
Abstract
Introduction
Alzheimer’s disease (AD) is the most common cause of dementia worldwide, making it a major public health issue. Anti-amyloid and anti-tau antibodies are the most advanced therapeutic approach at present. Three drugs (lecanemab, donanemab and aducanumab) are on track to be marketed in the coming months. In this systematic review, we review all Phase 2 and Phase 3 clinical trials conducted in this indication and the particularities of the molecules tested.
Methods
The PubMed and ClinicalTrials.gov databases were searched through February 2023 for Phase 2 and 3 clinical trials involving passive anti-amyloid or anti-tau immunotherapies with published results. This review has been compiled in compliance with the PRISMA checklists.
Results
Of the 165 studies found and after eliminating duplicates, 40 studies had their results published on PubMed and/or ClinicalTrials.gov. Eight anti-amyloid molecules and four anti-tau molecules were the subject of Phase 2 studies, seven anti-amyloids were the subject of Phase 3 trials, and two molecules were granted early marketing approval by the US Food and Drug Administration (FDA). The results were compiled in summary tables showing the primary endpoints used, results, age of the study population and specific adverse events for these molecules.
Discussion
Passive immunotherapy in AD is largely dominated by anti-amyloid antibodies, which are more numerous and more advanced in the pipeline. Lecanemab, donanemab and aducanumab are distinguished by their relative efficacy in terms of cognitive and functional evaluation but also by a decrease in amyloid and tau proteins in the brain. These three molecules have in common that they bind to N-terminal ends of amyloid fibrils and plaques. The findings of their studies raise the question of which criteria to apply when choosing which patient will receive them when marketed, such as the apoliprotein E gene’s fourth allele (APOE4) genetic status of patients. The large number of negative studies may also raise the question of the criteria for defining the disease and the possible interest in redefining it on biological grounds to offer a more personalized medicine to patients suffering from neurodegenerative diseases.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
This systematic review covers all anti-amyloid and anti-tau agents in AD that have reached Phase II or Phase III clinical development |
Eight anti-amyloid and four anti-tau agents met these criteria |
The characteristics of each antibody are summarized, with an original classification based on binding affinity |
Three tables summarize antibody characteristics, as well as Phase II and Phase III studies, to provide hands-on support for physicians in their clinical practice |
Only three anti-amyloid antibodies met the efficacy criteria, based on cognitive and/or functional criteria. However, their clinical relevance is questionable |
These treatments have many specific adverse effects: amyloid-related imaging abnormalities (ARIA) and a high cost of US $30,000 to $50,000 |
The authors propose a new classification of AD based on biology, to adapt treatments to the biomarkers and have a better benefit/risk ratio |
Introduction
Dementia is the association of cognitive-behavioral disorders with an inability to carry out daily activities and most often occurs in connection with a neuro-evolutionary disease, the main ones being Alzheimer’s disease and dementia with Lewy bodies (DLB). It is a serious and frequent condition with, according to the World Health Organization (WHO), 50 million cases worldwide, 10 million new cases per year and projections that foresee 82 million patients in 2030 and 152 million in 2050, making it a major public health issue [1]. In France, there were an estimated 1.1 million patients in 2020, and the figure is expected to rise to 1.3 million in 2023 [2]. The repercussions are major in terms of societal cost, impact on caregivers, comorbidity and mortality. Dementia was the sixth leading cause of death in the US in 2019 [3]. AD is the leading cause of dementia worldwide.
The typical presentation of AD is an early episodic memory impairment defined by a gradual and progressive change in memory function at disease onset reported by patients or informants for a period > 6 months. This definition is accurate in 86–94% of patients, but there are also atypical forms of neuropathologically confirmed AD. In the remaining 6–14% of patients, posterior cortical degeneration may cause visual and visuospatial impairments while frontal degeneration causes more prominent behavioral symptoms or language disorders, such as the logopenic variant of primary progressive aphasia [4, 5].
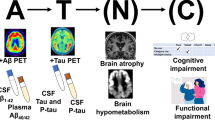
The pathophysiology of AD is still insufficiently understood. It has been possible to identify abnormalities in brain proteins, the pathological forms of which are found in abnormal quantities and more or less early in the disease, sometimes being present months or even years before the first clinical manifestations [6]. Three main hypothesis are studied: amyloid hypothesis, tubulin-associated unit (tau) hypothesis and inflammation [7] (see Fig. 1).

Amyloid and tau pathway (created with BioRender.com)
Amyloid protein is derived from Aß, a neuronal membrane protein. There is evidence that this protein could have antimicrobial properties [8]. In physiological conditions, monomers of Aß could be neuroprotective and even have a role in intracellular signaling and synaptic function [9]. The transmembrane amyloid precursor protein (APP) is degraded by a family of enzymes called secretases. In the absence of disease, the alpha pathway is the main one, releasing soluble extracellular domain of APP and, C-terminal fragments. In AD, the beta pathway and, to a lesser extent, the gamma pathway, which both cleave Aß into Aß1-42, have been observed to predominate. This abnormal degradation product is insoluble and, with their accumulation, these insoluble proteins aggregate into oligomers, then fibrils and plaques. These plaques in the brain have been observed before any clinical manifestation [6]. There is evidence of genetic mutations or characteristics mediating amyloid associated with AD, such as amyloid beta precursor protein (APP), presenilin 1 and 2 (PSEN1/2) or sortilin Related Receptor 1 (SORL1), in both early and late onset AD [9].
The tau hypothesis owes its name to the tau protein, which in its normally phosphorylated form is a stabilizing protein of the microtubules of the cellular cytoskeleton. However, in AD, a phenomenon of hyperphosphorylation occurs, leading to a change in conformation that renders the protein dysfunctional and causes its accumulation in the form of neurofibrillary tangles [10]. Amyloid lesions often appear to precede the tau lesions [11].
A process of brain inflammation, mediated by microglia, has also been observed. However, it remains difficult to determine whether this inflammation is the cause or a consequence of AD, and it is likely that the answer could be both [12]. It has also been suggested that AD may have an infectious cause, with a possible link between infection and the onset of AD [13], or with germs such as Porphyromonas gingivalis found in patients with AD [14].
Diagnosis is based on medical examination, neuropsychological assessment, brain imaging and measurement of the proteins underlying the pathophysiology in the cerebrospinal fluid including ratio of Aß 42/40. The use of metabolic imaging is becoming more common with the democratization of amyloid positron emission tomography (PET) scans.
Although AD is a common disease and there is a wealth of research on the subject, there is currently no drug treatment that modifies the evolution of the disease (i.e., disease-modifying treatment, DMT). Some symptomatic treatments have been validated but their effect is small and focused only on cognition. Two classes of drugs coexist: central anticholinesterase drugs (donepezil, rivastigmine, galantamine) and an N-methyl-d-aspartate (NMDA) antagonist (memantine) [15]. There are non-pharmacological treatments that have been shown to be effective, such as cognitive remediation by a physiotherapist, a speech therapist or a neuropsychologist [16, 17].
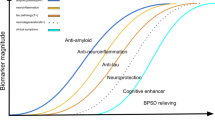
However, several drug classes are being tested to modify the course of the disease (see Fig. 2), led by amyloid-targeting and tau-targeting immunotherapies, but also metabolic, anti-infectious and anti-inflammatory pathways. Aducanumab, lecanemab and donanemab are currently in early approval in the USA or in the process of being approved worldwide.

Anti-amyloid and anti-tau antibodies pipeline (created with BioRender.com)
Here, we propose a systematic review of clinical trials on passive amyloid-targeting and tau-targeting immunotherapies, which are the most successful therapeutic approaches at the present time, with a focus on the state of the art of amyloid-targeting immunotherapies. Finally, we present an expert opinion on the reasons that could lead us to think that the classification of cognitive neurodegenerative diseases based on symptoms rather than biology might explain some of the difficulties encountered in treating these diseases.
Methods
This review was conducted using PRISMA guidelines. We included Phase 2 or 3 randomized controlled interventional clinical trials in AD in which the drug was a monoclonal antibody directed against Aß1-42 or tau proteins. We excluded preclinical trials and trials whose results were not published on ClinicalTrials.gov or PubMed (see Fig. 3). The databases were searched on February 1, 2023.

Systematic review flowchart (created with BioRender.com)
For PubMed, the query used was “Alzheimer’s disease (MeSH)” or “neurocognitive disorders” or “cognitive dysfunction” associated with one of the following terms: “antibodies,” “immunization,” “Alzheimer vaccines” or one of the monoclonal antibodies already identified in the previous reviews [18,19,20] (see Table 1). The filters used excluded animal models and required a randomized controlled trial. Full query was: ((alzheimer disease[MH] OR Neurocognitive Disorders[MH] OR Cognitive Dysfunction[MH]) AND ("aducanumab"[TW] OR "lecanemab"[TW] OR "bapineuzumab"[TW] OR "crenezumab"[TW] OR "Donanemab"[TW] OR "gantenerumab"[TW] OR "ponezumab"[TW] OR "Solanezumab"[TW] OR "gosuranemab"[TW] OR "zagotenemab"[TW] OR antibodies[MH] OR immunization[MH] OR Alzheimer vaccines[MH] OR "semorinemab"[TW] OR " Tilavonemab" [TW])) NOT ("animals"[MH] NOT "humans"[MH]) NOT (Mice[MeSH] OR "Models, Animal"[MH]) AND randomizedcontrolledtrial[Filter].
For the ClinicalTrials.gov database, we searched for all Phase 2 or 3 trials concerning these molecules, excluding trials that did not concern AD and those that did not have published results.
Only the first author (AE) manually searched the databases and selected data as specified in the protocol (see Table 2).
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors (see Table 3).
Results
Ninety-three publications were identified in PubMed and 72 in the ClinicalTrials.gov database. After screening and verification of the publications corresponding to the criteria, 40 trials were finally retained. Of these trials, only one (DIAN-TU) was a trial involving two different molecules.
In the tables below, we list the Phase 3 trials only or the Phase 2 trials for treatments without Phase 3 trials with the available results. With only six trials with results available for anti-tau against 34 for their amyloid counterpart, the latter are by far the largest family. Moreover, there are no anti-tau Phase 3 trials.
We show the results by molecules, first anti-amyloid then anti-tau, sorted by their binding affinity. Only Phase 3 trials, where they exist, are listed in the tables; details of all 40 trials are available in the supplemental appendix (see S1).
Monomer-Specific: Solanezumab
Solanezumab is a humanized immunoglobulin (Ig) G1 that binds exclusively to monomers at their central region [21]. It is administered intravenously. After three Phase 2 trials and the pivotal DIAN-TU trial, three Phase 3 trials were conducted with extension: EXPEDITION, EXPEDITION 2 and EXPEDITION 3. The primary endpoint was cognitive and functional with evaluation of the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADA-Cog) score in their 11 (EXPEDITION) and 14 (EXPEDITION 2 and 3)-item versions and Alzheimer’s Disease Cooperative Study – Activities of Daily Living (ADCS-ADL). These objectives were not significantly met, and the open-label extension (OLE) trials were stopped early despite a large enrollment of patients: EXPEDITION and EXPEDITION 2 had included > 1000 patients each and EXPEDITION 3 2129 patients. Solanezumab is part of the first generation of passive immunization following the failure of active immunizations, along with ponezumab, crenezumab and bapineuzumab.
Few amyloid-related imaging abnormalities (ARIA) were reported: < 1% of ARIA-edema (ARIA-E) at most and < 6% of ARIA-microhemorrhage (ARIA-H). A majority of patients were APOE4-positive, with nearly 60% in EXPEDITION and EXPEDITION 2 and 70% in EXPEDITION 3 [22, 23]. In DIAN-TU, between 26 and 32% of patients were APOE4 carriers [24].
Targeting Monomers, Oligomers and Fibrils: Ponezumab, Crenezumab and Gantenerumab
Ponezumab
Ponemuzab is a humanized IgG2a [25]. It is the only IgG2 among the antibodies tested in Phase 2/3 in AD. This IgG2 is mutated so that it does not trigger an immunological cascade and therefore acts solely by recruitment of phagocytic cells. It is also administered intravenously.
It binds exclusively to Aß40 monomers at the C-terminal level of Aß40 monomers, oligomers and plaques. It does not recognize Aß42, unlike all the other anti-amyloid immunotherapies presented here [26].
Two Phase 2 trials are registered on ClinicalTrials.gov concerning this molecule, with only one having published results. The primary endpoint was adverse effects of the molecule at different doses, but efficacy was not considered sufficient to warrant Phase 3 trials [27]. Practically no ARIA-E was reported in the safety data, but the rate of ARIA-H reached up to 25% depending on the arm. It should be noted that the placebo arm still had 15% of ARIA-H reported. These figures must be weighed against the majority of APOE4-positive patients (66.5%) [27].
Crenezumab
Crenezumab is a humanized IgG4 that binds preferentially to the central site of amyloid monomers, oligomers and fibrils [21]. It is notable for having been tested in Phase 2 in seven randomized trials by intravenous and subcutaneous routes. Two Phase 3 trials and one OLE were started: CREAD and CREAD 2, each involving about 800 patients. They were stopped for futility, and their OLE was stopped.
Few adverse events were reported with crenezumab. The majority of patients carried an APOE4 allele, 71–72% for CREAD and 65–66% for CREAD2 [28].
Gantenerumab
Gantenerumab is a human IgG1 that binds preferentially to monomers, oligomers and fibrils and has the particularity of targeting the N-terminal, medial and C-terminal portions [29]. Another of its characteristics is that it has only been tested by the subcutaneous route. The main Phase 3 trial, entitled Scarlet Road and including nearly 800 patients, was stopped early because of futility [30]. It followed a Phase 2 trial and a pivotal Phase 2/3 trial with solanezumab: DIAN-TU. The primary endpoint in the Scarlet Road Phase 3 trial was cognitive and functional via the Clinical Dementia Rating Scale-Sum of Boxes (CDR-SB). Another trial was conducted with a safety focus. A significant number of ARIA were reported, with 20–25% ARIA in the pivotal trial and up to 28.71% ARIA-E in the open-label extension. Many severe adverse events (SAEs) were reported, with 31.77% SAEs in the drug arm in the controlled trial and 37.96% in the OLE. Similarly, more ARIA were reported in the gantenerumab group compared to solanezumab in the DIAN-TU pivotal trial.
These figures must be set against a possible imbalance in the APOE4 populations. In the Scarlet Road trial, 61.5% of patients were APOE4 heterozygous in the 225 mg group compared with 50.4% in the placebo group, and 41% in the 105 mg group, but 38% were homozygous in the 105 mg group compared with 19.9% in the placebo group (no data available for the 225 mg group) [30]. For DIAN-TU, the figures are more homogeneous, with between 26 and 32% of patients carrying APOE4 [24].
New Phase 2 trials are about to begin, including the addition of Roche’s “brain shuttle” technology under the name trontinemab or RO7126209 [31, 32].
Targeting Oligomers and Protofibrils: The Case of Lecanemab
Lecanemab
Along with aducanumab, lecanemab is one of the few molecules to have shown a statistically significant effect in AD, notably in the CLARITY AD trial on the CDR-SB [33]. It is a humanized IgG1 that preferentially targets N-terminal oligomers and protofibrils [29]. A subcutaneous route was tested in Phase 1, but Phases 2 and 3 preferred an IV route. Ten to 20% of ARIA were reported in the intervention groups, although they remained mostly asymptomatic [33]. Sixty-eight percent of patients were APOE4 carriers, 15% of whom were homozygous. The Phase 3 trial AHEAD is still under recruitment and should add weight to the CLARITY results for a possible marketing authorization.
Lecanemab, along with aducanumab, donanemab and gantenerumab, is part of a second generation of drugs developed later than solanezumab, ponezumab and bapineuzumab.
Those Targeting Fibrils and Plaques: Aducanumab, Bapineuzumab
Aducanumab
Aducanumab (Aduhelm®) has received an anticipated marketing approval by the FDA in June 2021 [34]. It is a human IgG1 that targets fibrils and plaques [29]. After a Phase 2 trial named EVOLVE, the marketing authorization in the US was given following two large-scale Phase 3 trials (more than 1600 patients per trial): EMERGE and ENGAGE. These two trials were stopped prematurely because of futility, but statistical significance was reached for EMERGE in the high-dose subgroup [35]. Furthermore, a decrease in brain amyloid load was observed in the sub-studies with PET scan but without demonstrating that this decrease in amyloid load was associated with a decrease in symptoms or an improvement in the patients’ quality of life.
The two trials are quite similar in terms of safety, each having a significant number of ARIA. There were 16% ARIA-H and 26% ARIA-E in the low-dose trials compared to 20% and 35%, respectively, in the high-dose trials. Most of these ARIA were asymptomatic. The number of adverse events classified as severe was lower, around 13%. Most of these patients (between 67 and 71%) carried the APOE4 allele.
The early granting of marketing approval despite the discrepancies between the trials and the negative opinion of the FDA scientific committee is currently being criticized in the US [36].
Bapineuzumab
Bapineuzumab is a humanized IgG1 monoclonal antibody. It also binds to fibrils and plaques [21]. After five Phase 2 trials and one OLE, two Phase 3 trials have been completed, all subsequent trials and open-label extension Phases having been stopped prematurely following the failure of the initial trials. These two trials, named 301 and 302, were unique in that one of them was conducted in an all homozygous APOE4 carrier population and the other in an all non-APOE4 carrier population. Although neither trial showed a significant difference in its cognitive primary objective, the safety trend was clearly towards ARIA-E in the APOE4 carrier populations, with a rate of ARIA-E of 15% in the carrier trial compared with 4.2% for the same dose and 9.4% at higher doses of APOE4 in the non-carrier trial [37].
Fixing only Amyloid Plaques: Donanemab
Donanemab
Donanemab is a humanized IgG1 with affinity against plaques at their N-terminal end [38, 39]. No Phase 3 trial has published results to date, with three trials still ongoing. The only Phase 2 trial with published results, TRAILBLAZER, did not show any statistical difference on cognitive and functional criteria. However, a higher rate of ARIA was observed in the treatment group than in the placebo group, for ARIA-E 26.7% versus 1.8% and ARIA-H 22.1% versus 6.4%, respectively. In this trial, between 72.5 and 74.2% of patients were APOE4 carriers, 19% and 22% of them being homozygous [40]. As of May 2023, Eli Lilly and Company announced positive results for the TRAILBLAZER-ALZ 2 trial [41]. Peer-reviewed publication of TRAILBLAZER 2 has been published on July 17, 2023[42]. Donanemab showed a slowing of AD progression on the iADRS scale (a composite scale combining ADAS-Cog 13 and ADCS-iADL). A particular feature of this clinical trial was the stratification of populations according to their tau protein load. ARIA prevalence seems to be between that of aducanumab and lecanemab, with 24% of treated patients showing ARIA-E and 19.7% of treated patients showing ARIA-H.
Anti-tau Antibodies
Semorinemab
Semorinemab is a humanized IgG4 that targets the N-terminal end of all isoforms of the tau protein [43]. Two Phase 2 trials have been carried out without showing any significant effect on cognitive criteria. There are no Phase 3 trials reported on ClinicalTrials.gov at present. Almost three quarters of the patients in the Tauriel Phase 2 trial were APOE4 carriers [43].
Gosuranemab and Tilavonemab: Extracellular Forms of Tau
Gosuranemab
Gosuranemab is a humanized IgG4 targeting the N-terminal end of extracellular isoforms of tau protein [44]. The Phase 2 Tango trial was stopped at the end of the placebo-controlled period because of lack of efficacy [45], resulting in premature termination of the OLE and clinical trials. There is no information on the number of patients with the APOE4 allele in the Tango trial.
Tilavonemab
Tilavonemab is also a humanized IgG4 targeting the aggregated extracellular soluble and insoluble isoforms of tau on their N-terminal portion [46]. The trials failed to show efficacy in CDR-SB [47], and the OLE was stopped prematurely because of the lack of efficacy of the original trial. The proportion of patients with APOE4 in the study population is not available [48].
Zagotenemab: Specific for Neurofibrillary Tangles
Zagotenemab is a humanized IgG1, derived from MC-1, an tau conformation specific antibody directed against tau protein in its neurofibrillary form at its N-terminus on amino acids located at positions 7–9 [49]. Zagotenemab was tested in Phase 2 on the ADCS-iADL functional scale but did not show significant improvement. The number of patients positive for the APOE4 allele has not been published.
Discussion
Study Bias
With the current new results related to lecanemab, donanemab and aducanumab, we have performed a systematic review of all anti-amyloid and anti-tau immunotherapies tested in AD in Phase 2 and 3. Almost all studies were negative, apart from the Phase 3 trial with lecanemab and donanemab. The trials are consistent in terms of the target population, with a reproducible age range and AD at a mild cognitive impairment (MCI) or mild stage. The differences occur essentially in the percentage of patients carrying the APOE4 gene. We will come back to the reasons for this choice later in the discussion. We have chosen to highlight the specific adverse events, the ARIA, in view of their frequency and their importance in the benefit-risk balance of this new therapeutic class. On average, ARIA will occur in one in ten patients. There seems to be not much excess risk in ARIA-H given that 8.8% of patients will spontaneously develop hemosiderosis or microhemorrhage, but patients could be five times more likely to encounter ARIA-E. We would have liked to include the number of symptomatic ARIA, but the data are not widely available, and the reporting is not very reproducible. In this discussion, we look at the reasons for the negative results despite the efficacy at the cellular level as evidenced by ARIA. We then discuss the medico-economic issues before concluding on why a poor biological definition of AD could contribute to the failure of these drugs and delivering an opinion on why it could be relevant to redefine neurodegenerative diseases according to biomarkers at the level of each patient to propose a personalized medicine to each patient.
This review has several biases. Some manufacturers give a code name to molecules in development. We have included these when we were aware of them, when Phase 1 and 2 results were published in PubMed, but we may have omitted molecules whose trial results had not been published. Another reporting bias is present when studies have missing outcomes, notably concerning the APOE4 status of patients or the number and type of adverse events that occurred. To limit this bias, we chose to systematically consult the data published on ClinicalTrials.gov whenever they were available. We chose not to include studies that had not reached Phase 2, so we may have missed some therapies that were still in early development. We are also confronted with publication bias, as many clinical trials do not have results published on ClinicalTrials.gov.
However, we welcome the fact that most of the results, although negative, have been published by the pharmaceutical industry. One datum that we wanted to include, but which is often missing, is the rate of symptomatic patients among those with ARIA. Indeed, given the high prevalence (up to one third of patients depending on the series), it would have been relevant to know the number of patients that could be detected by interviewing them. We thus calculated the SAE ratio between patients on treatment and those on placebos to look for excess risk with significant consequences.
Anti-amyloid drugs are the focus of most of the research on AD. In addition to the interest that can be justified by the positive results of lecanemab and donanemab, the tau hypothesis may also explain part of this focus. While amyloid plaques are diffuse in the brain, abnormal tau proteins initially aggregate within neurons, making them more difficult to reach with monoclonal antibodies, although trials have shown a decrease in tau proteins levels in CSF and in PET scan with tau-targeting and amyloid-targeting therapies. The TRAILBLAZER 2 trial on Donanemab stratified its population according to this tau protein load [42].
Positive Results but Poor Clinical Relevance
Moreover, positive results of the CLARITY-AD (lecanemab), TRAILBLAZER-ALZ 2 (donanemab) and EMERGE (aducanumab) trials raise questions. Concerning EMERGE, questions arise about the liability of the results. The two randomized trials, EMERGE and ENGAGE, were stopped for futility, and it was only a post hoc analysis that showed a beneficial effect in patients who received an aducanumab high dose, and this effect was not present in both trials. The effect size itself of the clinically relevant criteria assessed by the CDR-SB is questionable in both EMERGE and CLARITY. A study [50] on the effect size needed to be clinically perceived estimates the variation of the CDR-SB to be > 0.98 for mild cognitive impairment due to AD (MCI-AD) and 1.63 for mild dementia due to AD. Drug effect compared to placebo was – 0.39 (95% CI – 0.69; – 0.09) in EMERGE and -0.45 (95% CI – 0.67; – 0.23) in CLARITY. The size effect for donanemab is reported to be a 35% slowed decline on CDR [41], so we expect it to be similar to lecanemab’s, which is 32%. The impact on the decrease in brain amyloid burden assessed by PET cannot be denied, at around 20 centiloids for aducanumab and > 50 centiloids for lecanemab, but without any real clinical relevance for this surrogate endpoint. EMERGE, ENGAGE and CLARITY-AD all showed a decrease in brain tau protein load [33, 35]. This effect is especially relevant because increased tau protein has been shown to be more associated with cognitive impairment than amyloid protein [51]. This needs to be put in perspective with the fact that cortical atrophy is already present at the prodromal stage compared to the elderly population without AD of the same age or to other neurodegenerative diseases [52]. In view of this early atrophy, the possibility of recovery seems low and questions the possibility of having a clinically perceptible result at 18 months regardless of the treatment, especially since the cognitive impairment is very difficult to perceive because not clinically visible outside the use of neuropsychological tests.
ARIA are Frequent and Potentially Serious Adverse Effects
These treatments are criticized for their specific adverse effect profile. Although most patients remain asymptomatic, edema or microhemorrhage may occur in up to 30% of patients [33]. In view of the frequency of ARIA, beyond the benefit-risk balance, there is also the risk of loss of blindness and follow-up bias if the investigators were able to identify the treatment arm of 30% of patients receiving antibodies. We can see that there are almost twice as many ARIA in the trials with aducanumab (about 30%) than in the one with lecanemab (about 15%), although the latter showed a more pronounced effect. One explanation for this difference may be the epitopes targeted by the antibodies. Since lecanemab targets soluble aggregates or protofibrils, it may be less likely to target amyloid present in the walls of cerebral vessels and thus cause less edema or microhemorrhage. In addition, aducanumab targeting plaques, which are more final lesions, may cause more ARIA but not be early enough to induce a significant benefit on the disease. Three deaths have already been reported in patients receiving lecanemab, in the context of anticoagulation or thrombolysis in stroke, raising the question of excluding APOE4 homozygous patients, who are at greater risk of ARIA. This is the choice made by the US Veterans Health Administration [53, 54], and it could also be proposed to exclude patients with amyloid angiopathy preceding antibodies. Eli Lilly also reported three deaths with donanemab but no details have been shared.
Treatments’ Price, Controversy and Market Authorization
The necessary monitoring of these treatments, mainly by MRI, adds to the already high costs of these treatments and of the health infrastructure needed to administer them. Aducanumab’s cost, initially set at $56,000 per patient per year, has contributed to its failure on the US market. The price was later fixed at $28,000. To pay for this treatment, the Medicare system would have spent more than NASA’s budget, five times more than the most expensive drug currently covered by Medicare [36]. The independent Institute for Clinical and Economic Review (ICER) estimated the fair price of aducanumab to be between $3000 and $8400 per patient per year [55]. The anticipated approval given by the FDA has led to accusations of collusion between the US agency and Biogen. Lecanemab was priced in the US at $26,500 by Eisai and Biogen, closer to the fair price estimated by ICER of between $8500 and $21,500 [56]. While aducanumab still is in accelerated approval, lecanemab’s accelerated approval has been converted to traditional approving in June 2023 [57] despite safety concerns [58]. Following TRAILBLAZER’s results, Eli Lilly & Co. also applied for FDA approval for donanemab [59].
How to Quantify Cognition?
Furthermore, we have discussed clinical relevance, but this is intrinsically limited by the scales used, whereas cognition is impalpable. The CLARITY-AD trial did have the merit of looking for improvement in parameters not directly related to the patient. Results suggest that, for patients on lecanemab, caregiver burden as determined by the Zarit scale would decrease by 38% [60]. Beyond cognition, we can therefore assume a smaller increase in the quality of life of patients and their caregivers [61].
Lecanemab, donanemab and aducanumab are the only ones out of the eight anti-amyloids and four anti-tau to have shown efficacy. Their common characteristics are that they bind to fibrils (or protofibrils) and plaques on their N-terminal end, but this is also the case for bapineuzumab. Yet, bapineuzumab has been evaluated in a particular methodological way by restricting its trials to both purely APOE4 carrier and non-carrier populations. Patients who carry the gene, particularly those who are homozygous, have earlier onset and more rapid progression of AD. It can be assumed that the recruitment of numerous patients carrying this gene in the various trials may allow the sponsors to have a more rapid progression of the disease and to expect to demonstrate an effect despite the short duration of the trials (between 1½ and 3 years in most cases) in a disease that evolves over a period of about 10 years. Temporality is an important issue in this context. As stated above, most Phase 3 trials have lasted between 1½ and 3 years, whereas the median survival time from age of onset of the disease may be between 7 and 10 years [62], and the time to progress from mild to severe dementia would be 10 years, when the patient is followed closely [63]. In response, we can only call for longer trials than those currently being conducted. Longer trials would also answer questions that are currently unanswered. Among these questions is the duration of treatment. What is the optimal duration of treatment with anti-amyloid or anti-tau monoclonal antibodies? Is there still a benefit to treating a patient with moderate to severe dementia? The methodology of these trials also raises the question of the safety of switching from one antibody to another, of combining them or of combining an anti-amyloid antibody with an anti-tau antibody. It is important that patient monitoring continues with Phase IV trials for lecanemab, donanemab and aducanumab to answer these questions.
Some authors interpret the ineffectiveness of the majority of anti-amyloid drugs as calling into question the amyloid hypothesis of AD [64]. They argue that some patients carry amyloid lesions in the cerebrospinal fluid or on the PET scan, but do not present cognitive disorders, and that while some later develop AD others remain asymptomatic. The amyloid load is also poorly correlated with the clinical severity of the patients, and the genes that promote or aggravate AD are not linked to amyloid peptides. Finally, there are many other established risk factors for AD, vascular, metabolic and maybe even infectious, with the presence of Porphyromonas gingivalis statistically associated with patients with AD [14]. An association between APOE4 status and the presence of herpes simplex virus (HSV)-1 has also been demonstrated, further strengthening the possibility of a link between AD and infectious diseases [65]. We can also mention the fact that for relatives, being a caregiver of an AD carrier is a high-risk factor for developing AD, which would not be incompatible with this last hypothesis.
Treatments are being developed to address these issues. Active immunotherapy is making a comeback after its failure using antigen and not antibodies in the amyloid hypothesis [66]. Another lead concerning the tau hypothesis is a promising approach with an aggregation inhibitor which, although negative on its primary endpoint, had several positive secondary endpoints, both cognitive and biological [67]. We also mention the metabolic pathways, with studies on semaglutide currently in Phase 3 [68, 69].
Could a Biological Approach to Help Defining the Disease Improve AD Management?
It may also be suspected that the failure of most trials may be due to an inadequate definition of the disease. The current definition of the disease focuses on a clinical definition that does not necessarily correspond to the thanatological series [70]. As a result, we observe patients diagnosed with AD with lesions of the Lewy body type, hippocampal sclerosis (LATE) or rarer ones such as argyrophilic grain disease, a limbic tauopathy. We also observe disparities in biomarkers in patients with genuine AD. It would be possible to redefine neuro-evolutionary diseases based on both the clinical and neuropsychological phenotypes but also separated into subtypes according to the biomarkers present. We therefore propose to consider the biological aspects of these diseases in the following way, based on neuropathology [71, 72]:
-
Type 1: Abeta and Tau
-
o
A. Abeta and Tau isolated
-
o
B. Abeta and Tau + TDP43 (LATE)
-
o
C. Abeta and Tau + Lewy bodies
-
o
-
Type 2: Abeta without Tau
-
o
A. Abeta isolated
-
o
B. Abeta + TDP43 (LATE)
-
o
C. Abeta + Lewy bodies.
-
o
This redefinition of the disease suggests that we could target treatments according to the proteins identified in patients rather than to the clinical phenotype. Thus, in a patient suffering from cognitive or behavioral impairment and carrying amyloid and tau proteins, a combination of antibodies could be proposed, and in a patient carrying TDP-43 and amyloid, an anti-TDP could be proposed (if antibodies are developed for this indication) in addition to an anti-amyloid, and thus personalized medicine could be proposed to our patients.
References
Dementia [Internet]. https://www.who.int/news-room/fact-sheets/detail/dementia. Accessed 1 Dec 2022.
Dumurgier J, Sabia S. Epidemiology of Alzheimer’s disease: latest trends. Rev Prat. 2020;70:149–51.
Alzheimer’s disease facts and figures. Alzheimers Dement. 2022;18:700–89.
Dubois B, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS–ADRDA criteria. Lancet Neurol. 2007;6:734–46.
Gorno-Tempini ML, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–14.
Thal DR, et al. Phases of Aβ-deposition in the human brain and its relevance for the development of AD. Neurology. 2002;58:1791–800.
Caselli RJ, et al. Alzheimer disease: scientific breakthroughs and translational challenges. Mayo Clin Proc. 2017;92:978–94.
Vijaya Kumar DK, et al. Amyloid-β Peptide Protects Against Microbial Infection In Mouse and Worm Models of Alzheimer’s Disease. Sci Transl Med. 2016;8:340ra72.
Hampel H, et al. The Amyloid-β Pathway in Alzheimer’s Disease. Mol Psychiatry. 2021;26:5481–503.
Morris M, et al. The many faces of tau. Neuron. 2011;70:410–26.
Milà-Alomà M, et al. Amyloid beta, tau, synaptic, neurodegeneration, and glial biomarkers in the preclinical stage of the Alzheimer’s continuum. Alzheimers Dement J Alzheimers Assoc. 2020;16:1358–71.
DeTure MA, Dickson DW. The neuropathological diagnosis of Alzheimer’s disease. Mol Neurodegener. 2019;14:32.
Levine, Kristin S et al. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 2023; 111(7):1086–1093.e2. https://doi.org/10.1016/j.neuron.2022.12.029.
Dominy SS, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5:eaau3333.
Tan C-C, et al. Efficacy and safety of donepezil, galantamine, rivastigmine, and memantine for the treatment of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;41:615–31.
Amieva H, et al. Group and individual cognitive therapies in Alzheimer’s disease: the ETNA3 randomized trial. Int Psychogeriatr. 2016;28:707–17.
Sitzer DI, et al. Cognitive training in Alzheimer’s disease: a meta-analysis of the literature. Acta Psychiatr Scand. 2006;114:75–90.
Plotkin SS, Cashman NR. Passive immunotherapies targeting Aβ and tau in Alzheimer’s disease. Neurobiol Dis. 2020;144: 105010.
Lu L, et al. Anti-Aβ agents for mild to moderate Alzheimer’s disease: systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2020;91:1316–24.
Cummings J, et al. Alzheimer’s disease drug development pipeline: 2022. Alzheimers Dement N Y N. 2022;8: e12295.
Bouter Y, et al. Abeta targets of the biosimilar antibodies of Bapineuzumab, Crenezumab, Solanezumab in comparison to an antibody against N-truncated Abeta in sporadic Alzheimer disease cases and mouse models. Acta Neuropathol (Berl). 2015;130:713–29.
Doody RS, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:311–21.
Honig LS, et al. Trial of solanezumab for mild dementia due to Alzheimer’s disease. N Engl J Med. 2018;378:321–30.
Salloway S, et al. A trial of gantenerumab or solanezumab in dominantly inherited Alzheimer’s disease. Nat Med. 2021;27:1187–96.
Leurent C, et al. Immunotherapy with ponezumab for probable cerebral amyloid angiopathy. Ann Clin Transl Neurol. 2019;6:795–806.
La Porte SL, et al. Structural basis of C-terminal β-amyloid peptide binding by the antibody ponezumab for the treatment of Alzheimer’s disease. J Mol Biol. 2012;421:525–36.
Landen JW, et al. Multiple-dose ponezumab for mild-to-moderate Alzheimer’s disease: Safety and efficacy. Alzheimers Dement Transl Res Clin Interv. 2017;3:339–47.
Ostrowitzki S, et al. Evaluating the safety and efficacy of crenezumab vs placebo in adults with early alzheimer disease: two phase 3 randomized placebo-controlled trials. JAMA Neurol. 2022;79:1113.
Söderberg L et al. Lecanemab, aducanumab, and gantenerumab-binding profiles to different forms of amyloid-beta might explain efficacy and side effects in clinical trials for Alzheimer’s disease. Neurother J Am Soc Exp Neurother. 2023;20(1):195–206.
The SCarlet RoAD Investigators. A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther. 2017;9:95.
Trontinemab | ALZFORUM [Internet]. https://www.alzforum.org/therapeutics/trontinemab. Accessed 9 Mar 2023.
Hoffmann-La Roche. A Phase Ib/IIa, Randomized, Double Blind, Placebo-Controlled, Multiple Ascending Dose, Parallel-Group Study to Investigate the Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of RO7126209 Following Intravenous Infusion in Patients With Prodromal or Mild to Moderate Alzheimer’s Disease [Internet]. clinicaltrials.gov; 2023 Feb. Report No.: NCT04639050. Available from: https://clinicaltrials.gov/ct2/show/NCT04639050
van Dyck CH et al. Lecanemab in early Alzheimer’s disease. N Engl J Med 2023; 388(1):9–21. https://doi.org/10.1056/NEJMoa2212948.
Commissioner O of the. FDA Grants Accelerated Approval for Alzheimer’s Drug [Internet]. FDA. FDA; 2021. https://www.fda.gov/news-events/press-announcements/fda-grants-accelerated-approval-alzheimers-drug. Accessed 22 Nov 2022.
Budd Haeberlein S, et al. Two randomized phase 3 studies of aducanumab in early Alzheimer’s disease. J Prev Alzheimers Dis Internet. 2022. https://doi.org/10.14283/jpad.2022.30.
Maloney and Pallone Release Staff Report on Review, Approval, and Pricing of Biogen’s Alzheimer’s Drug Aduhelm [Internet]. Democr. Energy Commer. Comm. 2022. https://democrats-energycommerce.house.gov/newsroom/press-releases/maloney-and-pallone-release-staff-report-on-review-approval-and-pricing-of. Accessed 3 Mar 2023.
Salloway S, et al. Two phase 3 trials of bapineuzumab in mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370:322–33.
Gueorguieva I, et al. Donanemab Population Pharmacokinetics, Amyloid Plaque Reduction, and Safety in Participants with Alzheimer's disease. Clin Pharmacol Ther. 2023; 113(6):1258–67. https://doi.org/10.1002/cpt.2875.
DeMattos RB, et al. A plaque-specific antibody clears existing β-amyloid Plaques in Alzheimer’s disease mice. Neuron. 2012;76:908–20.
Mintun MA, et al. Donanemab in early Alzheimer’s disease. N Engl J Med. 2021;384:1691–704.
Lilly’s Donanemab Significantly Slowed Cognitive and Functional Decline in Phase 3 Study of Early Alzheimer’s Disease | Eli Lilly and Company [Internet]. https://investor.lilly.com/news-releases/news-release-details/lillys-donanemab-significantly-slowed-cognitive-and-functional. Accessed 13 May 2023.
Sims JR et al. Donanemab in Early Symptomatic Alzheimer Disease: The TRAILBLAZER-ALZ 2 Randomized Clinical Trial. JAMA [Internet]. 2023. https://jamanetwork.com/journals/jama/fullarticle/2807533. Accessed 25 July 2023.
Teng E, et al. Safety and efficacy of semorinemab in individuals with prodromal to mild alzheimer disease: a randomized clinical trial. JAMA Neurol. 2022;79:758.
Qureshi IA, et al. A randomized, single ascending dose study of intravenous BIIB092 in healthy participants. Alzheimers Dement Transl Res Clin Interv. 2018;4:746–55.
Top-line results from tango, a phase 2 study of gosuranemab in participants with mild cognitive impairment due to alzheimer’s disease and mild alzheimer’s disease | Cochrane Library [Internet]. [cited 2022 Nov 29]. https://doi.org/10.1002/central/CN-02348792/full.
Yanamandra K, et al. Anti-tau antibody reduces insoluble tau and decreases brain atrophy. Ann Clin Transl Neurol. 2015;2:278–88.
AbbVie. A Phase 2 Multiple Dose, Multicenter, Randomized, Double-Blind, Placebo-Controlled Study to Evaluate the Efficacy and Safety of ABBV-8E12 in Subjects With Early Alzheimer’s Disease [Internet]. clinicaltrials.gov; 2022 Aug. Report No.: NCT02880956. Available from: https://clinicaltrials.gov/ct2/show/NCT02880956
AbbVie. An Extension Study of ABBV-8E12 in Early Alzheimer’s Disease [Internet]. clinicaltrials.gov; 2022 Aug. Report No.: NCT03712787. Available from: https://clinicaltrials.gov/ct2/show/NCT03712787
Jicha GA, et al. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–32.
Andrews JS, et al. Disease severity and minimal clinically important differences in clinical outcome assessments for Alzheimer’s disease clinical trials. Alzheimers Dement Transl Res Clin Interv. 2019;5:354–63.
Malpas CB, et al. Alzheimer’s Disease Neuroimaging Initiative. Tau and Amyloid-β cerebrospinal fluid biomarkers have differential relationships with cognition in mild cognitive impairment. J Alzheimers Dis JAD. 2015;47:965–75.
Blanc F, et al. Cortical thickness in dementia with lewy bodies and alzheimer’s disease: a comparison of prodromal and dementia stages. PLoS ONE. 2015;10: e0127396.
Tanne JH. Lecanemab: US Veterans Health Administration will cover cost of new Alzheimer’s drug. BMJ. 2023;380: p628.
Lecanemab-irmb (LEQEMBI) Criteria for Use February 2023.
ICER Publishes Final Evidence Report and Policy Recommendations on Aducanumab for Alzheimer’s Disease [Internet]. ICER. https://icer.org/news-insights/press-releases/icer-publishes-final-evidence-report-and-policy-recommendations-on-aducanumab-for-alzheimers-disease/. Accessed 3 Mar 2023.
ICER Publishes Evidence Report on Lecanemab for Alzheimer’s Disease [Internet]. ICER. https://icer.org/news-insights/press-releases/icer-publishes-evidence-report-on-lecanemab-for-alzheimers-disease/. Accessed 19 Mar 2023.
Commissioner O of the. FDA Converts Novel Alzheimer’s Disease Treatment to Traditional Approval [Internet]. FDA. FDA; 2023. https://www.fda.gov/news-events/press-announcements/fda-converts-novel-alzheimers-disease-treatment-traditional-approval. Accessed 9 Aug 2023.
Reardon S. FDA approves Alzheimer’s drug lecanemab amid safety concerns. Nature. 2023;613:227–8.
Constantino AK. Eli Lilly expects FDA decision on Alzheimer’s treatment donanemab by the end of the year [Internet]. CNBC. 2023. https://www.cnbc.com/2023/07/17/alzheimers-eli-lilly-expects-fda-decision-on-donanemab-by-year-end.html. Accessed 9 Aug 2023.
Mead S, Fox NC. Lecanemab slows Alzheimer’s disease: hope and challenges. Lancet Neurol. 2023;22:106–8.
Wolk DA, et al. A step forward in the fight against dementia—are we there yet? JAMA Neurol. 2023;80:429–30.
Todd S, et al. Survival in dementia and predictors of mortality: a review. Int J Geriatr Psychiatry. 2013;28:1109–24.
Blanc F, et al. Long-term cognitive outcome of Alzheimer’s disease and dementia with Lewy bodies: dual disease is worse. Alzheimers Res Ther. 2017;9:47.
Panza F, et al. A critical appraisal of amyloid-β-targeting therapies for Alzheimer disease. Nat Rev Neurol. 2019;15:73–88.
Linard M, et al. Herpes simplex virus, early neuroimaging markers and incidence of Alzheimer’s disease. Transl Psychiatry. 2021;11:414.
Gilman S, et al. Clinical effects of A immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–62.
TauRx Announces Results from Phase 3 Alzheimer’s Disease Study,… [Internet]. TauRx Pharm. https://taurx.com/news-insights/taurx-announces-results-from-phase-3-alzheimers-disease-study-lucidity-assuring-path-for-regulatory-submissions. Accessed 27 Mar 2023.
Novo Nordisk A/S. A Randomised Double-blind Placebo-controlled Clinical Trial Investigating the Effect and Safety of Oral Semaglutide in Subjects With Early Alzheimer´s Disease (EVOKE) [Internet]. clinicaltrials.gov; 2023 Mar. Report No.: NCT04777396. Available from: https://clinicaltrials.gov/ct2/show/NCT04777396
Novo Nordisk A/S. A Randomised Double-blind Placebo-controlled Clinical Trial Investigating the Effect and Safety of Oral Semaglutide in Subjects With Early Alzheimer´s Disease (EVOKE Plus) [Internet]. clinicaltrials.gov; 2023 Mar. Report No.: NCT04777409. Available from: https://clinicaltrials.gov/ct2/show/NCT04777409
Sarto J, et al. Evolution of clinical-pathological correlations in early-onset alzheimer’s disease over a 25-year period in an academic brain bank. J Alzheimers Dis JAD. 2022;87:1659–69.
Agrawal S, et al. The association of Lewy bodies with limbic-predominant age-related TDP-43 encephalopathy neuropathologic changes and their role in cognition and Alzheimer’s dementia in older persons. Acta Neuropathol Commun. 2021;9:156.
Robinson JL, et al. Pathological combinations in neurodegenerative disease are heterogeneous and disease-associated. Brain J Neurol. 2023;146(6):2557–69. https://doi.org/10.1093/brain/awad059.
Medical Writing and Editorial Assistance
Support in the preparation of this article was provided by Dr. Pierre Anthony, Geriatrics Department, General Hospital Centre, CM2R, Geriatric Day Hospital, Colmar, France, through revision of the manuscript.
There was no specific funding for this systematic review nor for the assistance provided to it.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole and have given their approval for this version to be published.
Funding
No funding or sponsorship was received for this study or publication of this article.
Author information
Authors and Affiliations
Contributions
All authors participated in the interpretation of the study results, the drafting and/or critical revision of the manuscript and approval of the final version. Nicolas Collongues and Frédéric Blanc were involved in the development of the concept for this review, Arthur Esquer was involved in development of the protocol, the review and approval of all studies retrieved for inclusion.
Corresponding author
Ethics declarations
Conflict of Interest
Arthur Esquer declares that he has no competing interests. Frédéric Blanc was the national coordinator for France for the Eisai Delphia (E2027), Axovant Headway-DLB and Roche Graduate clinical trials; he had received honoraria from Roche, Eisai, and Biogen for oral presentations, and from Eisai for a participation in a board meeting. Nicolas Collongues has received honoraria for consulting or presentation from Alexion, Biogen Idec, Bristol-Myers Squibb, Horizon Therapeutics, Merck Serono, Novartis, Roche and Sanofi-Genzyme and is a member of the Editorial Board of the Journal de la Ligue Française contre la Sclérose en plaques and the Neurology and Therapy Journal.
Ethical Approval
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Esquer, A., Blanc, F. & Collongues, N. Immunotherapies Targeting Amyloid and Tau Protein in Alzheimer’s Disease: Should We Move Away from Diseases and Focus on Biological Targets? A Systematic Review and Expert Opinion. Neurol Ther 12, 1883–1907 (2023). https://doi.org/10.1007/s40120-023-00541-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00541-1