolescents than in children. In contrast, the prevalence of PSH after brain injury due to other causes is approximately 6%. Therefore, PSH is a complication primarily observed in TBI. However, of the 62 cases collected in this review, 20.97% of PSH cases occurred after ischemic stroke, 20.97% after hypoxic–ischemic encephalopathy (HIE), and 19.35% after TBI, which differed from previous reports (Fig. 2). This may be because the detection rate of PSH has increased as more uniform diagnostic criteria have been established. Although the incidence of TBI has remained unchanged in recent years, the incidence of TBI in youth due to motor vehicle collisions has decreased and the incidence of TBI due to falls in the elderly has increased [5]. The mean age of patients with PSH is 32 years. The reduced incidence of TBI in young adults may be related to the changing epidemiology of PSH. In addition, PSH is more common in patients with lower Glasgow Coma Scale (GCS) scores than in those with higher GCS scores, consistent with a report by Hughes et al. [6].

Epidemiology of paroxysmal sympathetic hyperactivity (N = 62). Cases were grouped into six categories (HIE, TBI, ischemic stroke, inflammatory diseases, tumors, intracerebral hemorrhage) according to the principal diagnosis. The left cases were categorized as others, and then the ratio of each category to the total number of cases was calculated separately
Clinical Characteristics of PSH
Typical clinical manifestations of PSH include simultaneous paroxysmal excitation of sympathetic nerves (heart rate, blood pressure, respiratory rate, body temperature, sweating) and postural activity, pupil dilation, and nystagmus. However, there is no clear evidence stating that a certain number, frequency, or duration of autonomic symptoms must be present to diagnose PSH, and patients with only one of the typical clinical presentations (e.g., hyperthermia) may also have PSH [7]. PSH can occur at all stages after ABI [8], with most clinical features appearing within 2 weeks, some appearing as the first symptom [9, 10], and a proportion of patients with symptoms appearing months later during rehabilitation or long-term care. Most cases of PSH are caused by non-injurious stimuli, such as turning, back patting, body rubbing, and emotional arousal, and are also commonly observed during sputum aspiration of patients with tracheal intubation [11]. The duration of the episodes varies, ranging from minutes to hours. The recovery time can vary from days to months, and some patients have residual motor (postural) symptoms or recurrence of symptoms [12]. The most common clinical features in the case data collected in this review were tachycardia (88.7%) and hypertension (79.0%) (Fig. 3). This is slightly different from previous reports in which PSH after TBI and intracerebral hemorrhage predominantly manifested as hypertension (94%), hyperthermia (80%), and hyperhidrosis (80%) [13].

Clinical features of paroxysmal sympathetic hyperactivity. The six core symptoms of 62 cases were counted according to the 2014 PSH International Consensus. Some cases showing multiple core symptoms were categorized separately
Neuroimaging
Focal brain injury is unlikely to lead to PSH development. Global, diffuse, or multifocal brain damage is usually present [14]. PSH is associated with diffuse axonal injury (DAI) and damage to the periventricular white matter, corpus callosum, mesencephalon, and upper brain stem [15]. In total, 72% of patients with moderate or severe TBI have DAI, which is a risk factor for PSH development [16, 17]. In addition, PSH occurs more frequently when white matter bundles involving the posterior part of the corpus callosum and the posterior limb of the internal capsule are disconnected [18].
Risk Factors for PSH
Early diagnosis of PSH remains difficult, given the low specificity of its diagnostic tools and unclear pathogenesis. Delayed diagnosis may lead to unnecessary investigations and medications, further prolonging hospital stay. Failure to manage PSH early may lead to hypertension, hyperthermia, cardiac damage, secondary brain damage, and even death [19]. Therefore, exploring the risk factors for PSH may help provide predictors for the early identification of PSH. A recent study showed a significant increase in plasma catecholamine concentrations during PSH episodes [20], suggesting an additional role for catecholamine levels in the early diagnosis of PSH. Additional risk factors, such as age, early onset of fever [21], degree of DAI [22], lower GCS scores, and tracheostomy, are associated with PSH development [13].
Challenges of Diagnostic Criteria
Patients with PSH usually receive acute-phase sedation after ABI to minimize secondary brain damage, which can mask the clinical features of PSH and prevent early diagnosis. Moreover, PSH is essentially a diagnosis of exclusion. Of the six main clinical signs, elevated temperature is usually the first problem, and sepsis must be ruled out first. Elevated blood pressure and dystonic features should rule out intracranial abnormalities, such as hydrocephalus and elevated intracranial pressure. There has been a lack of uniform diagnostic criteria for PSH until an international consensus in 2014 established diagnostic criteria and developed diagnostic tools [4]. The consensus selected 11 pathological features of PSH and proposed a clinical scoring system, the PSH Assessment Measure (PSH-AM), to improve diagnostic consistency. The PSH-AM consists of two separate constructs: (1) the Clinical Characteristics Scale, which scores the severity of sympathetic and motor hyperactivity, and (2) the Diagnostic Likelihood Instrument, which scores the likelihood of diagnosing PSH. Combining the two subtotal scores yields the total PSH-AM score, which assesses the likelihood of PSH diagnosis [4]. The pediatric PSH scale is broadly similar to that of adults, but is further divided by age and assesses diastolic and systolic blood pressure separately [23]. The feasibility and reliability of these tools have been validated, and the Clinical Characteristics Scale can be used to monitor the severity of episodes over time [7, 24]. However, the PSH-AM still has limitations, such as the difficulty in quantifying the severity of sweating and dystonia and in dynamically monitoring both items. In addition, PSH does not include parasympathetic excitability [25, 26], and some parasympathetic excitability symptoms, such as oligohydropsia, dry mouth, and dry eye, can easily be overlooked, affecting the diagnosis of PSH.
Pathophysiological Mechanisms of PSH
The pathophysiological mechanisms of PSH are not fully understood, and there are several current views as follows:
-
1.
Disconnection theory: Following focal or diffuse brain injury, cortical inhibitory (dorsolateral prefrontal cortex, amygdala, and basal ganglia) and sympathetic control (brainstem, hypothalamus, and mesencephalon) centers are dissociated, in turn deregulating normally inhibited spinal excitatory circuits and amplifying sympathetic responses to internal or external stimuli. However, this theory failed to explain the “paroxysmal” principle and was therefore not widely accepted.
-
2.
Excitatory/inhibitory ratio (EIR): At the cerebral level, brainstem centers regulate inhibitory drives to the spinal reflex areas. At the spinal level, spinal centers provide upward feedback regarding sensory and perceptual stimuli and output sympathetic and motor efferents, thereby maintaining a balance between inhibitory and excitatory interneuronal activities [27]. In the EIR model of PSH, impaired descending inhibition leads to excitation of spinal circuits, with non-injurious stimuli increasing motor and sympathetic output (spinal) and possibly perceiving it as noxious (central), followed by cessation of paroxysmal spasms due to the restoration of inhibitory factors (Fig. 4) .
-
3.
Neuroendocrine: Following TBI, the activation of the hypothalamic–pituitary-adrenal axis allows an uncontrolled outflow of adrenergic energy, leading to an increase in circulating catecholamines and causing paroxysmal disease. During paroxysm, the levels of adrenocorticotropic hormone, epinephrine, norepinephrine, and dopamine are significantly elevated, leading to sympathetic excitation. The vulnerability of the pituitary axis was further emphasized in a case report in which a patient with PSH after TBI had hypothyroidism [26].
-
4.
Neutrophil extracellular traps (NETs): Neutrophils infiltrate the paraventricular nucleus after TBI, thereby inducing NET release. NETs promote microglial activation and interleukin-1β release via the LL37-P2 × 7/MST1 pathway, thereby promoting sympathetic excitation by altering the levels or functions of neurotransmitters, such as Glu and GABA, and their receptors [28]. In addition, cerebral contusion, acute brain swelling, cerebral ischemia, and the subsequent release of excitatory amino acids at the cellular level after TBI may exacerbate PSH development. Secondary brain damage caused by the production of inflammatory cytokines and reactive oxygen species, hyperthermia, hypoxemia, and general deterioration may also be associated with PSH pathophysiology.

At the brain level, the inhibitory centers comprising the cerebral cortex, thalamus, and hypothalamus regulate the inhibitory drive to the reflex areas of the spinal cord. At the spinal level, spinal centers provide upward feedback regarding sensory and perceptual stimuli and sympathetic output. Impairment of inhibitory centers leads to injury to descending inhibitory circuits, and non-injurious stimulation also leads to sympathetic over-output, which is subsequently halted by the restoration of inhibitory circuits
Clinical Management Challenges of PSH
Early detection of PSH, such as after the onset of hypoxic encephalopathy or extensive brain injury, and systematic management before it becomes irreversible may reverse PSH [19]. Clinical strategies for the management of PSH are divided into three main points: (1) reduction of stimulation, (2) reduction of sympathetic excitatory afferents, and (3) inhibition of the effects of sympathetic hyperactivity on target organs. Commonly used symptomatic medications include opioids, nonselective β-blockers, α2 agonists, bromocriptine, baclofen, gabapentin, and long-acting benzodiazepines, which are primarily used to lower body temperature, control heart rate, maintain blood pressure, sedate, and relieve spasticity or decrease muscle tone. There have also been case reports on the effectiveness of intrathecal baclofen [29] and intrarectal administration [30]. A previous study also found that trazodone was effective in reducing PSH symptoms following a left temporal lobe subcortical hemorrhage in a 49-year-old woman [31]. In the retrieved case reports, the assessment of the effectiveness of PSH pharmacological treatments was mainly based on the reduction of sympathetic and motor hyperactivity events, with no quantitative data available for assessment, and the mechanisms by which these drugs improve PSH symptoms remain speculative. Daily PSH-AM score changes were monitored in only one case series to assess patient outcomes [24]. As any stimulation that can trigger a PSH attack should be minimized, and sedation may be necessary. Isoproterenol is effective in controlling PSH symptoms; however, the use of some of these drugs remains controversial. A retrospective study in pediatric patients found that when drugs were administered during an acute episode of PSH, the most effective drugs were benzodiazepines, whereas analgesics had little effect. Another pediatric case study reported that morphine was most useful during acute exacerbations, especially for PSH, in which medications, such as propranolol, were ineffective, and that the dose of morphine need not be increased incrementally [32].
Monotherapy for PSH has been inconsistently reported, with some studies suggesting that monotherapy is ineffective for PSH [33]; however, some cases have reported the effectiveness of monotherapy, such as propranolol [34], which is often used as the first choice for PSH. In clinical practice, combination medications are used in most cases; for example, the combination of guanfacine with gabapentin is safe and effective [35]. Combination therapy using dexmedetomidine and propranolol is widely used [36]. Optimizing the efficacy of these medications and reducing their side effects are the goals and challenges faced by clinical medications. Propranolol and dextromethorphan treat PSH in the acute phase and have prophylactic effects [37]. Other reports have stated that dextromethorphan can only be used for treatment and has no preventive effects [38]. This finding suggests that further studies are required to standardize the clinical use of medications for the treatment of PSH. Over time, patients with PSH may develop secondary complications, including dehydration, electrolyte disturbances, malnutrition, and muscle wasting. Hydration and nutrition should be adjusted to compensate for the increased fluid loss and metabolic demands.
Prognosis of PSH
The severity of PSH affects the length of hospitalization and increases the risk of delayed complications, including weight loss, dehydration, lung infections, heterotopic ossification, and muscle atrophy, and secondary brain damage, aggravating cerebral edema and increasing cranial pressure [11]. Patients with autonomic dysregulation after TBI are younger and have more severe brain injuries, longer intensive care unit and hospital stays, and poorer prognoses than those without autonomic dysregulation [7]. It has been suggested that PSH is an independent aggravator of the ABI that affects recovery [39].
However, there is no evidence to suggest that the worse prognosis of patients with PSH is due to more severe brain damage. PSH is only a reflection of disease severity and does not affect prognosis. The findings of the literature review revealed that most studies reported improvement in patients discharged from the hospital, and only a few studies reported death or complications [40]. However, few patients had a discharge follow-up GCS or disability scale score to specifically assess prognosis. Therefore, further studies are required to demonstrate the prognostic effect of PSH after adjusting for confounding factors. In addition, recurrence is also common in PSH, and factors associated with PSH recurrence have been demonstrated: the older the age and the lower the GCS score, the more likely PSH is to recur [38].
Conclusion
According to updated case analyses in recent years, PSH is more commonly observed in patients with stroke with tachycardia and hypertension as the main clinical manifestations, which is not fully consistent with previous data. To date, the PSH-AM tool is optimal for the early identification of PSH and stratification of symptom severity and has been increasingly recognized by clinicians. Although the pathophysiological mechanisms of PSH remain unclear, the EIR model is still generally accepted, and endocrine-related theories are receiving more attention. Given the complexity and diversity of PSH presentations, early recognition of the location and severity of ABIs is critical for its clinical management. Advancements in laboratory tests and neuroimaging may provide a more objective basis for the early diagnosis of PSH. Inconsistencies remain in the clinical management of PSH, and more clinical studies should be conducted to standardize the management criteria and parallel prevention and treatment to reduce secondary brain injury. Finally, further investigation to determine the relationship between PSH severity and long-term neurological prognosis in patients with ABI is required.
There are some limitations of this integrative review, and caution should be taken in data interpretation. To date, PSH is poorly understood, affecting case recognition and reporting. To ensure the accuracy of the literature, cases that met the diagnostic criteria of international consensus were included, which may result in the omission of some cases that used outdated terms. There is some missing information on collected cases that may affect the assessment of PSH prognosis. In the future, neurologists need to emphasize the early identification of PSH and conduct large sample clinical studies.
References
Penfield W. Diencephalic autonomic epilepsy. Arch Neurol Psychiatry. 1929;22(2):358–74.
Rabinstein AA. Paroxysmal sympathetic hyperactivity in the neurological intensive care unit. Neurol Res. 2007;29(7):680–2.
Perkes I, Baguley IJ, Nott MT, Menon DK. A review of paroxysmal sympathetic hyperactivity after acquired brain injury. Ann Neurol. 2010;68(2):126–35.
Baguley IJ, Perkes IE, Fernandez-Ortega JF, Rabinstein AA, Dolce G, Hendricks HT, et al. Paroxysmal sympathetic hyperactivity after acquired brain injury: consensus on conceptual definition, nomenclature, and diagnostic criteria. J Neurotrauma. 2014;31(17):1515–20.
Taylor CA, Bell JM, Breiding MJ, Xu L. Traumatic Brain Injury-Related Emergency Department Visits, Hospitalizations, and Deaths—United States, 2007 and 2013. MMWR Surveill Summ. 2017;66(9):1–16.
Meyer KS. Understanding paroxysmal sympathetic hyperactivity after traumatic brain injury. Surg Neurol Int. 2014;5(Suppl 13):S490–2.
van Eijck MM, Sprengers MOP, Oldenbeuving AW, de Vries J, Schoonman GG, Roks G. The use of the PSH-AM in patients with diffuse axonal injury and autonomic dysregulation: a cohort study and review. J Crit Care. 2019;49:110–7.
Godoy DA, Panhke P, Guerrero Suarez PD, Murillo-Cabezas F. Paroxysmal sympathetic hyperactivity: an entity to keep in mind. Med Intensiva (Engl Ed). 2019;43(1):35–43.
Yin J, Wang W, Wang Y, Wei Y. Paroxysmal sympathetic hyperactivity: the storm after acute basilar artery occlusion. Acta Neurol Belg. 2022;122(5):1349–50.
Yin J, Wang W, Wang Y, Li G, Kong Y, Li X, et al. Case report: stroke chameleon: acute large vessel occlusion in the posterior circulation with paroxysmal sympathetic hyperactivity as the first manifestation. Front Neurosci. 2022;16: 890678.
Cheng L, Mitton K, Walton K, Sivan M. Retrospective analysis of functional and tracheostomy (decannulation) outcomes in patients with brain injury in a hyperacute rehabilitation unit. J Rehabil Med Clin Commun. 2019;2:1000024.
Hamlin DW, Hussain N, Pathare A. Storms and silence: a case report of catatonia and paroxysmal sympathetic hyperactivity following cerebral hypoxia. BMC Psychiatry. 2020;20(1):473.
Li Z, Chen W, Zhu Y, Han K, Wang J, Chen J, et al. Risk factors and clinical features of paroxysmal sympathetic hyperactivity after spontaneous intracerebral hemorrhage. Auton Neurosci. 2020;225: 102643.
Singh T, Arora TK, Bedi P, Kashinath S. Paroxysmal sympathetic hyperactivity after cardiac arrest in a young male. Cureus. 2018;10(7): e3028.
Lv LQ, Hou LJ, Yu MK, Qi XQ, Chen HR, Chen JX, et al. Prognostic influence and magnetic resonance imaging findings in paroxysmal sympathetic hyperactivity after severe traumatic brain injury. J Neurotrauma. 2010;27(11):1945–50.
Lv LQ, Hou LJ, Yu MK, Qi XQ, Chen HR, Chen JX, et al. Risk factors related to dysautonomia after severe traumatic brain injury. J Trauma. 2011;71(3):538–42.
Hendricks HT, Heeren AH, Vos PE. Dysautonomia after severe traumatic brain injury. Eur J Neurol. 2010;17(9):1172–7.
Hinson HE, Puybasset L, Weiss N, Perlbarg V, Benali H, Galanaud D, et al. Neuroanatomical basis of paroxysmal sympathetic hyperactivity: a diffusion tensor imaging analysis. Brain Inj. 2015;29(4):455–61.
Sakai K, Kitagawa T, Suzuki K, Toh K, Yamamoto J. Paroxysmal sympathetic hyperactivity following acute diffuse brain swelling due to traumatic brain injury: a case report with good clinical outcome. Egypt J Neurosurg. 2022;37(1):7.
Fernandez-Ortega JF, Baguley IJ, Gates TA, Garcia-Caballero M, Quesada-Garcia JG, Prieto-Palomino MA. Catecholamines and paroxysmal sympathetic hyperactivity after traumatic brain injury. J Neurotrauma. 2017;34(1):109–14.
Kennedy E, Cohen M, Munafo M. Childhood traumatic brain injury and the associations with risk behavior in adolescence and young adulthood: a systematic review. J Head Trauma Rehabil. 2017;32(6):425–32.
Sato T, Watanabe M, Onoda Y, Oyanagi T, Kushimoto S. Heterotopic ossification in a patient with paroxysmal sympathetic hyperactivity following multiple trauma complicated with vitamin D deficiency: a case report. Surg Case Rep. 2020;6(1):293.
Pozzi M, Locatelli F, Galbiati S, Radice S, Clementi E, Strazzer S. Clinical scales for paroxysmal sympathetic hyperactivity in pediatric patients. J Neurotrauma. 2014;31(22):1897–8.
Godo S, Irino S, Nakagawa A, Kawazoe Y, Fujita M, Kudo D, et al. Diagnosis and management of patients with paroxysmal sympathetic hyperactivity following acute brain injuries using a consensus-based diagnostic tool: a single institutional case series. Tohoku J Exp Med. 2017;243(1):11–8.
Meyfroidt G, Baguley IJ, Menon DK. Paroxysmal sympathetic hyperactivity: the storm after acute brain injury. Lancet Neurol. 2017;16(9):721–9.
Abdelhakiem AK, Torres-Reveron A, Padilla JM. Effectiveness of pharmacological agents and validation of diagnostic scales for the management of paroxysmal sympathetic hyperactivity in hispanics. Front Neurol. 2020;11: 603011.
Zheng RZ, Lei ZQ, Yang RZ, Huang GH, Zhang GM. Identification and management of paroxysmal sympathetic hyperactivity after traumatic brain injury. Front Neurol. 2020;11:81.
Zhu K, Zhu Y, Hou X, Chen W, Qu X, Zhang Y, et al. NETs lead to sympathetic hyperactivity after traumatic brain injury through the LL37-Hippo/MST1 pathway. Front Neurosci. 2021;15: 621477.
Pucks-Faes E, Hitzenberger G, Matzak H, Verrienti G, Schauer R, Saltuari L. Intrathecal baclofen in paroxysmal sympathetic hyperactivity: impact on oral treatment. Brain Behav. 2018;8(11): e01124.
May CC, Oyler DR, Parli SE, Talley CL. Rectal propranolol controls paroxysmal sympathetic hyperactivity: a case report. Pharmacotherapy. 2015;35(4):e27-31.
Morinaga Y, Nii K, Hanada H, Sakamoto K, Inoue R, Mitsutake T. Efficacy of trazodone for treating paroxysmal sympathetic hyperactivity presenting after left temporal subcortical hemorrhage. Intractable Rare Dis Res. 2020;9(2):119–22.
Raithel DS, Ohler KH, Porto I, Bicknese AR, Kraus DM. Morphine: an effective abortive therapy for pediatric paroxysmal sympathetic hyperactivity after hypoxic brain injury. J Pediatr Pharmacol Ther. 2015;20(4):335–40.
Shald EA, Reeder J, Finnick M, Patel I, Evans K, Faber RK, et al. Pharmacological treatment for paroxysmal sympathetic hyperactivity. Crit Care Nurse. 2020;40(3):e9–16.
Nguembu S, Meloni M, Endalle G, Dokponou H, Dada OE, Senyuy WP, et al. Paroxysmal sympathetic hyperactivity in moderate-to-severe traumatic brain injury and the role of beta-blockers: a scoping review. Emerg Med Int. 2021;2021:5589239.
Miyoshi T, Mizushima C, Noborio Y, Kimoto Y, Nakaharu Y, Shimamoto S. Efficacy of combination therapy with gabapentin and guanfacine for paroxysmal sympathetic hyperactivity following hypoxic encephalopathy: a case report. J Int Med Res. 2021;49(4):3000605211009721.
Branstetter JW, Ohman KL, Johnson DW, Gilbert BW. Management of paroxysmal sympathetic hyperactivity with dexmedetomidine and propranolol following traumatic brain injury in a pediatric patient. J Pediatr Intensive Care. 2020;9(1):64–9.
Tang Q, Wu X, Weng W, Li H, Feng J, Mao Q, et al. The preventive effect of dexmedetomidine on paroxysmal sympathetic hyperactivity in severe traumatic brain injury patients who have undergone surgery: a retrospective study. PeerJ. 2017;5: e2986.
Peng Y, Zhu H, Chen H, Zhu Z, Zhou H, Zhang S, et al. Dexmedetomidine attenuates acute paroxysmal sympathetic hyperactivity. Oncotarget. 2017;8(40):69012–9.
Kirk KA, Shoykhet M, Jeong JH, Tyler-Kabara EC, Henderson MJ, Bell MJ, et al. Dysautonomia after pediatric brain injury. Dev Med Child Neurol. 2012;54(8):759–64.
Bekele N, Mesfin N, Hailu T, Tadesse A. Diagnosis and treatment of paroxysmal sympathetic hyperactivity in medical ICU, University of Gondar Hospital, Northwest Ethiopia: a case report. Int Med Case Rep J. 2020;13:591–5.
Funding
This research was funded by grants from Shanxi Provincial Health and Family Planning Commission (2017033, 2020080); Doctoral Fund of the First Hospital of Shanxi Medical University (YB161706, BS03201631, SD2215); Shanxi Applied Basic Research Program (201801D221426, 20210302124404) the rapid service fee was funded by the authors.
Author information
Authors and Affiliations
Contributions
Conceptualization: Sui-yi Xu and Qi Zhang; Methodology: Qi Zhang; Formal analysis and investigation: Qi Zhang; Writing—original draft preparation: Sui-yi Xu and Qi Zhang; Writing—review and editing: Chang-xin Li; Funding acquisition: Sui-yi Xu and Chang-xin Li; Resources: Sui-yi Xu and Chang-xin Li; Supervision: Chang-xin Li; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Conflict of Interest
Sui-yi Xu, Qi Zhang, and Chang-xin Li declare that they have no competing interests.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Xu, Sy., Zhang, Q. & Li, Cx. Paroxysmal Sympathetic Hyperactivity After Acquired Brain Injury: An Integrative Review of Diagnostic and Management Challenges. Neurol Ther 13, 11–20 (2024). https://doi.org/10.1007/s40120-023-00561-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00561-x