
Abstract
Introduction
Hereditary transthyretin amyloidosis (ATTRv amyloidosis) is a clinically heterogeneous disease caused by mutations in the transthyretin (TTR) gene. The most common mutation, Val30Met, can manifest as an early- or late-onset disease.
Methods
The Transthyretin Amyloidosis Outcomes Survey (THAOS) is an ongoing, global, longitudinal, observational survey of patients with transthyretin amyloidosis, including both inherited and wild-type disease and asymptomatic patients with TTR mutations. This is a descriptive analysis of symptomatic patients with ATTRv Val30Met amyloidosis with late- (age at least 50 years) vs. early-onset (age less than 50 years) disease in THAOS (data cutoff August 1, 2019).
Results
Of 1389 patients with ATTRv Val30Met amyloidosis, 491 (35.3%) had late-onset disease. Compared with early-onset, patients with late-onset were more likely to be male (66.2% vs. 53.6%) and have a longer mean (standard deviation [SD]) time from onset to diagnosis (3.8 [3.4] vs. 2.7 [4.1] years). Late-onset disease was associated with more severe neurological impairment at enrollment (median [10th, 90th percentile] derived Neuropathy Impairment Score in the Lower Limbs, 25.0 [4.0, 69.3] vs. 8.0 [0, 54.8]; Neurologic Composite Score, 42.0 [2.0, 155.0] vs. 21.0 [0, 102.0]). Cardiac findings were more prominent in late-onset disease. An overall interpretation of electrocardiogram as abnormal was reported in 72.1% of late-onset patients (vs. 44.3% early-onset). A left-ventricular septal thickness of at least 12 mm was reported in 69.7% of late-onset patients (vs. 14.6% early-onset). All differences were statistically significant (p < 0.001).
Conclusion
In THAOS, late-onset ATTRv Val30Met amyloidosis is common, presenting with more severe neurologic and cardiac findings at enrollment. Heterogeneity of disease may make it more difficult to diagnose. Increased recognition of late-onset ATTRv Val30Met amyloidosis could lead to more timely diagnosis and improve patient outcomes.
Trial Registration
ClinicalTrials.gov NCT00628745.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Why carry out this study? |
Hereditary transthyretin (ATTRv) amyloidosis is a clinically heterogeneous disease caused by mutations in the transthyretin (TTR) gene. |
The most common mutation, Val30Met, can manifest as an early- or late-onset disease. |
This analysis from THAOS was carried out to broaden the understanding of the characteristics of patients with late- vs. early-onset ATTRv Val30Met amyloidosis |
What was learned from the study? |
In THAOS, late-onset ATTRv Val30Met amyloidosis is relatively common, occurring in approximately one-third of participants and presenting with more severe neurologic and cardiac findings at enrollment. |
Compared with patients with early-onset ATTRv Val30Met amyloidosis, patients with late-onset disease were more likely to have a longer mean time from symptom onset to diagnosis, and to have experienced misdiagnosis. |
Increased recognition of late-onset ATTRv Val30Met amyloidosis could lead to more timely diagnosis and improve patient outcomes. |
Digital Features
This article is published with digital features, including a summary slide, to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.14556189.
Introduction
Hereditary transthyretin amyloidosis (ATTRv amyloidosis) is a progressive, rare, fatal, systemic disorder caused by the deposition of amyloid fibrils composed of misfolded transthyretin (TTR) in peripheral nerves and vital organs. ATTRv amyloidosis is a clinically heterogeneous disease caused by mutations in the TTR gene [1], leading to progressive debilitating sensorimotor polyneuropathy and autonomic dysfunction [2] and, in many cases, cardiac complications [1, 3].
More than 140 ATTRv genotypes have been identified [4, 5]. The most common mutation is Val30Met (p.Val50Met), which may manifest as an early-onset or late-onset disease [1].
Early-onset disease (at age less than 50 years) has been considered the more common form of disease in endemic regions [1, 6]. However, a distinct late-onset form of disease (at age 50 years or more) is also relatively common in some endemic (e.g., Sweden) and non-endemic regions [2, 6,7,8,9]. Data characterizing late-onset disease are limited, though information from patients from Brazil indicates that this form is more difficult to diagnose because of the reduced incidence of family history of disease, which is considered a hallmark of early-onset disease [9]. In patients from Japan, late-onset disease was associated with sensorimotor symptoms beginning in the lower extremities, with disturbance of both superficial and deep sensation and relatively mild autonomic symptoms, while early-onset disease was associated with predominant loss of superficial sensation, severe autonomic dysfunction, and atrioventricular nodal block, which required pacemaker implantation [10]. Amyloid fibrils have been shown to directly damage Schwann cells [11], leading to the predominant loss of small-fiber axons commonly seen in early-onset cases, and suggesting that the neuropathy may be impacted by vasculopathy, particularly in late-onset cases [12]. The amyloid fibril type (type A or type B) has been shown to be important for the disease phenotype in transthyretin amyloidosis (ATTR amyloidosis), and type A fibrils are generally associated with a later onset of disease, more pronounced motor neuropathy, and amyloid cardiomyopathy in patients with ATTRv Val30Met amyloidosis [13, 14]. Type A fibrils are also more common in wild-type ATTR amyloidosis [15, 16], which presents with cardiomyopathy [17].
The Transthyretin Amyloidosis Outcomes Survey (THAOS) is an ongoing, global, longitudinal, observational survey of patients with ATTR amyloidosis, including both inherited and wild-type disease, and asymptomatic carriers with TTR mutations. THAOS was established in 2007 to study differences in disease presentation, diagnosis, and natural history in a large, geographically dispersed, heterogeneous patient population [18]. This is an analysis of the characteristics of patients in THAOS with symptomatic ATTRv Val30Met amyloidosis with late-onset (age at least 50 years) compared with early-onset (age less than 50 years) disease.
Methods
Study Design and Patient Characteristics
The design of THAOS (NCT00628745) has been published previously [18]. All THAOS study sites received ethical or institutional review board approval prior to patient enrollment and followed the International Council for Harmonisation Good Pharmacoepidemiology Practice guidelines and the principles of the Declaration of Helsinki. Each patient provided written informed consent.
Characteristics of symptomatic patients in THAOS with ATTRv Val30Met amyloidosis were assessed, with patients with late-onset disease (aged at least 50 years at symptom onset) compared with patients with early-onset disease (aged less than 50 years at symptom onset) (data cutoff date August 1, 2019).
Patient demographics including sex, geographical location, race/ethnicity, family history, medical history, age at enrollment and symptom onset, family history of ATTR amyloidosis, and time from symptom onset to diagnosis are collected at enrollment in THAOS and were examined in this analysis. Neurologic impairment was measured by a derived Neuropathy Impairment Score in the Lower Limbs (NIS-LL; ranges from 0 to 88) and the Neurologic Composite Score (NCS; ranges from 0 to 294). For both measures, higher scores indicate greater impairment [19]. Walking disability was measured by the modified polyneuropathy disability (mPND) score (ranges from I to IV), where I indicates sensory disturbance in lower limbs but preserved walking capacity; II indicates difficulties in walking but no need of a walking stick; IIIa indicates one stick or one crutch required for walking; IIIb indicates two sticks or two crutches required for walking; and IV indicates patient confined to a wheelchair or bed. Cardiac findings were assessed via electrocardiogram (ECG) and echocardiogram (echo). Overall interpretation of ECG as abnormal was based on investigator judgement.
Quality of life was assessed using the EQ-5D-3L questionnaire, a self-administered health status instrument. Respondents rated their current health state on five dimensions (mobility, self-care, usual activities, pain or discomfort, and anxiety or depression), with each dimension having three levels of function (1 = no problem, 2 = some problem, and 3 = extreme problem).
Statistical Analysis
Data are presented as mean (standard deviation [SD]) and percentages unless stated otherwise. p values were calculated using Student’s t test for continuous variables, Fisher’s exact test for dichotomous variables, and chi-squared test for variables with more than two categories. p values < 0.05 were considered statistically significant, with no adjustment for multiplicity.
Results
Patient Characteristics
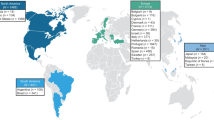
Of the 1389 patients with ATTRv Val30Met amyloidosis in THAOS, 491 (35.3%) had late-onset disease, and 898 (64.7%) early-onset disease (Table 1). Countries in which over 50% of enrolled patients with ATTRv Val30Met amyloidosis were late-onset were the USA with 19 of 21 patients (90.5%), Italy with 20 of 24 patients (83.3%), and Sweden with 135 of 185 patients (73.0%). Late-onset disease constituted less than 30% of enrolled patients with ATTRv Val30Met amyloidosis in Argentina with 5 of 28 patients (17.9%), Portugal with 125 of 675 patients (18.5%), and Brazil with 40 of 137 patients (29.2%) (Fig. 1). Patients with late-onset were more likely to be male, with a mean age at enrollment of 68.1 years compared with 40.4 years for early-onset, and a longer time from symptom onset to diagnosis (p < 0.001 for all; Table 1). Patients with late-onset were also more likely to have been misdiagnosed than those with early-onset, and a lower percentage had a known family history of disease (p < 0.001 for both; Table 1). The most common misdiagnoses in patients with late-onset disease were idiopathic neuropathy (29 of 139 patients with a misdiagnosis [20.9%]), neuropathy (24 patients [17.3%]), lumbar disc disease (16 patients [11.5%]), and chronic inflammatory demyelinating polyneuropathy (13 patients [9.4%]). The most common misdiagnoses in patients with early-onset disease were neuropathy (12 of 90 patients with a misdiagnosis [13.3%]), idiopathic neuropathy (9 patients [10.0%]), and irritable bowel syndrome (8 patients [8.9%]).

Illustration of incidence of late- and early-onset ATTRv Val30Met amyloidosis in different countries in THAOS. Countries with more late-onset disease than early-onset disease are marked in dark blue; countries with more early-onset than late-onset disease are marked in light blue. Countries contributing ≤ 5 patients with Val30Met not shown. Belgium: late-onset n = 1, early-onset n = 0; Bulgaria: late-onset n = 5, early-onset n = 0; Cyprus: late-onset n = 0, early-onset n = 1; Malaysia: late-onset n = 2, early-onset n = 0; the Netherlands: late-onset n = 3, early-onset n = 2; Turkey: late-onset n = 1, early-onset n = 0
Symptom Profile
Medical History
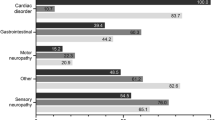
Nearly all patients in both groups had a medical history of neurologic disorders (Table 2). However, there were differences in the incidence of types of neurologic disorder (Table 3). Symptoms of motor neuropathy were more common (62.1% of patients with late-onset vs. 43.3% with early-onset disease; p < 0.001), and symptoms of autonomic neuropathy less common (73.1% late-onset vs. 86.0% early-onset; p < 0.001) in patients with late-onset disease. There was also a greater incidence of a history of cardiovascular disorders, and a lower incidence of genitourinary/renal/reproductive disorders and gastrointestinal disorders in patients with late-onset disease (p < 0.01 for all; Table 2).
Neurologic Impairment
Median (10th, 90th percentile) derived NIS-LL was higher in patients with late- vs. early-onset disease (25.0 [4.0, 69.3] compared with 8.0 [0, 54.8], respectively; p < 0.001; Table 4). Median (10th, 90th percentile) NCS was also higher in patients with late- vs. early-onset disease (42.0 [2.0, 155.0] compared with 21.0 [0, 102.0], respectively; Table 4), as was each component of the NCS (p < 0.001 for all; Table 4). Patients with late-onset disease were also more likely to experience more severe walking disability, as assessed by mPND score (p < 0.001; Table 5).
Cardiac Findings
Cardiac findings were also more prominent in patients with late-onset disease. A greater proportion of late-onset had an overall interpretation of ECG as abnormal (72.1% vs. 44.3% early-onset) and left-ventricular septal thickness of at least 12 mm (69.7% vs. 14.6%). Patients with late-onset disease also had a greater mean left-ventricular septal thickness (p < 0.001 for all; Fig. 2). There were also differences in other ECG and echo measures (Table 6).

Electrocardiographic (ECG) (a) and echocardiographic (b, c) findings at enrollment in patients with late- and early-onset ATTRv Val30Met amyloidosis. LV left ventricular
Quality of Life
There was no difference between mean (SD) EQ-5D-3L scores in patients with late- and early-onset disease when grouped by age at enrollment into 50–64 years (0.7 [0.3] late-onset vs. 0.6 [0.2] early-onset) and at least 65 years (0.6 [0.2] vs. 0.7 [0.2]).
Discussion
This analysis from THAOS broadens the understanding of the characteristics of patients with late-onset ATTRv Val30Met amyloidosis across many countries. The presentation of late-onset disease is relatively common, occurring in around one-third of patients in THAOS overall. Most of the patients in this analysis were from Sweden and Portugal, reflecting the endemic loci of ATTRv Val30Met amyloidosis in those countries. Although early-onset is the most common form of the disease in the majority of countries, late-onset was more common in Sweden, Italy, and the USA. In Spain and Japan, late-onset was as common as early-onset.
Patients with late-onset disease tended to have more severe neurologic and cardiac findings compared with patients with early-onset disease. Previous investigations from Japan and Brazil have also suggested that patients with late-onset ATTRv Val30Met amyloidosis experience a more severe and faster-progressing disease in comparison with early-onset [9, 20, 21]. In Japan and Brazil, patients with late-onset disease experience faster-progressing neuropathy, as measured by imbalance, deep sensory loss, NCS, and shorter times to requirement of a walking aid, as assessed by PND [9, 20, 21]. Results from a cohort of patients in THAOS from Brazil indicated that muscle weakness and balance abnormality were reported at enrollment by more patients with late-onset disease compared with early-onset disease, with similar percentages of patients reporting temperature or pain insensitivity [9]. These results are in line with the present analysis, which benefits from an additional 3 years of data and the inclusion of the global THAOS population. Differences in amyloid composition and deposition have been seen in patients with late-onset vs. those with early-onset disease [22, 23], which may point to an explanation for the distinct patterns of clinical presentation reported in this study and previously [9, 20, 21]. Studies in patients from Sweden and Japan indicate that type B fibrils (full-length, arranged in parallel) are mainly found in early-onset cases, while type A fibrils (a mixture of full-length and fragments of TTR arranged haphazardly, resembling wild-type fibrils) are more commonly associated with late-onset ATTRv Val30Met amyloidosis, non-Val30Met-mutations, and in patients with wild-type ATTR amyloidosis [13, 15, 16, 23, 24]. Type A fibrils have been associated with more pronounced motor neuropathy [13, 14, 23]. Late-onset disease has been associated with a greater effect on larger myelinated fibers than early-onset disease, which is predominantly associated with small-fiber loss [7]. In late-onset disease, type A fibrils may affect the larger motor nerves, resulting in the more severe walking disability observed in the present study and as previously reported [21]. There have also been differences reported in the site of deposition, with greater amyloid deposition in the dorsal root ganglia than in the sympathetic ganglia in late-onset disease, and vice versa in early-onset disease [22]. Another factor potentially contributing to the more severe neuropathy seen in late-onset disease could be the longer delays in diagnosis that were seen more frequently in these patients [9, 25]. As a rare disease, physicians may not recognize the symptoms associated with ATTRv amyloidosis. Accurate diagnosis, particularly of late-onset disease, can be confounded by the perception that ATTRv amyloidosis affects only younger people who have relatives diagnosed with ATTRv amyloidosis [14]. There may also be a relationship between baseline disease severity and natural disease progression, with one clinical trial showing that higher baseline disease severity was associated with a faster rate of neurological progression in the following 12 months [26]. Given its progressive, irreversible, degenerative nature, early diagnosis and treatment of ATTRv amyloidosis has the potential to improve patient outcomes [14, 27].
Reports from Brazil and Sweden indicate that cardiac findings such as ECG abnormalities [9], interventricular hypertrophy [9, 28], and cardiomyopathy [28] occur more frequently in patients with late-onset disease. Late-onset patients also have a shorter survival time [20, 21], with cardiomyopathy observed as a major cause of death [21]. In this analysis, the cardiac findings of rhythm abnormalities, pathologic Q waves, and increased QRS interval and wall thicknesses were significantly more common in patients with late-onset disease. Substantial cardiomyopathy at presentation and increased interventricular septal thickness have been associated with patients with type A fibrils [23]. One study reported that more amyloid was observed in the heart and pituitary gland in patients with late-onset ATTRv Val30Met amyloidosis (compared with early-onset disease), with the main recorded cause of death being heart failure due to cardiac amyloidosis [22]. In one small group of patients with late-onset ATTRv Val30Met amyloidosis over half of the cardiac amyloid deposits were shown to be composed of wild-type TTR [24]. This suggests that it may be a combination of both increased type A fibrils and aging that contributes to the increased cardiac findings seen in patients with late-onset ATTRv Val30Met amyloidosis. Delays in diagnosis could also have contributed to the more severe cardiac findings in patients with late-onset disease. However, cardiac disease is generally less pronounced in patients with early-onset disease, although arrhythmias are quite common in these patients, and increased levels of amyloid deposition in the myocardium is associated with cardiomyopathy in patients with late-onset disease [22].
While a greater proportion of patients with late-onset disease had cardiac findings, the greater age of these patients does mean that they may have been more likely to have developed underlying cardiovascular disease unrelated to ATTRv amyloidosis. Further, data were more limited for many of the cardiac assessments. This may have been a reflection of the different specialties of enrolling centers, with many focusing on neurological assessments and performing fewer routine cardiac assessments. This could have led to a bias in which the likelihood of cardiac abnormalities being detected was reduced. These limitations are offset by the ability to assess large numbers of patients with a rare disease from a global population, providing data on a broad range of patients.
While the reasons for the different age of onset in patients with ATTRv Val30Met amyloidosis are still not fully elucidated, reports have identified several different genetic factors that may influence age of disease onset, including sex [29], C1Q polymorphisms [30, 31], single nucleotide haplotype polymorphisms in the TTR gene [32, 33], mitochondrial haplogroup polymorphisms [34], RBP4 and AR gene variation [35], and mitochondrial DNA copy number [36]. It may be that environmental factors are also involved.
Conclusions
As a result of heterogeneity of disease and lack of awareness, late-onset ATTRv Val30Met amyloidosis may be more difficult to diagnose than early-onset disease. This analysis of patients with late-onset ATTRv Val30Met amyloidosis in THAOS further characterizes this patient group, providing valuable information that further defines the nature and severity of the neurological and cardiac disease findings in these patients. Results from this analysis indicate that patients with late-onset ATTRv Val30Met amyloidosis are more likely to be male, have a history of cardiovascular disease, overall interpretation of ECG as abnormal, and left-ventricular thickness of at least 12 mm as well as greater mean ventricular septal thickness. Neurologically, they are more likely to have higher median derived NIS-LL and NCS, and more severe walking disability. Increased recognition of late-onset ATTRv Val30Met amyloidosis could lead to earlier diagnosis and improve patient outcomes [14, 27].
References
Planté-Bordeneuve V, Said G. Familial amyloid polyneuropathy. Lancet Neurol. 2011;10:1086–97.
Ando Y, Nakamura M, Araki S. Transthyretin-related familial amyloidotic polyneuropathy. Arch Neurol. 2005;62:1057–62.
Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8:31.
Rowczenio D, Wechalekar A. Mutations in hereditary amyloidosis. http://amyloidosismutations.com/mut-attr.php. 2015. Accessed Dec 18, 2020.
Conceição I, Coelho T, Rapezzi C, et al. Assessment of patients with hereditary transthyretin amyloidosis—understanding the impact of management and disease progression. Amyloid. 2019;26:103–11.
Ueda M, Yamashita T, Misumi Y, Masuda T, Ando Y. Origin of sporadic late-onset hereditary ATTR Val30Met amyloidosis in Japan. Amyloid. 2018;25:143–7.
Koike H, Misu K, Sugiura M, et al. Pathology of early- vs. late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63:129–38.
Conceição I, De Carvalho M. Clinical variability in type I familial amyloid polyneuropathy (Val30Met): comparison between late- and early-onset cases in Portugal. Muscle Nerve. 2007;35:116–8.
Pinto MV, Pinto LF, Dias M, et al. Late-onset hereditary ATTR V30M amyloidosis with polyneuropathy: characterization of Brazilian subjects from the THAOS registry. J Neurol Sci. 2019;403:1–6.
Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs. late-onset form. Arch Neurol. 2002;59:1771–6.
Koike H, Nishi R, Ikeda S, et al. The morphology of amyloid fibrils and their impact on tissue damage in hereditary transthyretin amyloidosis: an ultrastructural study. J Neurol Sci. 2018;394:99–106.
Koike H, Ikeda S, Takahashi M, et al. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016;87:2220–9.
Arvidsson S, Pilebro B, Westermark P, Lindqvist P, Suhr OB. Amyloid cardiomyopathy in hereditary transthyretin v30m amyloidosis—impact of sex and amyloid fibril composition. PLoS ONE. 2015;10:e0143456.
Adams D, Suhr OB, Hund E, et al. First European consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29(Suppl 1):S14-26.
Ihse E, Rapezzi C, Merlini G, et al. Amyloid fibrils containing fragmented ATTR may be the standard fibril composition in ATTR amyloidosis. Amyloid. 2013;20:142–50.
Ihse E, Suhr OB, Hellman U, Westermark P. Variation in amount of wild-type transthyretin in different fibril and tissue types in ATTR amyloidosis. J Mol Med (Berl). 2011;89:171–80.
Ruberg FL, Grogan M, Hanna M, Kelly JW, Maurer MS. Transthyretin amyloid cardiomyopathy: JACC state-of-the-art review. J Am Coll Cardiol. 2019;73:2872–91.
Planté-Bordeneuve V, Suhr OB, Maurer MS, White B, Grogan DR, Coelho T. The Transthyretin Amyloidosis Outcomes Survey (THAOS) registry: design and methodology. Curr Med Res Opin. 2013;29:77–84.
Coelho T, Vinik A, Vinik EJ, Tripp T, Packman J, Grogan DR. Clinical measures in transthyretin familial amyloid polyneuropathy. Muscle Nerve. 2017;55:323–32.
Adams D, Coelho T, Obici L, et al. Rapid progression of familial amyloidotic polyneuropathy: a multinational natural history study. Neurology. 2015;85:675–82.
Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83:152–8.
Koike H, Nakamura T, Nishi R, et al. Common clinicopathological features in late-onset hereditary transthyretin amyloidosis (Ala97Gly, Val94Gly and Val30Met). Amyloid. 2019;26:24–5.
Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281:337–47.
Koike H, Ando Y, Ueda M, et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287:178–84.
Falcao de Campos C, Parreira S, Conceição I. Misdiagnosis in late vs. early onset hATTR amyloidosis patients: experience from a reference centre. Amyloid. 2019;26:37–8.
Gundapaneni BK, Sultan MB, Keohane DJ, Schwartz JH. Tafamidis delays neurological progression comparably across Val30Met and non-Val30Met genotypes in transthyretin familial amyloid polyneuropathy. Eur J Neurol. 2018;25:464–8.
Parman Y, Adams D, Obici L, et al. Sixty years of transthyretin familial amyloid polyneuropathy (TTR-FAP) in Europe: where are we now? A European network approach to defining the epidemiology and management patterns for TTR-FAP. Curr Opin Neurol. 2016;29(Suppl 1):S3–13.
Suhr OB, Lindqvist P, Olofsson BO, Waldenstrom A, Backman C. Myocardial hypertrophy and function are related to age at onset in familial amyloidotic polyneuropathy. Amyloid. 2006;13:154–9.
Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2020. https://doi.org/10.1007/s00415-019-09688-0.
Andreou S, Panayiotou E, Michailidou K, et al. Epidemiology of ATTRV30M neuropathy in Cyprus and the modifier effect of complement C1q on the age of disease onset. Amyloid. 2018;25:220–6.
Dias A, Santos D, Coelho T, et al. C1QA and C1QC modify age-at-onset in familial amyloid polyneuropathy patients. Ann Clin Transl Neurol. 2019;6:748–54.
Soares ML, Coelho T, Sousa A, et al. Haplotypes and DNA sequence variation within and surrounding the transthyretin gene: genotype-phenotype correlations in familial amyloid polyneuropathy (V30M) in Portugal and Sweden. Eur J Hum Genet. 2004;12:225–37.
Alves-Ferreira M, Coelho T, Santos D, et al. A trans-acting factor may modify age at onset in familial amyloid polyneuropathy ATTRV30M in Portugal. Mol Neurobiol. 2018;55:3676–83.
Olsson M, Hellman U, Planté-Bordeneuve V, Jonasson J, Lang K, Suhr OB. Mitochondrial haplogroup is associated with the phenotype of familial amyloidosis with polyneuropathy in Swedish and French patients. Clin Genet. 2009;75:163–8.
Santos D, Coelho T, Alves-Ferreira M, et al. Variants in RBP4 and AR genes modulate age at onset in familial amyloid polyneuropathy (FAP ATTRV30M). Eur J Hum Genet. 2016;24:756–60.
Santos D, Santos MJ, Alves-Ferreira M, et al. mtDNA copy number associated with age of onset in familial amyloid polyneuropathy. J Neurol Neurosurg Psychiatry. 2018;89:300–4.
Acknowledgements
We thank all THAOS patients and investigators for their important contributions to this study. We also thank Marcus Vinicius Pinto, Rajiv Mundayat, and Denise Rill for their contributions to earlier versions of this work.
Additional THAOS investigators contributing to this analysis are: Fabio Adrian Barroso: FLENI, Ciudad Autonoma de Buenos Aires, Argentina; Marcelo Rugiero: Hospital Italiano de Buenos Aires, Buenos Aires, Argentina; Johan Van Cleemput: Afdeling Klinische Cardiologie, O&N I, Leuven, Belgium; Ivaylo Tarnev: Alexandrovska University Hospital Clinic of Neurology, Sofia, Bulgaria; Theodoros Kyriakides: Cyprus Institute of Neurology and Genetics, Nicosia, Cyprus; Arnt Kristen: Medical University of Heidelberg, Heidelberg, Germany; Hartmut Schmidt: Universitätsklinikum Münster – Transplant Hepatology, Münster, Germany; Felix Darstein: Johann-Gutenberg-Universität, Mainz, Germany; Burkhard Gess: University Hospital of RWTH Aachen, Aachen, Germany; Josep Maria Campistol Plana: Institut Clinic de Nefrologia i Urologia – ICNU, Hospital Clinic i Provincial de Barcelona, Barcelona, Spain; Juan Gonzalez Moreno: Hospital Son Llatzer, Palma de Mallorca, Spain; Jose Gonzalez Costello: Hospital Universitari de Bellvitge, Barcelona, Spain; Pablo Garcia Pavia: Hospital Universitario Puerta de Hierro, Majadahonda, Spain; Roberto Fernandéz Torrón: Hospital Universitario Donostia, Gipuzkoa – San Sebastian, Spain; Francisco Munoz Beamud: Hospital Juan Ramon Jimenez, Huelva, Spain; Violaine Planté-Bordeneuve, CHU Henri Mondor, Créteil, France; David Adams: CHU de Bicetre, Cedex, France; Olivier Lairez: CHU de Toulouse – Hopital Rangueil, Toulouse Cedex 09, France; Claudio Rapezzi: Comitato Etico Indipendente dell Azienda, Bologna, Italy; Giampaolo Merlini: Centro per lo Studio e la Cura delle Amiloidosi Sistemiche, Pavia, Italy; Marco Luigetti: Fondazione Policlinico Gemelli – Universita Cattolica del Sacro Cuore, Roma, Italy; Yoshiki Sekijima: Shinshu University School of Medicine, Matsumoto, Japan; Taro Yamashita: Kumamoto University, Kumamoto-city, Japan; Sonoko Misawa: Chiba University Hospital, Chiba-shi, Japan; Soon Chai Low: University Malaya Medical Centre, Kuala Lumpur, Malaysia; Hans Nienhuis: University Medical Center Groningen, Groningen, the Netherlands; Teresa Coelho: Unidade Clinica de Paramiloidose-Centro Hospitalar Porto, EPE-Hospital Geral Santo Antonio, Porto, Portugal; Isabel Conceição: Hospital Santa Maria, Lisboa, Portugal; Rayomand Press: Karolinska University Hospital, Huddinge, Stockholm, Sweden; Yesim Parman: Istanbul University, Istanbul Faculty of Medicine, Department of Neurology, Istanbul, Turkey; Mathew Maurer: Columbia University Medical Center, New York, NY, USA; Stephen Gottlieb: University of Maryland, Baltimore, MD, USA; Annabel Wang: University of California, Irvine, Orange, CA, USA; Brian Drachman: University of Pennsylvania – Perelman Center for Advanced Medicine, Philadelphia, PA, USA; Angela Dispenzieri: Mayo Clinic, Rochester, MN, USA; Sasa Zivkovic: University of Pittsburgh Medical Center, Pittsburgh, PA, USA; Daniel Lenihan: Washington University School of Medicine, St. Louis, MO, USA
Funding
The THAOS registry and this analysis were sponsored by Pfizer. Medical writing support was provided by Caitlin Watson, PhD, of Engage Scientific Solutions, and funded by Pfizer, who also provided funding for the journal’s Rapid Service Fee.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Authors’ Contributions
All authors contributed to the design and conduct of the analysis; interpretation of the data; and preparation, review, and approval of the manuscript.
Prior Presentation
A prior version of the analysis in this manuscript was presented at the 2019 Peripheral Nerve Society (PNS) Annual Meeting, June 22–26, 2019, Genoa, Italy.
Disclosures
Márcia Waddington-Cruz received research funding, consulting fees, and travel support for advisory boards and meetings from FoldRx Pharmaceuticals and Pfizer. Jonas Wixner received consulting fees and travel support for lectures and advisory boards from Pfizer, Akcea Therapeutics, and Alnylam Pharmaceuticals. Yukio Ando declares receipt of consulting fee or honorarium, support for travel to meetings, and provision for writing assistance from Pfizer. Leslie Amass, Doug Chapman, and Jan Kiszko are full-time employees of Pfizer and hold stock and/or stock options with Pfizer.
Compliance with Ethics Guidelines
All THAOS study sites received ethical or institutional review board approval prior to patient enrollment and followed the International Council for Harmonisation Good Pharmacoepidemiology Practice guidelines and the principles of the Declaration of Helsinki. Each patient provided written informed consent.
Data Availability
Pfizer provides secure access to anonymized patient-level data to qualified researchers in response to scientifically valid research proposals. Further details can be found at https://www.pfizer.com/science/clinical-trials/trial-data-and-results.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
The members of the THAOS investigators group was processed under acknowledgements section.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Waddington-Cruz, M., Wixner, J., Amass, L. et al. Characteristics of Patients with Late- vs. Early-Onset Val30Met Transthyretin Amyloidosis from the Transthyretin Amyloidosis Outcomes Survey (THAOS). Neurol Ther 10, 753–766 (2021). https://doi.org/10.1007/s40120-021-00258-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-021-00258-z