
Abstract
Multiple sclerosis is an inflammatory neurodegenerative disease of the central nervous system (CNS) and the most frequent cause of non-traumatic disability in adults in the Western world. Currently, several drugs have been approved for the treatment of multiple sclerosis. While the newer drugs are more effective, they have less favourable safety profiles. Thus, there is a need to identify new targets for effective and safe therapies, particularly in patients with progressive disease for whom no treatments are available. One such target is granulocyte-macrophage colony-stimulating factor (GM-CSF) or its receptor. In this article we review data on the potential role of GM-CSF and GM-CSF inhibition in MS. We discuss the expression and function of GM-CSF and its receptor in the CNS, as well as data from animal studies and clinical trials in MS.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is a haematopoietic growth factor and proinflammatory cytokine with pleiotropic functions. The aim of this review is to highlight the role of GM-CSF in multiple sclerosis (MS) and the reasons it may be a therapeutic target. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Expression and Function of GM-CSF and Its Receptor in the Central Nervous System
Granulocyte-macrophage colony-stimulating factor is a glycosylated protein with a molecular weight of 23 kDa [1]. It is produced by many cell types (Table 1), including both CD4+ and CD8+ T cells, and its production is regulated differently in each cell type. For example, naive CD4+ cells do not secrete GM-CSF, while naive CD8+ cells do [2]. Researchers have recently identified a new T helper (Th) cell subset that produces GM-CSF and which is distinct from the other Th cells identified to date, such as Th1 and Th17 [3, 4]. GM-CSF is able to cross the blood–brain barrier (BBB), blood–spinal cord barrier and blood–testis barrier in mice. This occurs via a selective and saturable transport system in addition to simple diffusion and leakage [5, 6]. GM-CSF has been detected in the normal central nervous system (CNS) of humans and mice and is produced mainly by astrocytes. It may have a role in the regulation of microglial functions [7] and in the stimulation of microglial priming for antigen presentation [8].
The GM-CSF receptor (GMR) is expressed by many cell types, including monocytes, macrophages, antigen presenting cells (APCs), neurons, astrocytes, and oligodendrocytes, which suggests that GM-CSF is involved in the physiological regulation of these cells [9]. Some studies have shown that for GMR to be activated, it must form a dodecamer structure through head-to-head assembling of two ligand–receptor hexamers; this induces intracellular signalling [10, 11]. GM-CSF concentration controls which signalling pathways are activated. Low GM-CSF concentrations lead to a selective survival-only signalling pathway, while higher GM-CSF concentrations result in dodecamer assembly and activation of survival and proliferation signalling pathways. Therefore, the GMR is considered to be a therapeutic target in the treatment of autoimmune diseases [12, 13].
GM-CSF expression is also regulated by a number of cytokines, as shown in Table 2.
GM-CSF in MS
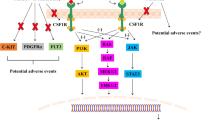
To date, the exact pathogenic action of GM-CSF in MS is incompletely elucidated. High GM-CSF concentrations have been found in the cerebrospinal fluid (CSF) of active-phase MS patients, suggesting that GM-CSF may play a role in MS pathogenesis [56]. The possible mechanisms by which GM-CSF may exert pathogenic effects in MS are shown in Fig. 1.

Diagram showing the potential mechanisms of GM-CSF involvement in MS. APC antigen presenting cell, CD cluster of differentiation, IFN interferon, IL interleukin, MHC major histocompatibility complex, NK natural killer cell, RNS reactive nitrogen species, ROS reactive oxygen species, TGF transforming growth factor, Th T helper cell, TNF tumor necrosis factor, Treg regulatory T cell
GM-CSF enhances the migration of monocytes to the CNS through the BBB. GM-CSF is also known to activate macrophages to adopt a phenotype similar to those found in active MS lesions [57]. Th cells producing GM-CSF have an enhanced ability to migrate to the CNS and are more abundant in the CSF of MS patients than in healthy controls [3]. Studies have shown varying results in terms of GM-CSF expression by T cells in peripheral blood in MS patients compared to controls, with the levels in the former ranging from being similar to the controls [3, 58] to increased levels in CD4+ T cells [59, 60] and CD8+ T cells [60]. The exact causes of elevated GM-CSF expression in MS are uncertain. A polymorphism in the interleukin (IL)-2 receptor alpha (IL-2RA) gene, which is one of the genetic risk factors for MS [59], dictates higher IL-2-induced GM-CSF in T cells of MS patients. Moreover, it has recently been shown that microglia and astrocytes have a higher expression of GMR in acute and chronic MS lesions [61]. The mechanism of immunoregulation of some MS therapies, such as interferon beta (IFN-β) and B-cell depleting therapy, may be due to the reduction of GM-CSF production or the removal of cells producing GM-CSF [60, 62].
GM-CSF-producing memory B cells are increased in the peripheral blood of MS patients and have myeloid cell-stimulating properties [62]. GM-CSF may also exert an autocrine effect on B-cell survival [34], which may be relevant to MS.
A newly identified subset of T cells producing predominantly GM-CSF but no or minimal other Th1 and Th17 signature cytokines may also be involved in MS pathogenesis. These cells have distinct migration properties, with an enhanced ability to migrate to inflamed CNS tissues [3]. Most of the Th cells in the CSF and blood of MS patients have recently been shown to be non-classical Th1 cells producing GM-CSF and Th cells producing only GM-CSF [63].
GM-CSF in Experimental Autoimmune Encephalomyelitis
The role of GM-CSF in MS is supported by studies in the experimental model of experimental autoimmune encephalomyelitis (EAE), where detailed analysis and manipulation of the immunopathology is more feasible than in human tissue. Despite the previous suspicion that EAE depends on IFN-γ and IL-17 for initiation and maintenance, recent experiments have found these two cytokines are redundant [64], while GM-CSF is crucial for the encephalitogenicity of CD4+ T cells infiltrating the CNS.
GM-CSF Knockout Mice are Resistant to EAE
Granulocyte-macrophage colony-stimulating factor knockout mice (GM-CSF−/−) backcrossed onto an EAE-susceptible (NOD/Lt) background are essentially protected from clinical signs of EAE after immunization with myelin-oligodendrocyte glycoprotein, an encephalitogenic peptide that induces severe disease in wild-type (WT) controls [65]. CNS infiltration was reduced in the GM-CSF−/− mice and did not persist, unlike in their WT counterparts. Resistance to EAE has also been observed in other mouse strains following knockout of GM-CSF or the GMR, as well as chimeras lacking GM-CSF expression in the bone marrow [16, 66, 67]. Furthermore, recombinant GM-CSF restores susceptibility to disease [67].
GM-CSF Synthesis by CD4+ T Cells is Necessary for Encephalitogenicity
Although myelin-specific Th1 and Th17 cells can induce EAE after passive transfer, knockout and antibody-blocking experiments confirm that the dominant cytokines (IFNγ, IL-17) are redundant and that the encephalitogenicity of these cells is proportional to the GM-CSF they produce [47]. Transfer of IL-17−/−, IFNγ−/−, or double knockout CD4+ T-cells cultured in conditions favouring GM-CSF expression results in EAE with similar kinetics to that seen after transfer of WT cells, whereas the transfer of GM-CSF−/− cells does not cause disease [16]. Cells polarized in vitro towards a Th1 or Th17 phenotype upregulate GM-CSF expression following entry to the CNS [16]. What is more, transferring myelin-specific CD4+ T cells into GM-CSF−/− recipients leads to disease indistinguishable from that seen in WT recipients [41], while transferring WT polyclonal CD4+ cells does not lead to EAE if they are co-transferred with myelin-targeted GM-CSF−/− T cells [68]. These results prove that GM-CSF is indispensable in EAE and that CNS-infiltrating Th cells are the source of GM-CSF.
Overexpression of GM-CSF Exacerbates EAE
Treatment with recombinant GM-CSF exacerbates disease and increases the frequency of relapses in EAE. Passive transfer experiments showed that myelin-specific CD4+ T cells transduced with adenovirus-expressing GM-CSF caused a more severe disease than cells transfected with virus lacking GM-CSF [68]. Transgenic mice constitutively over-expressing GM-CSF in CD4+ T cells spontaneously developed CNS lesions, even in the absence of sensitization with myelin peptides [69]. Although the lung and liver were also infiltrated by cells in this study [69], they did not demonstrate pathology or symptoms, suggesting the CNS is particularly susceptible to the effects of dysregulated GM-CSF.
Antibodies to GM-CSF Prevent Disease if Given Prior to Induction and Ameliorate Ongoing Disease
Anti-GM-CSF antibodies prevent the onset of chronic EAE if given prior to active induction and reduce CNS lesion load [65, 66]. They also reduce the number and severity of relapses in relapsing EAE [70]. However, cessation of treatment leads to disease with a similar evolution as that seen in untreated controls. Furthermore, treatment with antibodies targeting either GM-CSF [69] or its receptor [70] given after clinical disease onset results in partial (in the case of receptor antibody) or complete (in the case of cytokine antibody) clinical recovery and a significant reduction in CNS lesion burden. These results have implications for the treatment of MS when therapy is needed in established disease.
GM-CSF Levels in the CNS Increase at Disease Onset
The expression of GM-CSF is very low in the spinal cords of mice at the time of disease induction, but it increases dramatically when disease becomes apparent [41, 64].
What Does GM-CSF do in EAE?
Several research groups have demonstrated that the absence of GM-CSF impairs myelin-specific T-cell priming [65, 66] and prevents epitope spreading [70]. These effects are unlikely to be due to failure of autocrine effects of GM-CSF given the absence of GMRs from T cells. Rather, they are believed to be due to impaired antigen presentation by APCs, particularly monocyte-derived dendritic cells (moDCs), for T-cell priming in the periphery and reactivation in the CNS [71].
GM-CSF is a key factor in the differentiation and mobilization of inflammatory monocytes from the bone marrow prior to the onset of EAE [16, 67, 69]. The migration of these cells to the CNS is also believed to be influenced by EAE as the knocking out GM-CSF or its receptor reduces the number of granulocytes and moDCs in the CNS after induction [65, 66, 70] and overexpression increases recruitment [47]. Conditional GMR deletion restricted to the inflammatory monocytes prevents the onset of EAE if knockout occurs at disease induction and it ameliorates disease if knockout occurs after clinical signs develop [72]. However, the crucial role of GM-CSF appears not to be in the recruitment of inflammatory monocytes per se but rather in the further differentiation of these cells into moDCs and tissue macrophages within the CNS. For example, bone marrow chimeras have shown GMR expression is not an absolute requirement for the migration of these cells into the CNS [73]. GM-CSF can modulate cytokine and chemokine expression by mature myeloid-derived cells and upregulate costimulatory molecules and major histocompatibility complex (MHC) class II molecules[70,71,72, 74]. As the expression of GM-CSF restricted to CNS-infiltrating CD4+ T cells alone is sufficient to cause disease [65, 68], its essential role may be effector cell differentiation within the CNS, establishing a complex pro-inflammatory feedback system involving APCs, neutrophils, microglia and endothelial cells [71, 75].
GM-CSF is Necessary for Maintaining Disease
Preventing GM-CSF signalling results in milder disease with recovery in chronic models of EAE and fewer, milder relapses in relapsing–remitting models of EAE [16, 48, 65]. This action is reflected on a cellular level by pre-symptomatic lymphocyte and myeloid infiltration that persists in WT mice but regresses in GM-CSF−/− mice or mice treated with blocking antibodies [16].
However, results from animal models must be interpreted with caution. One problem intrinsic to EAE is that different host genotypes and methods of induction result in distinctive phenotypes, and factors found to be crucial in one model may not be in another. For example, C3HeB/FeJ mice develop an “atypical” EAE with neutrophilic brainstem and spinal cord infiltration and predominant ataxia, as well as the “classic” inflammation in the spinal cord and ascending paralysis. Using passive transfer of WT and GM-CSF−/− T-cells into WT and IL-17R−/− hosts, Pierson and Goverman demonstrated that either GM-CSF or IL-17 is required for brainstem inflammation, but neither is required for spinal cord disease [76]. This finding is in stark contrast to the plethora of similar experiments in different mouse strains demonstrating that GM-CSF, but not IL-17, is necessary for classic EAE [16, 48]. These differences may be accounted for by variation in methodologies and the mouse genotypes used, or by inadequate synthesis of IL-17 by alternative mouse lines, with subsequent failure to compensate for the absence of GM-CSF [76].
In conclusion, many experiments have demonstrated a critical role of GM-CSF in EAE. Although the importance of GM-CSF varies according to the model used, and the extrapolation of EAE results to MS can be problematic, the totality of the EAE evidence justifies pursuing work targeting GM-CSF, its receptor and signalling pathways in MS.
Clinical Trials Targeting GM-CSF in MS
Several agents targeting GM-CSF are being developed or currently under study in clinical trials. There are several ongoing or completed clinical trials targeting GM-CSF or GMR in rheumatoid arthritis (RA), psoriasis and MS. These have been reviewed elsewhere [77].
GSK3196165 (or MOR103) is a high-affinity recombinant human immunoglobulin (Ig) G1 antibody. It was developed by MorphoSys AG (Planegg, Germany) and in-licensed by GlaxoSmithKline (GSK; Brentford, UK) [78]. It blocks the interaction of GM-CSF with its receptor and thus averts subsequent signal transduction [79]. Its access into the CNS may be facilitated by the altered BBB in MS, which can potentially favour IgG penetration.
The study of MOR103 in RA included 96 patients with active disease [78]. The primary outcome measure was the adverse event (AE) rate and safety profile, while secondary endpoints included Disease Activity Score 28-joint assessment (DAS28), American College of Rheumatology (ACR) core set measures, European League Against Rheumatism (EULAR) response criteria and magnetic resonance imaging (MRI) of synovitis [78]. Three different dosages of MOR103 (0.3, 1.0 and 1.5 mg/kg) were compared to placebo, and significant clinical improvement was shown, most pronounced at 1.0 mg/kg. There were no safety and tolerability issues, and AEs were generally mild or moderate, with the most common AE being nasopharyngitis [78].
Based on these positive results regarding tolerability and efficacy in patients with RA, a phase 1b trial of MOR103 in patients with MS was performed (NCT01517282) [80]. The aim of this trial was to determine the safety, immunogenicity and pharmacokinetics (PK) of MOR103 in people with MS with clinical or MRI activity. This 20-week, double-blind, placebo-controlled phase 1b dose-escalation trial recruited 31 MRI-active relapsing–remitting or secondary progressive MS patients. Participants were randomized to receive an intravenous infusion of placebo (n = 6) or active treatment with 0.5 (n = 8), 1.0 (n = 8) or 2.0 (n = 9) mg/kg antibody every 2 weeks for 10 weeks [80]. The antibody was overall well-tolerated, and no evidence of immunogenicity was found. Most treatment-emergent AEs were mild to moderate, with again the most frequent AE being nasopharyngitis. There were no AE-related trial drop-outs, infusion-related reactions or deaths [80]. The differences in AE between the trial arms were small. No clinically significant changes were observed in other clinical assessments or laboratory safety assessments. Nine patients from different groups experienced MS exacerbations: three in the placebo arm, five in the 0.5 mg/kg group, one in the 5 mg/kg group and zero in the 2.0 mg/kg group. MRI activity (T1 Gd-enhancing lesions and/or new or enlarging T2 lesions) were observed in all treatment groups. PK assessments showed dose linearity with low or no drug accumulation over time [80].
The results of this trial show that the antibody has a good tolerability profile, with no unexpected safety concerns in the treatment of MS. This finding is particularly important since immune therapies can have different or unexpected AEs in different autoimmune conditions. Although GM-CSF inhibition seems to be generally safe to date [79], active monitoring for potential side effects is needed.
Studies in GM-CSF-deficient mice show a potential increased risk of infections [81]. An imbalance in GM-CSF regulation can induce colitis [82], and inhibition of GM-CSF expression may exacerbate existing intestinal inflammation [83]. People with high-titre GM-CSF autoantibodies or the mutations of GMR may be at risk for pulmonary alveolar proteinosis [84].
The good safety and tolerability findings from the phase I trial of anti-GM-CSF antibody in MS are consistent with prior safety data reported for patients with RA [78] and warrant further clinical studies.
In planning further studies, one should bear in mind the complex relationship between GM-CSF effects and the specific immunological status in a given patient. Studies of GM-CSF as an adjuvant in clinical trials of vaccination with autologous tumor cells, peptides and/or dendritic cells in patients with cancer have shown heterogeneous results in terms of the induction of vaccine-specific immune response and clinical response [85]. The authors of this study [85] noted that a dose-dependent effect was probable, with repeated low doses of GM-CSF increasing the vaccine-induced immune response and with an opposite effect with higher dosages. Indirect evidence of a possible role the clinical implications of GM-CSF in MS comes from a case report of a 51-year-old woman with stage-three melanoma who received subcutaneous recombinant human (rh) GM-CSF injections for 3 years in a phase two clinical trial [86]. During the trial, yearly MRI scans showed subtle stable demyelination. The patient was in remission of her melanoma at the end of the trial, and 7 months after discontinuing GM-CSF she had her first MS clinical relapse [86]. Development of anti-GM-CSF antibodies during rhGM-CSF therapy as a cause of the relapse was deemed unlikely, as white cell counts suggested persistent biological effect of GM-CSF [86]. Therefore, the enhanced activity could conceivably be due to persistent GM-CSF effects rather than to its withdrawal.
Further studies of anti-GM-CSF treatments in MS are warranted, and the identification of patients more likely to benefit is of crucial importance.
Conclusion
Granulocyte-macrophage colony-stimulating factor is increasingly recognized as a pivotal cytokine in the pathogenesis of MS. Its production by multiple immune cell types is increased in MS, and its levels in the CSF correlate with disease activity. Moreover, GM-CSF is implicated in the pathological mechanisms of EAE. The blockade of GM-CSF signalling prevents or suppresses EAE. These studies strongly support interventions to block GM-CSF in MS, and results from phase I studies indicate a favourable safety profile of antibodies against GM-CSF. GM-CSF is, therefore, a plausible therapeutic strategy.
References
Whetton AD, Dexter TM. Myeloid haemopoietic growth factors. Biochim Biophys Acta. 1989;989(2):111–32.
Min L, Mohammad Isa SA, Shuai W, Piang CB, Nih FW, Kotaka M, et al. Cutting edge: granulocyte-macrophage colony-stimulating factor is the major CD8+ T cell-derived licensing factor for dendritic cell activation. J Immunol. 2010;184(9):4625–9.
Noster R, Riedel R, Mashreghi MF, Radbruch H, Harms L, Haftmann C, et al. IL-17 and GM-CSF expression are antagonistically regulated by human T helper cells. Sci Transl Med. 2014;6(241):241ra80.
Sheng W, Yang F, Zhou Y, Yang H, Low PY, Kemeny DM, et al. STAT5 programs a distinct subset of GM-CSF-producing T helper cells that is essential for autoimmune neuroinflammation. Cell Res. 2014;24(12):1387–402.
McLay RN, Banks WA, Kastin AJ. Granulocyte macrophage-colony stimulating factor crosses the blood–testis barrier in mice. Biol Reprod. 1997;57(4):822–6.
McLay RN, Kimura M, Banks WA, Kastin AJ. Granulocyte-macrophage colony-stimulating factor crosses the blood–brain and blood–spinal cord barriers. Brain. 1997;120(Pt 11):2083–91.
Malipiero UV, Frei K, Fontana A. Production of hemopoietic colony-stimulating factors by astrocytes. J Immunol. 1990;144(10):3816–21.
Re F, Belyanskaya SL, Riese RJ, Cipriani B, Fischer FR, Granucci F, et al. Granulocyte-macrophage colony-stimulating factor induces an expression program in neonatal microglia that primes them for antigen presentation. J Immunol. 2002;169(5):2264–73.
Sawada M, Itoh Y, Suzumura A, Marunouchi T. Expression of cytokine receptors in cultured neuronal and glial cells. Neurosci Lett. 1993;160(2):131–4.
Muto A, Watanabe S, Itoh T, Miyajima A, Yokota T, Arai K. Roles of the cytoplasmic domains of the alpha and beta subunits of human granulocyte-macrophage colony-stimulating factor receptor. J Allergy Clin Immunol. 1995;96(6 Pt 2):1100–14.
Lilly MB, Zemskova M, Frankel AE, Salo J, Kraft AS. Distinct domains of the human granulocyte-macrophage colony-stimulating factor receptor alpha subunit mediate activation of Jak/Stat signaling and differentiation. Blood. 2001;97(6):1662–70.
Greven DE, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, et al. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Ann Rheum Dis. 2015;74(10):1924–30.
Wicks IP, Roberts AW. Targeting GM-CSF in inflammatory diseases. Nat Rev Rheumatol. 2016;12(1):37–48.
Infante-Duarte C, Horton HF, Byrne MC, Kamradt T. Microbial lipopeptides induce the production of IL-17 in Th cells. J Immunol. 2000;165(11):6107–15.
Mosmann TR, Cherwinski H, Bond MW, Giedlin MA, Coffman RL. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J Immunol. 1986;136(7):2348–57.
Codarri L, Gyulveszi G, Tosevski V, Hesske L, Fontana A, Magnenat L, et al. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol. 2011;12(6):560–7.
Hamilton JA. Coordinate and noncoordinate colony stimulating factor formation by human monocytes. J Leukoc Biol. 1994;55(3):355–61.
Baldwin GC, Gasson JC, Kaufman SE, Quan SG, Williams RE, Avalos BR, et al. Nonhematopoietic tumor cells express functional GM-CSF receptors. Blood. 1989;73(4):1033–7.
Filonzi EL, Zoellner H, Stanton H, Hamilton JA. Cytokine regulation of granulocyte-macrophage colony stimulating factor and macrophage colony-stimulating factor production in human arterial smooth muscle cells. Atherosclerosis. 1993;99(2):241–52.
Campbell IK, Novak U, Cebon J, Layton JE, Hamilton JA. Human articular cartilage and chondrocytes produce hemopoietic colony-stimulating factors in culture in response to IL-1. J Immunol. 1991;147(4):1238–46.
Timoshanko JR, Kitching AR, Semple TJ, Holdsworth SR, Tipping PG. Granulocyte macrophage colony-stimulating factor expression by both renal parenchymal and immune cells mediates murine crescentic glomerulonephritis. J Am Soc Nephrol. 2005;16(9):2646–56.
Bagby GC Jr, Dinarello CA, Wallace P, Wagner C, Hefeneider S, McCall E. Interleukin 1 stimulates granulocyte macrophage colony-stimulating activity release by vascular endothelial cells. J Clin Invest. 1986;78(5):1316–23.
Soldi R, Primo L, Brizzi MF, Sanavio F, Aglietta M, Polentarutti N, et al. Activation of JAK2 in human vascular endothelial cells by granulocyte-macrophage colony-stimulating factor. Blood. 1997;89(3):863–72.
Churchill L, Friedman B, Schleimer RP, Proud D. Production of granulocyte-macrophage colony-stimulating factor by cultured human tracheal epithelial cells. Immunology. 1992;75(1):189–95.
Giacomini G, Tabibzadeh SS, Satyaswaroop PG, Bonsi L, Vitale L, Bagnara GP, et al. Epithelial cells are the major source of biologically active granulocyte macrophage colony-stimulating factor in human endometrium. Hum Reprod. 1995;10(12):3259–63.
Zucali JR, Dinarello CA, Oblon DJ, Gross MA, Anderson L, Weiner RS. Interleukin 1 stimulates fibroblasts to produce granulocyte-macrophage colony-stimulating activity and prostaglandin E2. J Clin Invest. 1986;77(6):1857–63.
Fitzgerald SM, Chi DS, Hall HK, Reynolds SA, Aramide O, Lee SA, et al. GM-CSF induction in human lung fibroblasts by IL-1beta, TNF-alpha, and macrophage contact. J Interferon Cytokine Res. 2003;23(2):57–65.
Dedhar S, Gaboury L, Galloway P, Eaves C. Human granulocyte-macrophage colony-stimulating factor is a growth factor active on a variety of cell types of nonhemopoietic origin. Proc Natl Acad Sci USA. 1988;85(23):9253–7.
Montagnani S, Postiglione L, Giordano-Lanza G, Meglio FD, Castaldo C, Sciorio S, et al. Granulocyte macrophage colony stimulating factor (GM-CSF) biological actions on human dermal fibroblasts. Eur J Histochem. 2001;45(3):219–28.
Uemura Y, Kobayashi M, Nakata H, Kubota T, Saito T, Bandobashi K, et al. Effects of granulocyte colony-stimulating factor and granulocyte-macrophage colony-stimulating factor on lung cancer: roles of cyclooxygenase-2. Oncol Rep. 2007;17(4):955–61.
Mascia F, Cataisson C, Lee TC, Threadgill D, Mariani V, Amerio P, et al. EGFR regulates the expression of keratinocyte-derived granulocyte/macrophage colony-stimulating factor in vitro and in vivo. J Invest Dermatol. 2010;130(3):682–93.
Hancock GE, Kaplan G, Cohn ZA. Keratinocyte growth regulation by the products of immune cells. J Exp Med. 1988;168(4):1395–402.
Braunstein S, Kaplan G, Gottlieb AB, Schwartz M, Walsh G, Abalos RM, et al. GM-CSF activates regenerative epidermal growth and stimulates keratinocyte proliferation in human skin in vivo. J Invest Dermatol. 1994;103(4):601–4.
Harris RJ, Pettitt AR, Schmutz C, Sherrington PD, Zuzel M, Cawley JC, et al. Granulocyte-macrophage colony-stimulating factor as an autocrine survival factor for mature normal and malignant B lymphocytes. J Immunol. 2000;164(7):3887–93.
Cooper MA, Fehniger TA, Turner SC, Chen KS, Ghaheri BA, Ghayur T, et al. Human natural killer cells: a unique innate immunoregulatory role for the CD56(bright) subset. Blood. 2001;97(10):3146–51.
Kubista B, Trieb K, Herbacek I, Micksche M. Effect of granulocyte-macrophage colony-stimulating factor on natural-killer cell mediated cytotoxicity. Int J Biochem Cell Biol. 2003;35(7):1056–60.
Bezbradica JS, Gordy LE, Stanic AK, Dragovic S, Hill T, Hawiger J, et al. Granulocyte-macrophage colony-stimulating factor regulates effector differentiation of invariant natural killer T cells during thymic ontogeny. Immunity. 2006;25(3):487–97.
Lukens JR, Barr MJ, Chaplin DD, Chi H, Kanneganti TD. Inflammasome-derived IL-1beta regulates the production of GM-CSF by CD4(+) T cells and gammadelta T cells. J Immunol. 2012;188(7):3107–15.
Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M. GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med. 2008;205(10):2281–94.
Lee SC, Liu W, Brosnan CF, Dickson DW. GM-CSF promotes proliferation of human fetal and adult microglia in primary cultures. Glia. 1994;12(4):309–18.
Ponomarev ED, Shriver LP, Maresz K, Pedras-Vasconcelos J, Verthelyi D, Dittel BN. GM-CSF production by autoreactive T cells is required for the activation of microglial cells and the onset of experimental autoimmune encephalomyelitis. J Immunol. 2007;178(1):39–48.
Kita H, Ohnishi T, Okubo Y, Weiler D, Abrams JS, Gleich GJ. Granulocyte/macrophage colony-stimulating factor and interleukin 3 release from human peripheral blood eosinophils and neutrophils. J Exp Med. 1991;174(3):745–8.
Arnaout MA, Wang EA, Clark SC, Sieff CA. Human recombinant granulocyte-macrophage colony-stimulating factor increases cell-to-cell adhesion and surface expression of adhesion-promoting surface glycoproteins on mature granulocytes. J Clin Invest. 1986;78(2):597–601.
Lee J, Kim Y, Lim J, Kim M, Han K. G-CSF and GM-CSF concentrations and receptor expression in peripheral blood leukemic cells from patients with chronic myelogenous leukemia. Ann Clin Lab Sci. 2008;38(4):331–7.
Lopez AF, Vadas MA, Woodcock JM, Milton SE, Lewis A, Elliott MJ, et al. Interleukin-5, interleukin-3, and granulocyte-macrophage colony-stimulating factor cross-compete for binding to cell surface receptors on human eosinophils. J Biol Chem. 1991;266(36):24741–7.
Kim WK, Kim D, Cui J, Jang HH, Kim KS, Lee HJ, et al. Secretome analysis of human oligodendrocytes derived from neural stem cells. PLoS One. 2014;9(1):e84292.
Grifka-Walk HM, Giles DA, Segal BM. IL-12-polarized Th1 cells produce GM-CSF and induce EAE independent of IL-23. Eur J Immunol. 2015;45(10):2780–6.
El-Behi M, Ciric B, Dai H, Yan Y, Cullimore M, Safavi F, et al. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat Immunol. 2011;12(6):568–75.
Jansen JH, Wientjens GJ, Fibbe WE, Willemze R, Kluin-Nelemans HC. Inhibition of human macrophage colony formation by interleukin 4. J Exp Med. 1989;170(2):577–82.
Hashimoto SI, Komuro I, Yamada M, Akagawa KS. IL-10 inhibits granulocyte-macrophage colony-stimulating factor-dependent human monocyte survival at the early stage of the culture and inhibits the generation of macrophages. J Immunol. 2001;167(7):3619–25.
Sagawa K, Mochizuki M, Sugita S, Nagai K, Sudo T, Itoh K. Suppression by IL-10 and IL-4 of cytokine production induced by two-way autologous mixed lymphocyte reaction. Cytokine. 1996;8(6):501–6.
Jacobsen SE, Ruscetti FW, Dubois CM, Lee J, Boone TC, Keller JR. Transforming growth factor-beta trans-modulates the expression of colony stimulating factor receptors on murine hematopoietic progenitor cell lines. Blood. 1991;77(8):1706–16.
Young A, Linehan E, Hams E, O’Hara Hall AC, McClurg A, Johnston JA, et al. Cutting edge: suppression of GM-CSF expression in murine and human T cells by IL-27. J Immunol. 2012;189(5):2079–83.
Duhen T, Campbell DJ. IL-1beta promotes the differentiation of polyfunctional human CCR6 + CXCR3 + Th1/17 cells that are specific for pathogenic and commensal microbes. J Immunol. 2014;193(1):120–9.
Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol. 2008;8(7):533–44.
Carrieri PB, Provitera V, De Rosa T, Tartaglia G, Gorga F, Perrella O. Profile of cerebrospinal fluid and serum cytokines in patients with relapsing-remitting multiple sclerosis: a correlation with clinical activity. Immunopharmacol Immunotoxicol. 1998;20(3):373–82.
Vogel DY, Kooij G, Heijnen PD, Breur M, Peferoen LA, van der Valk P, et al. GM-CSF promotes migration of human monocytes across the blood brain barrier. Eur J Immunol. 2015;45(6):1808–19.
Peelen E, Muris AH, Damoiseaux J, Knippenberg S, Broens K, Smolders J, et al. GM-CSF production by CD4+ T cells in MS patients: regulation by regulatory T cells and vitamin D. J Neuroimmunol. 2015;280:36–42.
Hartmann FJ, Khademi M, Aram J, Ammann S, Kockum I, Constantinescu C, et al. Multiple sclerosis-associated IL2RA polymorphism controls GM-CSF production in human TH cells. Nat Commun. 2014;5:5056.
Rasouli J, Ciric B, Imitola J, Gonnella P, Hwang D, Mahajan K, et al. Expression of GM-CSF in T cells is increased in Multiple sclerosis and suppressed by IFN-beta therapy. J Immunol. 2015;194(11):5085–93.
Imitola J, Rasouli J, Watanabe F, Mahajan K, Sharan AD, Ciric B, et al. Elevated expression of granulocyte-macrophage colony-stimulating factor receptor in multiple sclerosis lesions. J Neuroimmunol. 2018;317:45–54.
Li R, Rezk A, Miyazaki Y, Hilgenberg E, Touil H, Shen P, et al. Proinflammatory GM-CSF-producing B cells in multiple sclerosis and B cell depletion therapy. Sci Transl Med. 2015;7(310):310ra166.
Restorick SM, Durant L, Kalra S, Hassan-Smith G, Rathbone E, Douglas MR, et al. CCR6(+) Th cells in the cerebrospinal fluid of persons with multiple sclerosis are dominated by pathogenic non-classic Th1 cells and GM-CSF-only-secreting Th cells. Brain Behav Immun. 2017;64:71–9.
Kroenke MA, Chensue SW, Segal BM. EAE mediated by a non-IFN-gamma/non-IL-17 pathway. Eur J Immunol. 2010;40(8):2340–8.
McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, et al. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med. 2001;194(7):873–82.
Duncker PC, Stoolman JS, Huber AK, Segal BM. GM-CSF promotes chronic disability in experimental autoimmune encephalomyelitis by altering the composition of central nervous system-infiltrating cells, but is dispensable for disease induction. J Immunol. 2018;200(3):966–73.
King IL, Dickendesher TL, Segal BM. Circulating Ly-6C + myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood. 2009;113(14):3190–7.
Marusic S, Miyashiro JS, Douhan J, Konz RF, Xuan D, Pelker JW, et al. Local delivery of granulocyte macrophage colony-stimulating factor by retrovirally transduced antigen-specific T cells leads to severe, chronic experimental autoimmune encephalomyelitis in mice. Neurosci Lett. 2002;332(3):185–9.
Spath S, Komuczki J, Hermann M, Pelczar P, Mair F, Schreiner B, et al. Dysregulation of the cytokine GM-CSF induces spontaneous phagocyte invasion and immunopathology in the central nervous system. Immunity. 2017;46(2):245–60.
Ifergan I, Davidson TS, Kebir H, Xu D, Palacios-Macapagal D, Cann J, et al. Targeting the GM-CSF receptor for the treatment of CNS autoimmunity. J Autoimmun. 2017;84:1–11.
Croxford AL, Spath S, Becher B. GM-CSF in neuroinflammation: licensing myeloid cells for tissue damage. Trends Immunol. 2015;36(10):651–62.
Croxford AL, Lanzinger M, Hartmann FJ, Schreiner B, Mair F, Pelczar P, et al. The cytokine GM-CSF drives the inflammatory signature of CCR2+ monocytes and licenses autoimmunity. Immunity. 2015;43(3):502–14.
Greter M, Helft J, Chow A, Hashimoto D, Mortha A, Agudo-Cantero J, et al. GM-CSF controls nonlymphoid tissue dendritic cell homeostasis but is dispensable for the differentiation of inflammatory dendritic cells. Immunity. 2012;36(6):1031–46.
Hart PH, Whitty GA, Piccoli DS, Hamilton JA. Synergistic activation of human monocytes by granulocyte-macrophage colony-stimulating factor and IFN-gamma. Increased TNF-alpha but not IL-1 activity. J Immunol. 1988;141(5):1516–21.
Zhao J, Sun L, Li X. Commanding CNS invasion: GM-CSF. Immunity. 2017;46(2):165–7.
Pierson ER, Goverman JM. GM-CSF is not essential for experimental autoimmune encephalomyelitis but promotes brain-targeted disease. JCI Insight. 2017;2(7):e92362.
Shiomi A, Usui T, Mimori T. GM-CSF as a therapeutic target in autoimmune diseases. Inflamm Regen. 2016;36:8.
Behrens F, Tak PP, Ostergaard M, Stoilov R, Wiland P, Huizinga TW, et al. MOR103, a human monoclonal antibody to granulocyte-macrophage colony-stimulating factor, in the treatment of patients with moderate rheumatoid arthritis: results of a phase Ib/IIa randomised, double-blind, placebo-controlled, dose-escalation trial. Ann Rheum Dis. 2015;74(6):1058–64.
Steidl S, Ratsch O, Brocks B, Durr M, Thomassen-Wolf E. In vitro affinity maturation of human GM-CSF antibodies by targeted CDR-diversification. Mol Immunol. 2008;46(1):135–44.
Constantinescu CS, Asher A, Fryze W, Kozubski W, Wagner F, Aram J, et al. Randomized phase 1b trial of MOR103, a human antibody to GM-CSF, in multiple sclerosis. Neurol Neuroimmunol Neuroinflamm. 2015;2(4):e117.
Hirata Y, Egea L, Dann SM, Eckmann L, Kagnoff MF. GM-CSF-facilitated dendritic cell recruitment and survival govern the intestinal mucosal response to a mouse enteric bacterial pathogen. Cell Host Microbe. 2010;7(2):151–63.
Biondo M, Nasa Z, Marshall A, Toh BH, Alderuccio F. Local transgenic expression of granulocyte macrophage-colony stimulating factor initiates autoimmunity. J Immunol. 2001;166(3):2090–9.
Xu Y, Hunt NH, Bao S. The role of granulocyte macrophage-colony-stimulating factor in acute intestinal inflammation. Cell Res. 2008;18(12):1220–9.
Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol. 2010;135(2):223–35.
Parmiani G, Castelli C, Pilla L, Santinami M, Colombo MP, Rivoltini L. Opposite immune functions of GM-CSF administered as vaccine adjuvant in cancer patients. Ann Oncol. 2007;18(2):226–32.
Chong J, Cohen M, Waubant E. Multiple sclerosis onset after granulocyte macrophage colony-stimulating factor withdrawal. Mult Scler Relat Disord. 2018;20:178–80.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published”.
Disclosures
Cris S. Constantinescu was Chief Investigator in the Morphosys phase I trial of anti-GM-CSF in MS. Radu Tanasescu was Sub-Investigator in the Morphosys phase I trial of anti-GMCSF in MS. Jehan Aram and Anna Francis have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any noncommercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced digital features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.7376096.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Aram, J., Francis, A., Tanasescu, R. et al. Granulocyte-Macrophage Colony-Stimulating Factor as a Therapeutic Target in Multiple Sclerosis. Neurol Ther 8, 45–57 (2019). https://doi.org/10.1007/s40120-018-0120-1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40120-018-0120-1