
Abstract
ATTR amyloidosis is caused by systemic deposition of transthyretin (TTR) and comprises ATTRwt (wt for wild-type) amyloidosis, ATTRv (v for variant) amyloidosis, and acquired ATTR amyloidosis after domino liver transplantation. ATTRwt amyloidosis has classically been regarded as cardiomyopathy found in the elderly, whereas carpal tunnel syndrome has also become a major initial manifestation. The phenotypes of ATTRv amyloidosis are diverse and include neuropathy, cardiomyopathy, and oculoleptomeningeal involvement as the predominant features, depending on the mutation and age of onset. In addition to variant TTR, the deposition of wild-type TTR plays a significant role, even in patients with ATTRv amyloidosis. The formation of amyloid fibrils tends to occur in association with the basement membrane. The thickening or reduplication of the basement membrane surrounding endoneurial microvessels, which is similar to diabetic neuropathy, is observed in ATTRv amyloidosis, suggesting that common mechanisms, such as an accumulation of advanced glycation end products, may participate in the disease process. In addition to direct damage caused by amyloid fibrils, recent studies have suggested that the toxicity of nonfibrillar TTRs, such as TTR oligomers, participates in the process of tissue damage. Although liver transplantation has been performed for patients with ATTRv amyloidosis since 1990, late-onset patients were not eligible for this treatment. However, as the efficacy of orally administered tafamidis and diflunisal, which stabilize TTR tetramers, was suggested in the early 2010s, such late-onset patients have also become targets for disease-modifying therapies. Additionally, recent studies of small interfering RNA (patisiran) and antisense oligonucleotide (inotersen) therapies have demonstrated the efficacy of these gene-silencing agents. A strategy for monitoring patients that enables the choice of an appropriate treatment from comprehensive and long-term viewpoints should be established. As many patients with ATTR amyloidosis are aged and have heart failure, they are at increased risk of aggravation if they are infected by SARS-CoV2. The optimal interval of evaluation should also be considered, particularly in this COVID-19 era.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
ATTR amyloidosis comprises ATTRwt amyloidosis, ATTRv amyloidosis, and acquired ATTR amyloidosis after domino liver transplantation. |
ATTRwt amyloidosis has classically been regarded as cardiomyopathy found in the elderly, whereas carpal tunnel syndrome has also been recognized as the major manifestation of this disease. |
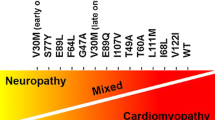
The phenotypes of ATTRv amyloidosis are diverse and include neuropathy, cardiomyopathy, and oculoleptomeningeal involvement as the predominant features, depending on the mutation and age of onset. |
In addition to variant TTR, the deposition of wild-type TTR plays a significant role even in ATTRv amyloidosis, particularly in patients with late-onset Val30Met ATTR amyloidosis. |
The reduplication of the basement membrane surrounding endoneurial microvessels, which is similar to diabetic neuropathy, suggests that common mechanisms, such as an accumulation of advanced glycation end products, may participate in the disease process. |
In addition to direct damage caused by amyloid fibrils, nonfibrillar TTRs, such as TTR oligomers, participate in the process of tissue damage. |
As new disease-modifying therapies, such as TTR stabilizers and gene-silencing agents, have become available, the need for early diagnosis of patients with ATTR amyloidosis is increasing. |
Digital Features
This article is published with digital features to facilitate understanding of the article. To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12841199.
Introduction
Transthyretin (TTR) is a protein mainly produced in the liver and functions as a transporter of thyroxin (T4) and retinol (vitamin A)-binding protein [1]. ATTR amyloidosis is a gain-of-toxic function protein-misfolding disease in which amyloidogenic TTR assembles into amyloid fibrils in extracellular spaces, leading to systemic organ dysfunction [2]. The nomenclature committee of the International Society of Amyloidosis has defined two major forms of ATTR amyloidosis: wild-type ATTR amyloidosis designated ATTRwt (wt for wild-type) amyloidosis and hereditary ATTR amyloidosis named ATTRv (v for variant) amyloidosis [3], although these are traditionally called senile systemic amyloidosis and familial amyloid polyneuropathy, respectively. In addition, another ATTR amyloidosis has been reported in patients who received livers from ATTRv amyloidosis donors [4], which is referred to as acquired ATTR amyloidosis after domino liver transplantation. Owing to the recent increase in awareness and progress in diagnostic techniques for this disease, the number of newly diagnosed patients has increased significantly, leading to an expansion of the clinical spectrum of ATTR amyloidosis [1, 5]. As the recent development of disease-modifying therapies for ATTR amyloidosis such as TTR stabilizers and gene-silencing agents is outstanding, the need for early diagnosis of patients with this disease is increasing. Hence, understanding the clinical spectrum and management based on the pathophysiology of this disease is important.
In this review, we discuss the clinical spectrum, pathogenesis, and disease-modifying therapies of ATTR amyloidosis, particularly focusing on recent developments in these aspects. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Clinical Spectrum
ATTRwt Amyloidosis
ATTRwt amyloidosis has classically been regarded as cardiomyopathy found in elderly individuals, particularly men [6]. Studies of autopsy specimens revealed that a significant proportion of the elderly population has wild-type TTR deposition in the heart, even in subjects without a history of underlying diseases [6, 7]. A recent development in diagnostic techniques revealed that cardiomyopathy resulting from ATTRwt amyloidosis is an important differential diagnosis of heart failure with preserved ejection fraction [8, 9]. ATTRwt amyloidosis may also present features suggestive of carpal tunnel syndrome that frequently precede those of cardiomyopathy [10]. Analyses of tenosynovial tissues obtained at carpal tunnel release surgery revealed that a significant proportion of patients diagnosed with idiopathic carpal tunnel syndrome had wild-type TTR amyloid deposits [11]. It should be noted that both cardiomyopathy and carpal tunnel syndrome due to ATTRwt amyloidosis tend to affect elderly male patients [10]. Other studies have also suggested an association between spinal canal stenosis and wild-type TTR deposition in ligaments [12, 13]. These findings may suggest the tendency of TTR to deposit in organs affected by shear stress [14].
In addition, a recent report described three patients with myopathy resulting from ATTRwt amyloidosis [15]. Myopathy was the predominant feature in these patients and the initial manifestation of ATTRwt amyloidosis in two of these patients. Notably, serum creatine kinase levels were normal or only slightly elevated in these patients. This report further expands the spectrum of ATTRwt amyloidosis and increases the need for physicians’ awareness of this disease at the time of differential diagnosis of myopathy [16].
ATTRv Amyloidosis
A concept regarding the clinical features of ATTRv amyloidosis has been established on the basis of patients with Val30Met (p.Val50Met, according to the Human Genome Variation Society recommendation) mutation from conventional endemic foci of Portugal and Japan [1, 2]. These factors consist of an onset age from the late 20s to early 40s, an almost 1:1 male-to-female ratio, predominant impairment of superficial sensations compared to deep sensations that is alternatively called sensory dissociation, marked autonomic dysfunctions, and cardiac conduction disturbances that require pacemaker implantation [17, 18]. Despite having the same Val30Met mutation, patients from Sweden, where another well-known endemic focus alongside Portugal and Japan is present, exhibit a later age of onset than conventional Portuguese and Japanese patients [19]. In addition, recent reports revealed the presence of late-onset (over 50 years of age) patients with the same Val30Met mutation even in nonendemic areas [17, 20]. The clinical features of these late-onset Val30Met patients from nonendemic areas are distinct from early-onset cases from endemic foci in Portugal and Japan because they tend to manifest loss of all sensory modalities, mild autonomic dysfunctions, and heart failure resulting from massive cardiac amyloid deposition [17, 21]. Extreme male preponderance, which is similar to ATTRwt amyloidosis, also characterizes late-onset Val30Met patients from nonendemic areas [20]. If the patients do not receive the disease-modifying therapies described later, the mean survival time in the early-onset Val30Met patients from endemic foci is 12.6 years [22], whereas it is much shorter (7.3 years) in the late-onset Val30Met patients from nonendemic areas [23].
Although Val30Met is considered the most common mutation among patients predominantly manifesting neuropathy [1, 18, 24], recent developments in diagnostic techniques led to the discovery of many patients with mutations other than Val30Met (i.e., non-Val30Met mutations) in the TTR gene, in which more than 130 mutations have been reported [1, 25]. Cardiomyopathy may be the predominant feature in some of the patients carrying non-Val30Met mutations [18, 26]. For example, the Val122Ile (p.Val142Ile) mutation is considered a common cause of heart disease in African Americans originating from West African countries [27, 28]. Although the main source of TTR that circulates systemically is the liver, it is also synthesized by the choroid plexus and retinal pigment epithelium, which may lead to the occurrence of oculoleptomeningeal amyloidosis [29]. In addition, a recent report revealed that myopathy could also become conspicuous in patients with ATTRv amyloidosis [15]. A patient who developed myopathy 3 years after liver transplantation for the treatment of ATTRv amyloidosis has also been described [15].
Acquired ATTR Amyloidosis After Domino Liver Transplantation
As TTR is produced mainly in the liver, liver transplantation has been established as a treatment for ATTRv amyloidosis [30]. Because the liver removed from patients with ATTRv amyloidosis functions well except for the production of variant TTR, it may be transplanted into a patient with severe liver disease because of the shortage of donor livers [4]. This domino liver transplantation was first performed in 1995 [4]. A total of 1254 domino liver transplantations were performed worldwide from 1995 to 2017 [31]. However, as time passes and the number of patients who have undergone domino liver transplantation increases, patients with symptoms suggesting amyloidosis have been reported [32,33,34]. Analysis of a patient who received an ATTRv amyloidosis liver 8 years before autopsy demonstrated that systemic amyloid deposition occurs even before the appearance of symptoms associated with amyloidosis [35].
The mean duration between domino liver transplantation and the first detection of amyloid deposition and symptom onset in recipients is approximately 8 years [31]. Notably, the clinical features of acquired ATTR amyloidosis after domino liver transplantation are different from those of donors [31]. Although liver transplantation is usually performed to treat early-onset Val30Met ATTR amyloidosis cases from endemic foci, recipients of their livers tend to complain of only sensory deficits but not autonomic symptoms, even though they are not elderly [32,33,34]. These features are similar to those in patients with late-onset Val30Met ATTR amyloidosis from nonendemic areas, rather than conventional early-onset patients from endemic foci with marked autonomic dysfunctions [17, 21]. These findings may support the view that late-onset Val30Met cases prevalent throughout the world are the prototype of ATTRv amyloidosis [36].
Pathogenesis
Misfolding and Aggregation of TTR
TTR is stable in its homotetrameric form, and the dissociation of tetramers into monomers causes misfolding of TTR, resulting in aggregation into amyloid fibrils [1, 37]. Most TTR mutations result in the production of variant TTR that is more unstable compared to wild-type TTR [38], whereas dissociation and subsequent aggregation may also occur even in wild-type TTR [39]. This wild-type TTR deposition plays a significant role in not only ATTRwt amyloidosis but also ATTRv amyloidosis. For example, wild-type TTR consists of more than half of cardiac amyloid deposits from late-onset Val30Met ATTR amyloidosis [40]. In patients with ATTRv amyloidosis who underwent liver transplantation, cardiac amyloidosis may progress even after transplantation as a result of wild-type TTR deposition, particularly in elderly male patients [41, 42].
In addition to the full-length TTR, C-terminal fragments, starting at approximately amino acid position 50, are also found in amyloid fibrils from patients with ATTRwt, late-onset Val30Met ATTR, and most non-Val30Met ATTR amyloidosis [43, 44]. The presence of C-terminal fragments suggests that proteolytic cleavage of TTR also destabilizes native tetrameric structures and accelerates TTR misfolding and subsequent aggregation [45].
Evolution of TTR into Amyloid Fibrils
Despite an accumulation of knowledge obtained from in vitro experiments, studies demonstrating the chronological sequence of how amyloid fibrils are formed from dissociated TTR monomers in vivo are still scarce [46]. Some studies examining materials obtained from animal models or patients with ATTRv amyloidosis indicated that the deposition of nonfibrillar TTR occurs before amyloid fibril formation [21, 46,47,48,49]. On electron microscopy, amorphous electron-dense extracellular materials probably containing nonfibrillar TTR have been demonstrated (Fig. 1) [1, 46]. A study of nerve biopsy samples from patients with ATTRv amyloidosis revealed that such amorphous materials were particularly abundant in the subperineurial space and in the vicinity of endoneurial microvessels [46]. As small dotty structures are often observed among these amorphous materials, nonfibrillar oligomeric intermediates are thought to be formed during the process of amyloid fibril formation [46].

Representative electron microscopic findings of amorphous materials forming amyloid fibrils. A cross section of a sural nerve biopsy specimen from a patient with early-onset Val30Met ATTR from an endemic area. Uranyl acetate and lead citrate stain. Amorphous electron-dense extracellular materials (asterisks) are abundant, particularly at the vicinity of a collection of collagen fibers (circular structures with a diameter of 50–70 nm). Aggregations of small dotty structures (arrows) are often observed among these amorphous materials, suggesting that nonfibrillar oligomeric intermediates are formed during the process of amyloid fibril formation. Mature amyloid fibrils are indicated by arrowheads. Scale bar 0.2 μm
A previous study of cardiac amyloid deposits suggested that TTR aggregation into fibrillar structures tends to occur in association with basement membrane because the expression of basement membrane components, such as collagen IV, laminin, and fibronectin, increased in parallel with the accumulation of amyloid fibrils [50]. Studies of nerve biopsy specimens also revealed that amyloid fibrils were frequently found at or around basement membranes surrounding endoneurial microvessels or Schwann cells (Fig. 2) [46, 51]. In nerve biopsy specimens from patients with ATTRv amyloidosis, thickening or reduplication of basement membranes, particularly those surrounding endoneurial microvessels, is occasionally observed (Fig. 3) [52]. The reduplication of basement membranes surrounding endoneurial microvessels has been considered a unique feature in diabetic neuropathy [53]. In diabetes mellitus, an accumulation of advanced glycation end products (AGEs) leads to the reduplication of the vascular basement membrane via alteration of the properties of collagen [54]. As AGEs have also been demonstrated to be associated with the distribution of amyloid deposits in patients with ATTRv amyloidosis [55], they may play a role in promoting amyloid fibril formation.

Amyloid fibrils formed in association with the basement membrane. A cross section of a sural nerve biopsy specimen from a patient with early-onset Val30Met ATTR from an endemic area. Uranyl acetate and lead citrate stain. Amyloid fibrils are found in association with basement membranes (arrowheads) surrounding an endoneurial microvessel. Aggregations of amyloid fibrils are indicated by arrows. Scale bar 0.5 μm

Reduplication of basement membranes surrounding endoneurial microvessels. A cross section of a sural nerve biopsy specimen from a patient with late-onset Val30Met ATTR from a nonendemic area. Uranyl acetate and lead citrate stain. The reduplication of basement membranes surrounding endoneurial microvessels, which is similar to that in diabetic neuropathy patients, is frequently observed, even where amyloid deposits are not present. Scale bar 2 μm
Tissue Damage Resulting from Amyloid Deposition
The deposition of amyloid in extracellular spaces has been thought to affect various organs in ATTR amyloidosis. The impact of amyloid deposition on neighboring tissues differs depending on the morphology of amyloid fibrils in individual cases [46, 51]. Amyloid fibrils in patients with conventional early-onset Val30Met ATTR amyloidosis from endemic foci tend to evolve into long and thick fibers possessing the textbook characteristics of amyloid deposits with a good affinity to Congo red accompanied by strong birefringence under polarized light (Fig. 4a–c) [40, 51]. By contrast, amyloid fibrils remain shorter and finer in patients with other forms of ATTR amyloidosis (i.e., late-onset Val30Met, non-Val30Met, and wild-type) (Fig. 4d) [40, 43, 46]. Amyloid deposits in these patients tend to show weak affinity to Congo red, resulting in weak birefringence under polarized light (Fig. 4e, f) [40, 43].

Differential characteristics of amyloid deposits between patients with conventional early-onset Val30Met ATTR amyloidosis from endemic foci (a–c) and patients with late-onset Val30Met ATTR amyloidosis from nonendemic areas (d–f). Biopsy specimens of the sural nerve (a, d) and autopsy specimens of the heart (b, c, e, f). Uranyl acetate and lead citrate staining specimens (a, d). Alkaline Congo red staining specimens (b, c, e, f). In early-onset patients from endemic foci, amyloid fibrils tend to be long and thick on electron microscopy (a). On light microscopy, amyloid deposits tend to be highly congophilic (b) and exhibit a strong apple-green birefringence (c) in early-onset patients from endemic foci. Atrophy and degeneration of myocardial cells result in the formation of amyloid rings (arrowhead). In late-onset patients from nonendemic areas, amyloid fibrils are generally short and thin on electron microscopy (d). Circular structures with a diameter of 50–70 nm are collagen fibers. On light microscopy, amyloid deposits are generally weakly congophilic (e) and exhibit a faint apple-green birefringence (f) in late-onset patients from nonendemic areas. Scale bars 0.2 μm (a, d) and 10 μm (b, d, e, f)
The influence of long and thick (i.e., large) amyloid fibrils in patients with early-onset Val30Met ATTR amyloidosis from endemic foci on surrounding tissues seems to be more conspicuous than that of short and fine (i.e., small) amyloid fibrils in other patients with ATTR amyloidosis [46, 51]. For example, large fibrils in the nerve seemed to pull neighboring tissues during their maturation, resulting in distortion and atrophy of Schwann cells, particularly those associated with small-diameter nerve fibers (Fig. 5a, b) [46, 51]. By contrast, the influence of small fibrils on neighboring tissues seemed to be less than that of large fibrils (Fig. 5c) [46, 51]. In the heart, a direct invasion of large fibrils induces atrophy and degeneration of myocardial cells, inducing cardiac conduction abnormalities in patients with early-onset Val30Met ATTR amyloidosis from endemic foci (Fig. 4b) [21], whereas an influence of short fibrils in the other types of ATTR amyloidosis on myocardial cells is less conspicuous even though massive amyloid deposition induces diastolic dysfunction (Fig. 4e) [21].

Differential impact on neighboring tissues according the size of amyloid fibrils. Cross sections of sural nerve biopsy specimens from a patient with early-onset Val30Met ATTR with long and thick (i.e., large) amyloid fibrils (a, b) and a patient with non-Val30Met ATTR amyloidosis with short and thin (i.e., small) amyloid fibrils (c). Uranyl acetate and lead citrate stain. Large amyloid fibrils seemed to pull surrounding tissues during their maturation, resulting in distortion and atrophy of Schwann cells, particularly those associated with small-diameter nerve fibers, such as unmyelinated fibers indicated by an arrow (a). A high-powered view of the box in a is shown in b. Large myelinated fibers indicated by an asterisk are relatively preserved even though they are apposed to large amyloid fibrils. The influence of small amyloid fibrils on neighboring tissues seems to be less conspicuous than that of large amyloid fibrils (c). An asterisk indicates an axon of an unmyelinated fiber. Scale bars 2 μm (a) and 0.5 μm (b, c)
Possible Impact of Nonfibrillar TTR
Although the concept that amyloid fibrils themselves induce damage to surrounding tissues is widely accepted, recent studies suggest that nonfibrillar TTR also affects tissues in patients with ATTR amyloidosis [2]. In particular, the toxicity of TTR oligomers formed during the process of amyloid fibril formation has been suggested [48, 56]. To support this view, studies using schwannoma or neuroblastoma cell lines demonstrated the toxic effects of nonfibrillar TTR [47, 57,58,59]. Animal models using Caenorhabditis elegans or Drosophila also demonstrated the neurotoxicity of TTR despite the absence of fibrillar amyloid deposition [59, 60]. As the amount of endoneurial amyloid deposition is smaller relative to the extent of nerve fiber loss in patients with late-onset Val30Met ATTR amyloidosis compared to patients with early-onset Val30Met ATTR amyloidosis [21], the toxicity of nonfibrillar TTR may participate in the process of neurodegeneration in some of the patients with ATTR amyloidosis, such as those with late-onset Val30Met ATTR amyloidosis.
Nonfibrillar TTR also seems to play an important role in enhancing the leakage of circulating TTR into extracellular spaces by affecting endothelial cells of blood vessels, which are always exposed to TTR in the bloodstream [51]. In vitro studies suggested that both wild-type TTR and variant TTR regulate endothelial cell functions [61, 62]. Studies of microangiopathy associated with diabetes mellitus have also provided evidence regarding the regulation of endothelial cell functions by TTR [63, 64]. A study of sural nerve biopsy specimens from patients with ATTRv amyloidosis demonstrated that abnormalities of endoneurial microvessels, including the disruption of blood–nerve barriers, occurred before the initiation of amyloid deposition [51]. Magnetic resonance neurography demonstrated swelling of the nerve trunk in patients with ATTRv amyloidosis, even asymptomatic carriers, indicating the presence of edema resulting from blood–nerve barrier disruption [65]. Cardiac magnetic resonance imaging also revealed gadolinium enhancement in patients with ATTRv amyloidosis [66]. These findings indicate that the leakage of serum components, including TTR produced by the liver, into extracellular spaces of various organs occurs as a result of endothelial cell dysfunction before the initiation of amyloid fibril formation.
Disease-Modifying Therapies
Liver Transplantation
Currently available therapeutic approaches for ATTR amyloidosis consist of a reduction in wild-type/variant TTR and stabilization of circulating TTR to prevent dissociation of tetramers into monomers [1, 2]. As the main source of circulating TTR is the liver, liver transplantation has been performed since 1990 in patients with ATTRv amyloidosis [30]. Long-term efficacy from the viewpoint of survival has been proven, particularly in conventional early-onset Val30Met patients from endemic foci [67]. A retrospective analysis of the data obtained from the Familial Amyloidotic Polyneuropathy World Transplant Registry suggested an early age of onset, short disease duration, and Val30Met mutation to be better predictors for survival [68]. However, the progression of cardiomyopathy and neuropathy resulting from wild-type TTR deposition may occur even after liver transplantation, particularly in late-onset male patients, resulting in poor prognosis after liver transplantation in these patients [41, 42, 69, 70].
TTR Stabilizers
As the dissociation of TTR tetramers into monomers is the crucial step for the subsequent process of protein misfolding and amyloid fibril formation [1, 2], an approach to stabilize the native quaternary structure of TTR tetramers using small molecules that bind to thyroxin-binding sites has been proposed as a potential approach for the treatment of not only ATTRv amyloidosis but also ATTRwt amyloidosis [71, 72]. In the early 2010s, randomized controlled trials suggested the efficacy of two orally administered TTR stabilizers (i.e., tafamidis and diflunisal) for ameliorating the progression of neuropathy in patients with ATTRv amyloidosis [73, 74]. As these TTR-stabilizing drugs can be administered orally, patients with late-onset ATTRv amyloidosis who were not eligible for liver transplantation also became targets for disease-modifying treatment. Another recent randomized controlled trial suggested the efficacy of tafamidis even for cardiomyopathy resulting from both ATTRv and ATTRwt amyloidosis [75].
Tafamidis is an analogue of thyroxine designed to stabilize TTR tetramers [25]. A phase III clinical trial involving 128 patients with early-stage Val30Met ATTR amyloidosis who were randomly assigned in a 1:1 ratio to receive tafamidis (tafamidis meglumine) 20 mg once daily or placebo for 18 months suggested that tafamidis delayed the progression of neuropathy, although the primary endpoint could not be achieved [73]. An open-label extension study for up to 6 years also demonstrated the slowing of neuropathy progression without any unexpected adverse events [76]. In particular, patients who continued to receive tafamidis had less progression of neuropathy than those who switched to tafamidis following 18 months of placebo, warranting the need for early intervention [76]. In addition to its efficacy on neuropathy, the efficacy of tafamidis on cardiomyopathy due to not only ATTRv amyloidosis but also ATTRwt amyloidosis was suggested by a recent phase III clinical trial involving 441 patients with ATTR amyloidosis [75]. This study included 335 patients with ATTRwt amyloidosis and 106 patients with ATTRv amyloidosis who were randomly assigned in a 2:1:2 ratio to receive 80 mg of tafamidis, 20 mg of tafamidis, or placebo for 30 months and demonstrated reduced mortality and cardiovascular-related hospitalizations.
Diflunisal is a nonsteroidal anti-inflammatory drug that also acts to stabilize TTR [72]. A study involving 130 patients with ATTRv amyloidosis who were randomly assigned in a 1:1 ratio to receive 250 mg of diflunisal twice daily or placebo for 2 years suggested that diflunisal can slow the progression of neuropathy, although 67 patients (27 diflunisal patients and 40 placebo patients) discontinued the treatment before completing the 2-year protocol [74]. The demographics of patients in this study were different from those in the phase III trial of tafamidis for neuropathy in patients with ATTRv amyloidosis [73] because it included patients with relatively late disease onset, various disease severities, and non-Val30Met mutation.
Gene-Silencing Drugs
Theoretically, preventing the production of TTR efficiently ameliorates systemic organ dysfunction in ATTR amyloidosis because it not only prevents amyloid fibril formation but also suppresses an increase in toxic amyloid precursors, such as TTR oligomers [2]. As described earlier, TTR seems to exert harmful effects even when fibrillar structures recognized as amyloid fibrils are not formed. Given that circulating variant TTR may induce microangiopathy, which plays a role as an initial lesion of organ damage [51], a strategy that eliminates circulating TTR is more reasonable than liver transplantation and TTR stabilizers [2]. This strategy became a reality with the development of gene-silencing therapeutics, including small interfering RNA (siRNA) and antisense oligonucleotide (ASO) [1]. In 2018, two randomized controlled trials of such gene-silencing agents (patisiran and inotersen) demonstrated an efficacy on neuropathy in patients with ATTRv amyloidosis [77, 78].
Patisiran is an RNA interference therapeutic comprising siRNA formulated in a lipid nanoparticle that is predominantly delivered to the liver and reduces TTR production [1]. In a phase III trial, 225 patients with ATTRv amyloidosis with polyneuropathy were randomly assigned in a 2:1 ratio to receive patisiran intravenously (0.3 mg/kg of body weight) or placebo once every 3 weeks [77]. The results of this study were excellent because all endpoints, including the scores related to somatic and autonomic neuropathies, quality of life score, and exploratory cardiac measures, were better in patients who received patisiran than in those who received placebo. In particular, even an improvement of primary outcome measures represented by neuropathy impairment and quality of life scores was seen in more than half of the patients receiving patisiran.
Inotersen is a second-generation ASO designed to reduce the production of TTR [79]. Parenterally administered ASO, in general, is rapidly transferred into various organs, with the highest concentration in the liver and kidneys [80]. A phase III trial involving 172 patients with ATTRv amyloidosis who were randomly assigned in a 2:1 ratio to receive weekly subcutaneous injections of inotersen (300 mg) or placebo for 15 months demonstrated significantly better primary endpoints represented by neuropathy impairment and quality of life scores [78]. Because glomerulonephritis and thrombocytopenia were reported as severe adverse events, close monitoring of renal function and platelet count is required in patients receiving inotersen.
Summary
ATTR amyloidosis comprises ATTRwt amyloidosis, ATTRv amyloidosis, and acquired ATTR amyloidosis after domino liver transplantation. ATTRwt amyloidosis has classically been regarded as cardiomyopathy found in elderly individuals, whereas carpal tunnel syndrome has also been recognized as a major manifestation of this disease [11]. In addition, a recent report suggested that myopathy has become a predominant feature in ATTRwt amyloidosis [15]. For ATTRv amyloidosis, the clinical phenotypes are diverse and include neuropathy, cardiomyopathy, and oculoleptomeningeal involvement as the predominant features depending on the mutation and age of onset [1, 2]. Patients from conventional endemic foci in Portugal and Japan have the Val30Met mutation and are characterized by an early age of onset, predominant loss of superficial sensation, marked autonomic dysfunctions, and cardiac conduction disturbances that require pacemaker implantation [17, 21]. Features in patients with Val30Met ATTR amyloidosis from nonendemic areas are distinct in terms of late age of onset, loss of all sensory modalities, mild autonomic dysfunctions, and heart failure resulting from massive cardiac amyloid deposition [17, 21]. Clinical features in patients with acquired ATTR amyloidosis after domino liver transplantation are similar to patients with late-onset Val30Met ATTR amyloidosis from nonendemic areas [31].
In addition to variant TTR, the deposition of wild-type TTR plays a significant role even in ATTRv amyloidosis, particularly in patients with late-onset Val30Met ATTR amyloidosis [40]. In addition to full-length TTR, C-terminal fragments are found in amyloid fibrils from patients with ATTRwt, late-onset Val30Met ATTR, and most non-Val30Met ATTR amyloidosis, suggesting the importance of proteolytic cleavage of TTR during the process of amyloid fibril formation in these types of ATTR amyloidosis [43, 44]. The formation of amyloid fibrils tends to occur in association with the basement membrane. Moreover, the reduplication of the basement membrane surrounding endoneurial microvessels, which is similar to diabetic neuropathy, is occasionally observed in patients with ATTRv amyloidosis [52], suggesting that common mechanisms, such as an increased production of AGEs, may participate in the disease process. Amyloid fibrils are usually large in patients with early-onset Val30Met ATTR amyloidosis from endemic foci, whereas they tend to be small in patients with other ATTR amyloidosis [46, 51]. The influence of large amyloid fibrils on surrounding tissues seems to be more conspicuous than that of small amyloid fibrils [46]. As the amount of endoneurial amyloid deposition is smaller relative to the extent of nerve fiber loss in patients with late-onset Val30Met ATTR amyloidosis compared to patients with early-onset Val30Met ATTR amyloidosis [21], the toxicity of nonfibrillar TTR, such as TTR oligomers, may participate in the process of neurodegeneration in these patients.
Currently available therapeutic approaches for ATTR amyloidosis consist of a reduction in wild-type/variant TTR and stabilization of circulating TTR to prevent the dissociation of tetramers into monomers [1, 2]. Although liver transplantation has been performed since 1990 in patients with ATTRv amyloidosis [30], late-onset patients were not eligible for this treatment. As the efficacy of orally administered TTR stabilizers was suggested in the early 2010s [73, 74], such late-onset patients also became targets for disease-modifying therapies. Additionally, recent phase III trials of intravenous administration of siRNA and subcutaneous administration of ASO have also demonstrated the efficacy of these gene-silencing agents [77, 78]. A strategy for monitoring patients that enables the choice of an appropriate treatment from comprehensive and long-term viewpoints should be established. As many patients with ATTR amyloidosis are aged and have heart failure, they are at increased risk of aggravation if they are infected by SARS-CoV2. The optimal interval of evaluation should be considered, particularly in this COVID-19 era.
References
Adams D, Koike H, Slama M, Coelho T. Hereditary transthyretin amyloidosis: a model of medical progress for a fatal disease. Nat Rev Neurol. 2019;15:387–404.
Koike H, Katsuno M. Ultrastructure in transthyretin amyloidosis: from pathophysiology to therapeutic insights. Biomedicines. 2019;7:11.
Benson MD, Buxbaum JN, Eisenberg DS, et al. Amyloid nomenclature 2018: recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid. 2018;25:215–9.
Stangou AJ, Heaton ND, Rela M, Pepys MB, Hawkins PN, Williams R. Domino hepatic transplantation using the liver from a patient with familial amyloid polyneuropathy. Transplantation. 1998;65:1496–8.
Koike H, Hashimoto R, Tomita M, et al. Diagnosis of sporadic transthyretin Val30Met familial amyloid polyneuropathy: a practical analysis. Amyloid. 2011;18:53–62.
Cornwell GG 3rd, Murdoch WL, Kyle RA, Westermark P, Pitkänen P. Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med. 1983;75:618–23.
Ueda M, Horibata Y, Shono M, et al. Clinicopathological features of senile systemic amyloidosis: an ante- and post-mortem study. Mod Pathol. 2011;24:1533–44.
González-López E, Gallego-Delgado M, Guzzo-Merello G, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36:2585–94.
Griffin JM, Maurer MS. Transthyretin cardiac amyloidosis: a treatable form of heart failure with a preserved ejection fraction. Trends Cardiovasc Med. https://doi.org/10.1016/j.tcm.2019.12.003.
Sekijima Y, Yazaki M, Ueda M, Koike H, Yamada M, Ando Y. First nationwide survey on systemic wild-type ATTR amyloidosis in Japan. Amyloid. 2018;25:8–10.
Sekijima Y, Uchiyama S, Tojo K, et al. High prevalence of wild-type transthyretin deposition in patients with idiopathic carpal tunnel syndrome: a common cause of carpal tunnel syndrome in the elderly. Hum Pathol. 2011;42:1785–91.
Westermark P, Westermark GT, Suhr OB, Berg S. Transthyretin-derived amyloidosis: probably a common cause of lumbar spinal stenosis. Ups J Med Sci. 2014;119:223–8.
Yanagisawa A, Ueda M, Sueyoshi T, et al. Amyloid deposits derived from transthyretin in the ligamentum flavum as related to lumbar spinal canal stenosis. Mod Pathol. 2015;28:201–7.
Marcoux J, Mangione PP, Porcari R, et al. A novel mechano-enzymatic cleavage mechanism underlies transthyretin amyloidogenesis. EMBO Mol Med. 2015;7:1337–49.
Pinto MV, Milone M, Mauermann ML, et al. Transthyretin amyloidosis: putting myopathy on the map. Muscle Nerve. 2020;61:95–100.
Koike H, Katsuno M. Expanding the spectrum of transthyretin amyloidosis. Muscle Nerve. 2020;61:3–4.
Koike H, Misu K, Ikeda S, et al. Type I (transthyretin Met30) familial amyloid polyneuropathy in Japan: early- vs late-onset form. Arch Neurol. 2002;59:1771–6.
Sekijima Y, Ueda M, Koike H, Misawa S, Ishii T, Ando Y. Diagnosis and management of transthyretin familial amyloid polyneuropathy in Japan: red-flag symptom clusters and treatment algorithm. Orphanet J Rare Dis. 2018;13:6.
Andersson R. Familial amyloidosis with polyneuropathy. A clinical study based on patients living in Northern Sweden. Acta Med Scand Suppl. 1976;590:1–64.
Koike H, Kawagashira Y, Iijima M, et al. Electrophysiological features of late-onset transthyretin Met30 familial amyloid polyneuropathy unrelated to endemic foci. J Neurol. 2008;255:1526–33.
Koike H, Misu K, Sugiura M, et al. Pathology of early- vs late-onset TTR Met30 familial amyloid polyneuropathy. Neurology. 2004;63:129–38.
Coelho T, Inês M, Conceição I, Soares M, de Carvalho M, Costa J. Natural history and survival in stage 1 Val30Met transthyretin familial amyloid polyneuropathy. Neurology. 2018;91:e1999–2009.
Koike H, Tanaka F, Hashimoto R, et al. Natural history of transthyretin Val30Met familial amyloid polyneuropathy: analysis of late-onset cases from non-endemic areas. J Neurol Neurosurg Psychiatry. 2012;83:152–8.
Koike H, Fukami Y, Nishi R, et al. Clinicopathological spectrum and recent advances in the treatment of hereditary transthyretin amyloidosis. Neurol Clin Neurosci. 2019;7:166–73.
Obici L, Adams D. Acquired and inherited amyloidosis: knowledge driving patients' care. J Peripher Nerv Syst. 2020;25:85–101.
Koike H, Nakamura T, Nishi R, et al. Widespread cardiac and vasomotor autonomic dysfunction in non-Val30Met hereditary transthyretin amyloidosis. Intern Med. 2018;57:3365–70.
Buxbaum JN, Ruberg FL. Transthyretin V122I (pV142I)* cardiac amyloidosis: an age-dependent autosomal dominant cardiomyopathy too common to be overlooked as a cause of significant heart disease in elderly African Americans. Genet Med. 2017;19:733–42.
Damrauer SM, Chaudhary K, Cho JH, et al. Association of the V122I hereditary transthyretin amyloidosis genetic variant with heart failure among individuals of African or Hispanic/Latino ancestry. JAMA. 2019;322:2191–202.
Uemichi T, Uitti RJ, Koeppen AH, Donat JR, Benson MD. Oculoleptomeningeal amyloidosis associated with a new transthyretin variant Ser64. Arch Neurol. 1999;56:1152–5.
Holmgren G, Steen L, Ekstedt J, et al. Biochemical effect of liver transplantation in two Swedish patients with familial amyloidotic polyneuropathy (FAP-met30). Clin Genet. 1991;40:242–6.
Misumi Y, Ueda M, Masuda T, et al. Characteristics of acquired transthyretin amyloidosis: a case series and review of the literature. Neurology. 2019;93:e1587–e15961596.
Stangou AJ, Heaton ND, Hawkins PN. Transmission of systemic transthyretin amyloidosis by means of domino liver transplantation. N Engl J Med. 2005;352:2356.
Goto T, Yamashita T, Ueda M, et al. Iatrogenic amyloid neuropathy in a Japanese patient after sequential liver transplantation. Am J Transplant. 2006;6:2512–5.
Barreiros AP, Geber C, Birklein F, Galle PR, Otto G. Clinical symptomatic de novo systemic transthyretin amyloidosis 9 years after domino liver transplantation. Liver Transpl. 2010;16:109.
Koike H, Kiuchi T, Iijima M, et al. Systemic but asymptomatic transthyretin amyloidosis 8 years after domino liver transplantation. J Neurol Neurosurg Psychiatry. 2011;82:1287–90.
Koike H, Sobue G. What is the prototype of familial amyloid polyneuropathy? J Neurol Neurosurg Psychiatry. 2014;85:713.
Kelly JW. Amyloid fibril formation and protein misassembly: a structural quest for insights into amyloid and prion diseases. Structure. 1997;5:595–600.
Sekijima Y, Wiseman RL, Matteson J, et al. The biological and chemical basis for tissue-selective amyloid disease. Cell. 2005;121:73–85.
Westermark P, Sletten K, Johansson B, Cornwell GG 3rd. Fibril in senile systemic amyloidosis is derived from normal transthyretin. Proc Natl Acad Sci USA. 1990;87:2843–5.
Koike H, Ando Y, Ueda M, et al. Distinct characteristics of amyloid deposits in early- and late-onset transthyretin Val30Met familial amyloid polyneuropathy. J Neurol Sci. 2009;287:178–84.
Yazaki M, Mitsuhashi S, Tokuda T, et al. Progressive wild-type transthyretin deposition after liver transplantation preferentially occurs onto myocardium in FAP patients. Am J Transplant. 2007;7:235–42.
Okamoto S, Wixner J, Obayashi K, et al. Liver transplantation for familial amyloidotic polyneuropathy: impact on Swedish patients' survival. Liver Transpl. 2009;15:1229–355.
Bergström J, Gustavsson A, Hellman U, et al. Amyloid deposits in transthyretin-derived amyloidosis: cleaved transthyretin is associated with distinct amyloid morphology. J Pathol. 2005;206:224–32.
Suhr OB, Lundgren E, Westermark P. One mutation, two distinct disease variants: unravelling the impact of transthyretin amyloid fibril composition. J Intern Med. 2017;281:337–47.
Dasari AKR, Arreola J, Michael B, Griffin RG, Kelly JW, Lim KH. Disruption of the CD loop by enzymatic cleavage promotes the formation of toxic transthyretin oligomers through a common transthyretin misfolding pathway. Biochemistry. 2020;59:2319–27.
Koike H, Nishi R, Ikeda S, et al. The morphology of amyloid fibrils and their impact on tissue damage in hereditary transthyretin amyloidosis: an ultrastructural study. J Neurol Sci. 2018;394:99–106.
Sousa MM, Cardoso I, Fernandes R, Guimarães A, Saraiva MJ. Deposition of transthyretin in early stages of familial amyloidotic polyneuropathy: evidence for toxicity of nonfibrillar aggregates. Am J Pathol. 2001;159:1993–2000.
Sousa MM, Fernandes R, Palha JA, Taboada A, Vieira P, Saraiva MJ. Evidence for early cytotoxic aggregates in transgenic mice for human transthyretin Leu55Pro. Am J Pathol. 2002;161:1935–48.
Ueda M, Ando Y, Hakamata Y, et al. A transgenic rat with the human ATTR V30M: a novel tool for analyses of ATTR metabolisms. Biochem Biophys Res Commun. 2007;352:299–304.
Misumi Y, Ando Y, Ueda M, et al. Chain reaction of amyloid fibril formation with induction of basement membrane in familial amyloidotic polyneuropathy. J Pathol. 2009;219:481–90.
Koike H, Ikeda S, Takahashi M, et al. Schwann cell and endothelial cell damage in transthyretin familial amyloid polyneuropathy. Neurology. 2016;87:2220–9.
Koike H. Pathology of familial amyloid polyneuropathy. Jiritushinkei. 2017;54:295–305.
Giannini C, Dyck PJ. Basement membrane reduplication and pericyte degeneration precede development of diabetic polyneuropathy and are associated with its severity. Ann Neurol. 1995;37:498–504.
Madonna R, Balistreri CR, Geng YJ, De Caterina R. Diabetic microangiopathy: pathogenetic insights and novel therapeutic approaches. Vasc Pharmacol. 2017;90:1–7.
Nyhlin N, Ando Y, Nagai R, et al. Advanced glycation end product in familial amyloidotic polyneuropathy (FAP). J Intern Med. 2000;247:485–92.
Dasari AKR, Hughes RM, Wi S, et al. Transthyretin aggregation pathway toward the formation of distinct cytotoxic oligomers. Sci Rep. 2019;9:33.
Monteiro FA, Sousa MM, Cardoso I, do Amaral JB, Guimarães A, Saraiva MJ. Activation of ERK1/2 MAP kinases in familial amyloidotic polyneuropathy. J Neurochem. 2006;97:151–61.
Fong VH, Vieira A. Pro-oxidative effects of aggregated transthyretin in human Schwannoma cells. Neurotoxicology. 2013;39:109–13.
Ibrahim RB, Yeh SY, Lin KP, et al. Cellular secretion and cytotoxicity of transthyretin mutant proteins underlie late-onset amyloidosis and neurodegeneration. Cell Mol Life Sci. 2020;77:1421–34.
Madhivanan K, Greiner ER, Alves-Ferreira M, et al. Cellular clearance of circulating transthyretin decreases cell-nonautonomous proteotoxicity in Caenorhabditis elegans. Proc Natl Acad Sci USA. 2018;115:E7710–E7719719.
Nunes RJ, de Oliveira P, Lages A, et al. Transthyretin proteins regulate angiogenesis by conferring different molecular identities to endothelial cells. J Biol Chem. 2013;288:31752–60.
Lee CC, Ding X, Zhao T, et al. Transthyretin stimulates tumor growth through regulation of tumor immune, and endothelial cells. J Immunol. 2019;202:991–1002.
Shao J, Yin Y, Yin X, et al. Transthyretin exerts pro-apoptotic effects in human retinal microvascular endothelial cells through a grp78-dependent pathway in diabetic retinopathy. Cell Physiol Biochem. 2017;43:788–800.
Fan G, Gu Y, Zhang J, et al. Transthyretin upregulates long non-coding RNA MEG3 by affecting PABPC1 in diabetic retinopathy. Int J Mol Sci. 2019;20:6313.
Kollmer J, Sahm F, Hegenbart U, et al. Sural nerve injury in familial amyloid polyneuropathy: MR neurography vs clinicopathologic tools. Neurology. 2017;89:475–84.
Martinez-Naharro A, Treibel TA, Abdel-Gadir A, et al. Magnetic resonance in transthyretin cardiac amyloidosis. J Am Coll Cardiol. 2017;70:466–77.
Yamashita T, Ando Y, Okamoto S, et al. Long-term survival after liver transplantation in patients with familial amyloid polyneuropathy. Neurology. 2012;78:637–43.
Ericzon BG, Wilczek HE, Larsson M, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99:1847–54.
Liepnieks JJ, Zhang LQ, Benson MD. Progression of transthyretin amyloid neuropathy after liver transplantation. Neurology. 2010;75:324–7.
Koike H, Hashimoto R, Tomita M, et al. Impact of aging on the progression of neuropathy after liver transplantation in transthyretin Val30Met amyloidosis. Muscle Nerve. 2012;46:964–70.
Peterson SA, Klabunde T, Lashuel HA, Purkey H, Sacchettini JC, Kelly JW. Inhibiting transthyretin conformational changes that lead to amyloid fibril formation. Proc Natl Acad Sci USA. 1998;95:12956–60.
Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Investig. 2004;84:545–52.
Coelho T, Maia LF, da Silva AM, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79:785–92.
Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310:2658–67.
Maurer MS, Schwartz JH, Gundapaneni B, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379:1007–166.
Barroso FA, Judge DP, Ebede B, et al. Long-term safety and efficacy of tafamidis for the treatment of hereditary transthyretin amyloid polyneuropathy: results up to 6 years. Amyloid. 2017;24:194–204.
Adams D, Gonzalez-Duarte A, O'Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:11–21.
Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379:22–31.
Benson MD, Kluve-Beckerman B, Zeldenrust SR, et al. Targeted suppression of an amyloidogenic transthyretin with antisense oligonucleotides. Muscle Nerve. 2006;33:609–18.
Geary RS, Norris D, Yu R, Bennett CF. Pharmacokinetics, biodistribution and cell uptake of antisense oligonucleotides. Adv Drug Deliv Rev. 2015;87:46–51.
Acknowledgements
Funding
This work was supported by grants from the Ministry of Health, Labor and Welfare (Research on rare and intractable diseases, H29-022) and the Ministry of Education, Culture, Sports, Science and Technology (17K09777) of Japan. No funding or sponsorship was received for the publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Haruki Koike and Masahisa Katsuno report consulting fees and travel fees from Pfizer and Alnylam.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Digital Features
To view digital features for this article go to https://doi.org/10.6084/m9.figshare.12841199.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Koike, H., Katsuno, M. Transthyretin Amyloidosis: Update on the Clinical Spectrum, Pathogenesis, and Disease-Modifying Therapies. Neurol Ther 9, 317–333 (2020). https://doi.org/10.1007/s40120-020-00210-7
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-020-00210-7