
Abstract
Introduction
Spinal muscular atrophy (SMA) can cause multiple system dysfunction, especially lipid metabolic disorders, for which management strategies are currently lacking. Microbes are related to metabolism and the pathogenesis of neurological diseases. This study aimed to preliminarily explore the alterations in the gut microbiota in SMA and the potential relationship between altered microbiota and lipid metabolic disorders.
Methods
Fifteen patients with SMA and 17 gender- and age-matched healthy controls were enrolled in the study. Feces and fasting plasma samples were collected. 16S ribosomal RNA sequencing and nontargeted metabolomics analysis were performed to explore the correlation between microbiota and differential lipid metabolites.
Results
No significant difference was found in microbial diversity (α- and β-diversity) between the SMA and control groups, with both groups having a relatively similar community structure. However, compared to the control group, the SMA group showed an increased relative abundance of the genera Ruminiclostridium, Gordonibacter, Enorma, Lawsonella, Frisingicoccus, and Anaerofilum and a decreased abundance of the genera Catabacter, Howardella, Marine_Methylotrophic_Group_3, and Lachnospiraceae_AC2044_group. The concurrent metabolomic analysis showed that the SMA group had 56 different kinds of lipid metabolite levels than did the control group. Additionally, the Spearman correlation suggested a correlation between the altered differential lipid metabolites and the above-mentioned altered microbiota.
Conclusions
The gut microbiome and lipid metabolites differed between the patients with SMA and the control subjects. The altered microbiota may be related with the lipid metabolic disorders in SMA. However, further study is necessary to clarify the mechanism of lipid metabolic disorders and develop management strategies to improve the related complications in SMA.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Spinal muscular atrophy (SMA) is a hereditary neuromuscular disorder with multiple system dysfunction, especially lipid metabolic disorders, for which the pathogenic mechanism is unknown and targeted management strategies are lacking. |
In this study, we sought to combine 16S amplicon sequencing and untargeted metabolomics analysis to explore the potential relationship between the gut microbiome and lipid dysmetabolism in SMA for the first time. |
What was learned from the study? |
The predominant microbiota of patients with SMA was not identical to that of the control group, and the Spearman correlation analysis indicated that the altered microbiota may be related to some lipid metabolite expression. |
The results need to be explored further in order to clarify the pathogenic mechanism of lipid metabolic disorders and develop strategies to improve the related complications in patients with SMA. |
Introduction
Spinal muscular atrophy (SMA), an autosomal recessive neuromuscular disease, is the most common genetic rare disease that causes death in infants under 2 years of age [1]. The most common type of SMA is 5qSMA, which is caused by homozygous deletion or compound heterozygous variant in the survival motor neuron 1 gene (SMN1) located in the chromosome 5q13 region, resulting in SMN protein deficiency in vivo, with a neonatal incidence of 1/10,000 [1]. Based on the gross motor milestones, SMA is clinically classified into type 0 (with death often occurring within 1 month of birth) and types 1–4 [1], with the onset of types 1–3 manifesting in childhood as progressive muscular atrophy and muscle weakness, possibly accompanied by multisystem organ damage [2] and relatively poor quality of life [3].
Previous studies have revealed that patients with SMA may have profound malnutrition and dysmetabolism issues [2], especially lipid metabolic disorders, and these are currently considered to be the most significant metabolic issue in these patients [2, 4]. Patients with SMA are susceptible to dyslipidemia, hepatic steatosis, and non-alcoholic fatty liver disease [2, 4]. Studies have found that the lipid dysmetabolism reported so far mainly refers to fatty acid catabolism disorders, especially β-oxidation, while the identity of the molecules involved in this process remains unclear [2, 4]. It is speculated that the metabolic dysfunctions of patients with SMA may be affected by SMN protein deficiency [2]; however, the specific mechanism of the pathogenesis has not yet been elucidated. Previous research also suggests that regulating the gut microbiota through special diets or drugs may help improve lipid metabolism or non-alcoholic fatty liver disease in patients with obesity or other diseases [5,6,7]. The microbes in the feces of patients with other neurological disorders, such as Alzheimer’s disease (AD), autism spectrum disorder, myasthenia gravis, and Parkinson’s disease (PD), are abnormal compared with the feces of the general population, possibly indicating a potential correlation with the abnormal expression of disease-related metabolic pathways [8,9,10,11]. Similarly, the possibility that the potential alterations in the intestinal microbes affect the patient’s lipid metabolism cannot be ruled out in patients with SMA as a chronic progressive neurologically related disease, which may provide research approaches for the management of lipid metabolic disorders in such patients.
The aim of the present study was to explore differences in the intestinal microbiota of patients with SMA compared with healthy subjects, investigate the abnormal expression of lipid metabolites in these patients, and preliminarily study the relationship between intestinal microbiota and abnormal lipid metabolism in such patients. Throughout the study, we tried to provide a clinical basis to further explore the pathological mechanism and the potential management of lipid metabolic disorders in SMA.
Methods
Compliance with Ethics Guidelines
The study was approved by the Ethical Committee of Children’s Hospital, Zhejiang University School of Medicine (Approval number: 2019-IRB-177). The study was conducted following the Declaration of Helsinki of 1964 and its subsequent amendments. Written informed consent was obtained from all participants and/or their guardians before starting the study.
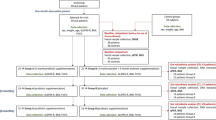
Participants
Initially, 20 patients with 5qSMA and 20 age- and gender-matched healthy controls from the outpatient physical examination center of our hospital were enrolled in the study. Of these, five patients and three control subjects did not fully meet the inclusion criteria; consequently, 15 patients and 17 controls were included in the final analysis. All participants were assessed between March 2021 and September 2022 by the researchers according to a uniform standard, mitigating potential assessor bias. The inclusion criteria for the patients with SMA were: (1) diagnosis of 5qSMA types by genetic testing; (2) age under 18 years with 5qSMA types II or III; (3) provision of informed consent to participate in the study; (4) exclusion of any other acute or chronic diseases which may affect gut microbiota or energy metabolism; and (5) simultaneous collection of both peripheral venous blood under an 8-h fasting regimen and fresh fecal samples within 24 h. All of the patients with SMA did not receive disease-modifying treatment, including Nusinersen, Risdiplam or Zolgensma®. Additionally, all of the enrolled study participants came from the eastern of China, and the dietary structure of the patients (Electronic Supplementary Material Table S1) was similar to that of the general Chinese population according to the Chinese Dietary Reference Intakes (2013, https://www.cnsoc.org/drpostand/); thus, the differences in race, lifestyle, and daily diets were minimized. Healthy children of similar age, gender, and body mass index (BMI) who concurrently visited the physical examination center in our hospital and whose physical examination results were suggestive of good health, were taken as the control group. The fat mass index (FMI) of both of the two groups was also examined using the Inbody 770 MF-BIA device (InBody USA, Cerritos, CA, USA). None of the participants had taken antibiotics, probiotics, dietary supplements, (es)-omprezaole, other medications or supplements for at least 6 months preceding fecal sample collection or suffered from obvious malnutrition. Additionally, at the time of our study, COVID-19 infection in China was sporadic, and children had gradually returned to school and resumed their normal daily activities. None of the enrolled children had a history of COVID-19 infection and, consequently, the results were likely to be less affected by the COVID-19 epidemic.
Specimen Collection, Preparation, and Storage
Fresh stool and peripheral blood specimens were obtained from patients and healthy controls using the uniform sterile fecal collection device and ethylenediaminetetraacetic acid anticoagulation tubes, respectively. Two samples of each fresh fecal collection (approx. 2 g for each participant) were immediately collected by researchers and each sample was placed in a separate sterile centrifuge tubes, immediately frozen, and stored at − 80 °C for preservation until the final examinations. A 2-ml sample of peripheral blood was centrifuged in a low-temperature ultracentrifuge at 6000 rpm, 4 °C for 10 min immediately after collection. The supernatant was separated and immediately re-centrifuged at 12,000 rpm, 4 °C for 10 min. The final supernatant acquired was divided over screw-cap cryotubes and stored at − 80 °C.
DNA Extraction and Amplification, and Library Construction and Sequencing
The MagPure Soil DNA LQ Kit (Magen Biotechnology Co., Ltd., Guangzhou, China) was used to extract the total genomic DNA. DNA concentrations and integrity were measured using the NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA) and agarose gel electrophoresis. Extracted DNA was stored at − 20 °C until further processing. The extracted DNA was used as a template for PCR amplification with the barcoded primers and Takara Ex Taq DNA polymerase (Takara Bio Inc., Shiga, Japan). V3–V4 variable regions of 16S ribosomal RNA (rRNA) genes were amplified with universal primers 343-F (primer sequence: 5′-TACGGRAGGCAGCAG-3′) and 798-R (primer sequence: 5′-AGGGTATCTAATCCT-3′) for bacterial diversity analysis. The PCR products were purified with Agencourt AMPure XP beads (Beckman Coulter Co., Brea, CA, USA) and quantified using a Qubit dsDNA assay kit (Invitrogen, Thermo Fisher Scientific). Sequencing was performed on an Illumina NovaSeq6000 system with two paired-end read cycles of 250 bases each (Illumina Inc., San Diego, CA; OE Biotech Company, Shanghai, China).
Bioinformatic Analysis of the Microbiota
Paired-end reads were preprocessed using Trimmomatic software [12] to detect and cut off ambiguous bases (N) as well as low-quality sequences with an average quality score of < 20 using a sliding window trimming approach. After trimming, paired-end reads were assembled using FLASH software [13]. Reads with 75% of bases > Q20 score were retained using QIIME software (version 1.8.0) [14]. Then, reads with chimera were detected and removed using VSEARCH [15]. Clean reads were subjected to the removal of primer sequences and clustering to generate operational taxonomic units (OTUs) using VSEARCH software with a 97% similarity cutoff [15]. The representative read of each OTU was selected using the QIIME package. The alpha diversity, including the Chao1 index [16] and Shannon index [17], was used to estimate the microbial diversity in fecal samples. The Bray–Curtis distance matrix performed by QIIME software was used for Bray–Curtis principal coordinate analysis (PCoA) and phylogenetic tree construction. Sequencing and analysis of the 16S rRNA gene amplicon were conducted by OE Biotech Co., Ltd. (Shanghai, China).
Liquid and Gas Chromatograph–mass Spectrometry Detection
An untargeted metabolomics analysis on peripheral blood specimens was prepared and detected following a unified and standardized operation process of liquid chromatography-mass spectrometry (LC–MS) and gas chromatography-mass spectrometry (GC–MS) analyses. LC–MS analysis was mainly performed on a Dionex Ultimate 3000 RS UHPLC system fitted with a Q-Exactive plus quadrupole-orbitrap mass spectrometer equipped with a heated electrospray ionization source (Thermo Fisher Scientific, Waltham, MA, USA). The derivative samples for the GC–MS analysis were analyzed on an Agilent 7890B gas chromatography system coupled to an Agilent 5977A MSD system (Agilent Technologies Inc., Santa Clara, CA, USA). Mass spectrometric data were acquired in a full-scan mode (m/z 50–500).
Computational Analyses of Metabonomic Data
The original LC–MS data were processed using Progenesis QI V2.3 (Nonlinear, Dynamics, Newcastle, UK) software for baseline filtering, peak identification, integral, retention time correction, peak alignment, and normalization. The GC–MS data were imported into the MS-DIAL software, which performs peak detection, peak identification, MS2Dec deconvolution, characterization, peak alignment, wave filtering, and missing value interpolation. All peak signal intensities were segmented and normalized according to the internal standards, with a relative standard deviation of > 0.3 after screening.
LC and GC data matrices were merged, and then the combined data were normalized into columns and used for the subsequent analysis. The combined matrix was imported into R (R Foundation for Statistical Computing, Vienna, Austria) to conduct the PCoA, with the aim to observe the overall distribution among the samples and the stability of the whole analysis process. Orthogonal partial least-squares-discriminant analysis (OPLS-DA) was utilized to distinguish the metabolites that differ between groups. The quality of the model was evaluated using sevenfold cross-validation and 200 response permutation testing to prevent overfitting. Variable importance of projection (VIP) values obtained from the OPLS-DA model was used to rank the overall contribution of each variable to group discrimination. A two-tailed Student’s t-test was further used to verify the significant differences in metabolites between groups. Differential metabolites were selected with VIP values of > 1.0 and P-values of < 0.05.
Correlation Analysis Between Microbiome and Metabolomics
Spearman’s correlation coefficient was used to analyze the potential correlation between the differential microorganisms and metabolites. Images and figures were integrated by a complete set of data analysis tools (https://cloud.oebiotech.cn/task/) and the Adobe Illustrator CC2018 (Adobe Inc., San Jose, CA, USA).
Results
Study Participant Characteristics
The demographic characteristics of the 15 patients with SMA and 17 healthy controls at baseline are shown in Table 1. All patients with SMA had no history of any other diseases, such as congenital/metabolic disorders or factors, which have the potential to contribute to the altered gut microbiota or the blood lipid levels. No obvious racial, dietary, or lifestyle differences were found in the study population, and the nutritional status (BMI, FMI) between the two groups was not significantly different (P > 0.05).
Characteristics of Gut Microbiome
Alteration of Fecal Microbiota Diversity
The results of α- and β-diversity of fecal microbiota in the two study groups are shown in Fig. 1. First, the Chao1 index showed no significant differences in the abundance and α-diversity of bacterial microbes between patients with SMA and the controls (Fig. 1a; P > 0.05); the Shannon index showed result that were similar to those of the Chao1 index (Fig. 1b; P > 0.05). The PCoA with binary Jaccard distance results suggested that there were no significant differences between the SMA and the control groups (Fig. 1c).


The gut microbiota sequencing overview between the patients with spinal muscular atrophy (SMA) and the healthy controls (CON). a, b Alpha diversity (Chao1 and Shannon index) between the SMA and the control group. c Principal coordinate analysis plot based on binary Jaccard distance. PC1 and PC2 are used to plot the coordinate axis and the percentages refer to the extent of variation explained. d, e The taxonomic distributions and relative abundance of the fecal microorganisms from both study groups at the phylum and the genus level, respectively (only the top 10 displayed). Different colors represent different microbes at the same level. The height of the bars represents relative abundance. f Difference in the relative abundance of microbiota genus between the SMA and the control group based on the Wilcoxon analysis
Community Structure and Fecal Microbiota Taxa
The microbial community structure and dominant microbiota of the SMA group and the matched control group were studied. The results of this analysis is shown mainly at the phylum and genus levels (Fig. 1d, e).
Based on the 16S rRNA results, the four most abundant phyla detected included Bacteroidetes, Firmicutes, Proteobacteria, and Actinobacteriota, accounting for 41.9%, 41.0%, 8.6%, and 8.0% of microbiota in the SMA group and 48.6%, 38.2%, 6.8%, and 5.7% of microbiota in the control group; there was no significant difference between the groups (Fig. 1d). At the genus level, the five dominant genera detected in patients with SMA were Bacteroides (29.9%), Faecalibacterium (7.4%), Bifidobacterium (5.9%), Escherichia-Shigella (6.0%), and Blautia (4.0%); in comparison, the five dominant groups in the healthy controls were Bacteroides (38.0%), Faecalibacterium (7.6%), Bifidobacterium (5.0%), Escherichia-Shigella (4.3%), and Prevotella (4.7%) (Fig. 1e). Analysis of the dominant microbiota in the two groups revealed that in comparison with the control group, the genera Gordonibacter, Ruminiclostridium, Enorma, Lawsonella, Frisingicoccus, and Anaerofilum were present at a significantly higher abundance (Fig. 1f). However, no significant difference was found in the Firmicutes/Bacteroidota (F/B) ratio between the SMA and control groups (P = 0.331).
Characteristics of Metabolism
Differences in Metabolic Profiles
Nontargeted metabolomic analysis was performed on the differences in metabolism between the SMA and control groups. A total of 983 differential metabolites, including 695 positive-mode features and 180 negative-mode features, were identified in the metabolic profiles of all samples between the SMA and control groups. The OPLS-DA score plot clearly distinguished the SMA and control groups (Fig. 2a), indicating that patients with SMA have metabolic abnormalities. The heatmap revealed a portion of the statistically significant differential metabolites (Fig. 2b), which were regulated to varying degrees in patients with SMA compared with the controls.


a The orthogonal partial least-squares-discriminant analysis (OPLS-DA) score chart of the difference in metabolites between the SMA and the control group. b The taxa heatmap of the expression of metabolites between the SMA and the control groups. The color gradient from blue to red is used to reflect the abundance from low to high, respectively
The Prediction of Unigenes and Functional Annotation
Of all the differential metabolites, 182 could be linked to their Kyoto Encyclopedia of Genes and Genomes metabolic pathways (all P < 0.05). All of the differential metabolites identified are classified as compounds, including lipids and lipid-like molecules, benzenoids, nucleosides, nucleotides and analogs, organic acids and derivatives, organic nitrogen or oxygen compounds, organoheterocyclic compounds, and phenylpropanoids and polyketides.
In particular, 56 kinds of lipids and lipid-like molecules were significantly different from the control (all P < 0.05). These differential lipid metabolites were linked to specific lipid metabolic pathways, such as linoleic and alpha-linolenic acid metabolism, arachidonic acid metabolism, fatty acid degradation and elongation, primary bile acid biosynthesis, biosynthesis of unsaturated fatty acids, and cholesterol metabolism. Additionally, some of the statistically significant metabolic enrichment pathways involved in lipid metabolism included linoleic and alpha-linolenic acid metabolism, synthesis and degradation of ketone bodies, cholesterol metabolism, propanoate metabolism, and a number of related pathways, such as pantothenate and CoA biosynthesis, glutathione metabolism, cAMP signaling, and mTOR signaling pathway (Fig. 3a).

a Kyoto Encyclopedia of Genes and Genomes metabolic pathway enrichment results. Each point represents a functional profile of the participants, which is presented in the same color as the same group. b Spearman correlation analysis of the relationship between the differential microbiota genus and differential lipid metabolites. Spearman’s rank correlation coefficient is indicated by the color of the cells. Positive or negative correlations are represented by shades of red or blue, respectively. *P < 0.05, **P < 0.01, ***P < 0.001
The Conjoint Analysis of Metabolomics and Metagenomics
Taking the change in both microbial composition and metabolic function into account, we then used both metagenomics and metabolomics to investigate the possible relationship between the lipid metabolites and microbes based on our findings. As shown on Fig. 3b, ten differential microbiota genera were significantly correlated with 33 lipid metabolites. In particular, the altered lipid levels and lipid-like molecules were found to be correlated with the specific microbes that were abundant, especially for Ruminiclostridium, Gordonibacter, and Catabacter. For example, the genus Ruminiclostridium was negatively associated with the levels of 3-hydroxy-3-methyl-2-oxo-butanoic acid, palmitaldehyde, 7-keto-8-aminopelargonic acid (KAPA), coriolic acid, 6-keto-PGF1α, and thromboxane B2 (r = − 0.63, P = 0.0001; r = − 0.59, P = 0.0004; r = − 0.51, P = 0.003; r = − 0.51, P = 0.003; r = − 0.55, P = 0.001; r = − 0.50, P = 0.003, respectively). The genus Gordonibacter was negatively associated with 8-HpODE (r = − 0.63, P = 0.0001), and genus Catabacter was negatively associated with KAPA (r = − 0.53, P = 0.002) and 3-oxooctanoyl-CoA (r = − 0.53, P = 0.002). These metabolites are involved in several lipid metabolic pathways, such as fatty acid degradation and elongation, and linoleic and arachidonic acid metabolism.
Discussion
In the present study we combined 16S rRNA amplicon sequencing and untargeted metabolomics analysis to explore the relationship between the gut microbiome and lipid dysmetabolism in SMA for the first time, which to our knowledge, is also the first study on the gut microbiome of patients with SMA worldwide. We found that patients with SMA had no significant alteration in microbial diversity compared to the matched controls, but the predominant microbiota was not identical to that of the healthy controls and the expression of some specific lipid metabolites was altered. The correlation analysis indicated that there may be a link between the two phenomena, which deserves further exploration to clarify the pathogenic mechanism of lipid metabolic disorders and develop strategies to improve the related complications in patients with SMA.
Previous studies revealed a relationship between the gut microbiome and the onset of various diseases. Intestinal microbiome disorders of different types and degrees were found in some nervous system diseases, such as AD [9], PD [8], and so on, which was considered to be a potential mechanism of causing disease or aggravating disease phenotypes due to the role of the brain–gut axis [18, 19]. Our study found no significant alteration in the diversity and community of the gut microbiome in patients with SMA after reducing the differences in region and dietary culture as much as possible. The F/B ratio, indicating an intestinal inflammatory state [20], was not significantly different between the SMA group and the control group. However, genera Gordonibacter, Ruminiclostridium, and Enorma were more dominant in the SMA group compared with the control group, suggesting that the intestinal microbiota in the SMA group remained different from that of controls even though genera Bacteroidetes and Firmicutes were the main microorganisms between the two groups. Considering that the subjects in our study were all children with SMA types 2 and 3 and with similar motor function, and that the disease course was relatively short, further study is necessary to expand the age range to observe whether the abundance and diversity of the intestinal microbiome will alter with disease prognoses.
The gut microbiota regulates a series of key metabolic functions of the body, and its dysbiosis plays an important role in modulating metabolic disturbances [21]. The bacteria with increased relative abundance found in our microbiome results belonged to the genera Clostridia, Actinobacteria, Lachnospiraceae, and Ruminococcaceae, while those with decreased relative abundance belonged to the genera Lachnospiraceae, Methylophagaceae, and Christensenellaceae. In persons with type 2 diabetes, obesity, and nervous system diseases, such as neonatal brain injury, AD, and amyotrophic lateral sclerosis (ALS), the abundance of short-chain fatty acid-producing bacteria (e.g., Clostridium, Helicobacter, Eubacterium, Ruminococcus, Lactobacillus, Bifidobacterium, and Proteobacteria) often decreases [9, 22,23,24,25]. However, the abundance of probiotics producing short-chain fatty acids did not decrease at all in our patients with SMA, suggesting that the alterations in intestinal microbiota may differ from that occurring in other diseases. Whether the alterations in gut microbiota are compensatory changes caused by metabolic disorders or other causes requires dynamic follow-up observation.
The management of functional damage to multiple systems in patients with SMA is being increasingly emphasized [26], especially lipid metabolic disorders [2, 4], with the availability of disease-modifying treatment drugs. Studies of SMA lipid metabolic disorders have revealed that defects in the transport and β-oxidation of fatty acids and the catabolism of some lipid substances are common manifestations in SMA [2, 4, 27, 28]. These defects have been reported to be probably associated with SMN protein deficiency [2, 4, 27], while the specific mechanism has not yet been clarified and, therefore, effective interventions are lacking. The results of our metabolomic analysis confirmed abnormal lipid metabolism in patients with SMA. Additionally, the potential disorders of lipid metabolic pathways, involving linoleic acid metabolism, alpha-linolenic acid metabolism, synthesis and degradation of ketone bodies, cholesterol metabolism, and propanoate metabolism, could occur in various other diseases, such as obesity, diabetes, and cardiovascular diseases [29,30,31,32,33,34], reflecting the intersection of SMA and other systemic diseases.
In addition to the microbiome and metabonomic analyses, the Spearman correlation indicated that alterations in the expression of lipid metabolites might be linked to those in differential microbiota, particularly the genera Ruminiclostridium and Gordonibacter. Ruminiclostridium has been found to modulate lipid metabolism, produce short-chain fatty acids, stimulate the development of beige adipocytes in white adipose tissue, reduce inflammation, and improve intestinal barrier function [35, 36]. Gordonibacter has been reported to be involved in various biosynthetic processes, including fat metabolism and fatty liver formation [37, 38]. KAPA is associated with biotin metabolism, and biotin mainly influences phospholipid metabolism, which can be generated by intestinal bacteria in general, and normal fat metabolism will be affected in case of deficiency [39]. Coriolic acid is involved in linoleic acid metabolism, which is an essential fatty acid for the human body and can help reduce blood cholesterol and prevent atherosclerosis [31, 32, 40]. Its abnormal metabolism may affect cholesterol metabolism and cause cardiovascular and cerebrovascular diseases [32, 40]. Thromboxane B2 and 6-keto-PGF1α are involved in arachidonic acid metabolism, which participates in various physiological processes and is an important substance for cell regulation [41]. Its metabolic disorder may affect the physiological functions of multiple systems. Additionally, palmitaldehyde participates in fatty acid degradation [42]. Thus, these bacteria we found may be linked to fatty acid degradation and elongation, linoleic acid metabolism, arachidonic acid metabolism, among others, by negatively regulating the lipid metabolite expression. These results indicate the potential influence of the intestinal microbiota in lipid metabolic disorders in SMA in addition to the effects of SMN protein deficiency. The potential effects of these microbiome alterations on lipid metabolism need to be further explored.
To our knowledge, the present study is the first to explore the microbiome and combined metabonomics in SMA. The results provide initial evidence that alterations to the intestinal microbiota may be connected with the lipid metabolism disorders in patients with SMA; this finding may help explore the management of metabolic complications in such patients. There are a number of limitations to mention. First, the number of cases in this study is limited because it is relatively difficult to simultaneously collect stool and peripheral blood samples from patients with SMA, and the disease course of patients with SMA is relatively short (all were children in the present study). The source area of patients with SMA was also limited. Additionally, as the alterations in both the gut microbiota and lipid metabolism could be affected by the severe muscle weakness and immobility, and chronic constipation may also influence the patients’ gut microbiota, future research is needed to explore the potential effects of multiple factors on the gut microbiome and lipid metabolism in SMA. Animal experiments or in vitro studies are also necessary.
Conclusions
In this study, we observed the characteristics of the gut microbiome and lipid metabolites of patients with SMA. The gut microbiome of patient with SMA was different from that of the healthy controls, and patients with SMA had abnormal lipid metabolism, which may be related with the gut microbiome disorders in SMA. These results suggest that the gut microbiome might play a role in the pathogeny of lipid metabolic disorders in SMA, which deserves further research to explore management strategies to improve the related complications in such patients.
References
Mercuri E, Sumner CJ, Muntoni F, et al. Spinal muscular atrophy. Nat Rev Dis Primers. 2022;8:52.
Li YJ, Chen TH, Wu YZ, et al. Metabolic and nutritional issues associated with spinal muscular atrophy. Nutrients. 2020;12:3842.
Yao M, Ma Y, Qian R, et al. Quality of life of children with spinal muscular atrophy and their caregivers from the perspective of caregivers: a Chinese cross-sectional study. Orphanet J Rare Dis. 2021;16:7.
Deguise MO, Baranello G, Mastella C, et al. Abnormal fatty acid metabolism is a core component of spinal muscular atrophy. Ann Clin Transl Neurol. 2019;6:1519–32.
Wanezaki S, Taniwaki T, Miyamoto J, et al. Dietary combination of fish oil and soy β-conglycinin inhibits fat accumulation and reduces blood glucose levels by altering gut microbiome composition in diabetic/obese KK-A y mice. J Oleo Sci. 2023;72:303–12.
Li Q, Liu W, Zhang H, et al. α-D-1,3-glucan from Radix Puerariae thomsonii improves NAFLD by regulating the intestinal flora and metabolites. Carbohyd Polym. 2023;299: 120197.
Maestri M, Santopaolo F, Pompili M, Gasbarrini A, Ponziani FR. Gut microbiota modulation in patients with non-alcoholic fatty liver disease: Effects of current treatments and future strategies. Front Nutr. 2023;10:1110536.
Kenna JE, Chua EG, Bakeberg M, et al. Changes in the gut microbiome and predicted functional metabolic effects in an Australian Parkinson’s disease cohort. Front Neurosci. 2021;15: 756951.
Marć MA, Jastrząb R, Mytych J. Does the gut microbial metabolome really matter? The connection between GUT metabolome and neurological disorders. Nutrients. 2022;14:3967.
Liu P, Jiang Y, Gu S, et al. Metagenome-wide association study of gut microbiome revealed potential microbial marker set for diagnosis of pediatric myasthenia gravis. BMC Med. 2021;19:159.
Chen YC, Lin HY, Chien Y, et al. Altered gut microbiota correlates with behavioral problems but not gastrointestinal symptoms in individuals with autism. Brain Behav Immun. 2022;106:161–78.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Reyon D, Tsai SQ, Khayter C, et al. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30:460–5.
Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods. 2010;7:335–6.
Rognes T, Flouri T, Nichols B, et al. VSEARCH: a versatile open source tool for metagenomics. PeerJ. 2016;4: e2584.
Chao A, Bunge J. Estimating the number of species in a stochastic abundance model. Biometrics. 2002;58:531–9.
Hill TC, Walsh KA, Harris JA, et al. Using ecological diversity measures with bacterial communities. FEMS Microbiol Ecol. 2003;43:1–11.
Willyard C. How gut microbes could drive brain disorders. Nature. 2021;590:22–5.
Cryan JF, O’Riordan KJ, Sandhu K, et al. The gut microbiome in neurological disorders. Lancet Neurol. 2020;19:179–94.
Crawford MS, Mohr AE, Sweazea KL. Novel organic mineral complex prevents high-fat diet-induced changes in the gut and liver of male sprague-dawley rats. J Nutr Metab. 2020;2020:8846401.
Pascale A, Marchesi N, Marelli C, et al. Microbiota and metabolic diseases. Endocrine. 2018;61:357–71.
Zeng Q, Shen J, Chen K, et al. The alteration of gut microbiome and metabolism in amyotrophic lateral sclerosis patients. Sci Rep. 2020;10:12998.
Zhao L, Zhang F, Ding X, et al. Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science. 2018;359:1151–6.
Li Y, Zhou W, Yuan W, et al. Composition of gut microbiome in neonates with severe hyperbilirubinemia and its effect on bilirubin brain injury. Chin J Appl Clin Pediatr. 2018;2:103–7.
John GK, Mullin GE. The gut microbiome and obesity. Curr Oncol Rep. 2016;18:45.
Mercuri E, Finkel RS, Muntoni F, et al. Diagnosis and management of spinal muscular atrophy: part 1: recommendations for diagnosis, rehabilitation, orthopedic and nutritional care. Neuromuscul Disord. 2018;28:103–15.
Zolkipli Z, Sherlock M, Biggar WD, et al. Abnormal fatty acid metabolism in spinal muscular atrophy may predispose to perioperative risks. Eur J Paediatr Neurol. 2012;16:549–53.
Watson KS, Boukhloufi I, Bowerman M, et al. The relationship between body composition, fatty acid metabolism and diet in spinal muscular atrophy. Brain Sci. 2021;11:131.
Naughton SS, Mathai ML, Hryciw DH, et al. Linoleic acid and the pathogenesis of obesity. Prostagland Other Lipid Mediat. 2016;125:90–9.
O’Reilly ME, Lenighan YM, Dillon E, et al. Conjugated linoleic acid and alpha linolenic acid improve cholesterol homeostasis in obesity by modulating distinct hepatic protein pathways. Mol Nutr Food Res. 2020;64: e1900599.
Cabout M, Alssema M, Nijpels G, et al. Circulating linoleic acid and alpha-linolenic acid and glucose metabolism: the Hoorn Study. Eur J Nutr. 2017;56:2171–80.
Naghshi S, Aune D, Beyene J, et al. Dietary intake and biomarkers of alpha linolenic acid and risk of all cause, cardiovascular, and cancer mortality: systematic review and dose-response meta-analysis of cohort studies. BMJ. 2021;375: n2213.
Yurista SR, Chong CR, Badimon JJ, et al. Therapeutic potential of ketone bodies for patients with cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol. 2021;77:1660–9.
Coppola S, Avagliano C, Calignano A, et al. The protective role of butyrate against obesity and obesity-related diseases. Molecules. 2021;26:682.
Wang P, Li D, Ke W, et al. Resveratrol-induced gut microbiota reduces obesity in high-fat diet-fed mice. Int J Obes. 2005;2020(44):213–25.
Song H, Shen X, Chu Q, et al. Vaccinium bracteatum Thunb. fruit extract reduces high-fat diet-induced obesity with modulation of the gut microbiota in obese mice. J Food Biochem. 2021;45:e13808.
Kang K, Sun Y, Pan D, et al. Distinctive gut microbial dysbiosis between chronic alcoholic fatty liver disease and metabolic-associated fatty liver disease in mice. Exp Ther Med. 2021;21:418.
Ngom II, Hasni I, Lo CI, et al. Taxono-genomics and description of Gordonibacter massiliensis sp. Nov., a new bacterium isolated from stool of healthy patient. New Microbes New Infect. 2020;33:100624.
McMahon RJ. Biotin in metabolism and molecular biology. Annu Rev Nutr. 2002;22:221–39.
Whelan J, Fritsche K. Linoleic acid. Adv Nutr. 2013;4:311–2.
Szczuko M, Kikut J, Komorniak N, et al. The role of arachidonic and linoleic acid derivatives in pathological pregnancies and the human reproduction process. Int J Mol Sci. 2020;21:9628.
Ferrell WJ, Yao KC. Metabolism of palmitaldehyde in human cardiac muscle-II Fatty chain elongation and shortening. Int J Biochem. 1982;14:635–40.
Acknowledgements
The authors want to thank all the participants of the study and thank Yan Ni for her contribution to the Methodology guidance of this study.
Funding
This study was supported by the National Natural Science Foundation (82,271,735), Key R&D Program of Zhejiang Province (2022C03167), Zhejiang Province Public Welfare Technology Application Research Project (LGC21H090001), Key Technologies Research and Development Program of Zhejiang Province (2021C03099), and Scientific Research Fund of Zhejiang University (XY2022045). The journal’s Rapid Service Fee was funded by the authors.
Author Contributions
Study conception and design: Shanshan Mao. Data collection: Yijie Feng, Yiqin Cui, Jianing Jin, Siyi Huang, Mei Yao, and Dongming Zhou. Statistical analysis: Yijie Feng. All authors were involved in discussing the results, drafting and critically reviewing the manuscript for intellectual content, and providing final approval of the submitted version. All authors agree to be accountable for this work.
Disclosures
Shanshan Mao, Yijie Feng, Yiqin Cui, Jianing Jin, Siyi Huang, Jia Wei, Mei Yao and Dongming Zhou report no relevant disclosures.
Compliance with Ethics Guidelines
The study was approved by the Ethical Committee of Children’s Hospital, Zhejiang University School of Medicine (Approval number: 2019-IRB-177). The study was conducted following the Declaration of Helsinki of 1964 and its subsequent amendments. Written informed consent was obtained from all participants and/or their guardians before starting the study.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Feng, Y., Cui, Y., Jin, J. et al. The Alterations of Gut Microbiome and Lipid Metabolism in Patients with Spinal Muscular Atrophy. Neurol Ther 12, 961–976 (2023). https://doi.org/10.1007/s40120-023-00477-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-023-00477-6