
Abstract
Cardiovascular diseases (CVDs) are the leading cause of death in the global world. The emergence of single-cell technologies has greatly facilitated the research on CVDs. Currently, those single-cell technologies have been widely applied in atherosclerosis, myocardial infarction, cardiac ischemia–reperfusion injury, arrhythmia, hypertrophy cardiomyopathy, and heart failure, which are extremely helpful in elucidating the underlying mechanisms of CVDs from physiological and pathological perspectives at DNA, RNA, protein, post-transcriptional, post-translational, and metabolite levels. In this review, we would like to briefly introduce the current single-cell technologies, and will focus on the utilization of single-cell genomics in various heart diseases. Single-cell technologies have great potential in exploration of CVDs, and widespread application of single-cell genomics will promote the understanding and therapeutic treatments for CVDs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Single-cell genomics is essential for cardiovascular research. |
Isolation of good-quality single cells is a prerequisite before performing single-cell genomics. |
Single-cell genomics has been widely applied in various cardiovascular diseases. |
Single-cell technologies aid the exploration of mechanisms, diagnostics, and therapeutic treatments for cardiovascular diseases. |
Introduction
Cardiovascular diseases (CVDs) are the leading cause of death globally. All the etiologies that damage the heart, muscle, valves, or blood arteries can develop CVDs. CVDs continue to be the primary cause of mortality and morbidity in the industrialized world, and it is responsible for 30% of global mortality due to their fast-increasing prevalence. According to a report by American Heart Association, an estimation of 80.7 million US adults (around one out of three) suffer at least one or more types of CVDs [1]. CVDs were the primary factor accounting for 36.3% of all mortalities in 2004, around one out of every 2.8 mortalities in the United States [1]. According to a mortality report from WHO, it is mentioned that CVDs are the leading cause of death in the global world, around 17.5 million lives were claimed to death in 2005, accounting for 30% of all mortalities around the world [2]. In order to understand the complicated mechanisms behind CVDs, single-cell technologies have great potential in the exploration of the physiological and pathological mechanisms under CVDs. Moreover, single-cell technologies pave the way for the development of therapeutic drugs for CVDs.
A single cell is the smallest unit in a tissue or an organ to detect the biological, physiological, and pathological processes in the body. In traditional technologies, most studies were carried out via bulk tissues or a bunch of cells from a tissue or an organ to demonstrate the features of DNA, RNA, protein expressions, and epigenetic regulations in physiological and pathological states. Since a huge amount of heterogeneity exists between every single cell, the real biological reactions and responses in each cell cannot be reflected correctly. The emergence of single-cell technologies exhibits a huge potential to meet the requirement for precision medicine and provides more accurate biomedical data to aid targeted therapy. During the past 10 years, single-cell technologies, including genomics, have been fast-developed and extensively applied in different aspects of biomedical research, which enables exploring unprecedented questions and understand underlying mechanisms of a particular disease via a single and typical cell from various tissues, organs, and disease models. Single-cell whole-genome DNA sequencing and single-cell RNA sequencing (scRNA-seq) are two major current technologies involved in single-cell genomics [3]. Moreover, other techniques based on single-cell levels are single-cell epigenetic sequencing, single-cell proteomics, single-cell metabolomics, etc. Each technique has its own specific features, addressing different perspectives regarding gene variation and mutation, DNA methylation, histone modifications, transcription, translation, and metabolism, which benefits the research for CVDs and other diseases. In order to detect a single novel or rare cell in a particular disease, such as CVDs, a combination of those technologies facilitates a more comprehensive understanding and aids the development of target therapies. In this review, the application of single-cell genomics in CVDs will be detailed as illustrated, which can help clinicians and scientists to have a rough idea about how these technologies have been utilized in current research.
Single-Cell Isolation
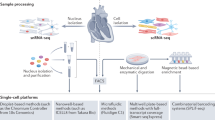
The most essential and prerequisite step before performing single-cell technology is to isolate a single cell from a typical tissue or organ. The quality of single-cell isolation ensures the accuracy and precision for the interpretation of the data afterwards. An ideal quantity and quality of cells are determined by multiple factors, such as cell viability, culture conditions, cell sizes and types, enzyme digesting time, storage and delivery of cell samples, etc. Here are the most frequently used methods for isolating single cells. The advantages and disadvantages are listed in Table 1.
Common Types of Single-Cell Genomics and Other Single-Cell Technologies
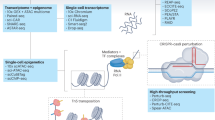
To obtain comprehensive mechanisms of a disease requires answers from levels of DNA, RNA, epigenetics, proteins, and metabolites. All these technologies mentioned here are equally important in cardiovascular research. Choosing different technologies is mostly determined by specific purposes of different studies. Moreover, different combinations of these methods can facilitate the understanding of CVDs from a broader perspective [4]. It is noteworthy that biological samples from animals require the approval from Animal Care and Use Committee (IACUC), and clinical samples from human patients require informed consents. All studies need to be approved by the Ethics Committee from the relevant institutes, and strictly follow the Helsinki Declaration. Here is a brief introduction to single-cell genomics and other technologies that have been frequently utilized in CVDs. The stepwise procedures for performing single-cell technologies can be seen in Fig. 1.

Application of single-cell technologies in CVDs. Figure made on BioRender.com
Single-Cell Whole Genome-wide Association Studies (GWAS) - Data Exploration at the DNA Level
Single-cell genomic DNA sequencing is a potential biological tool to discover genetic variations, point mutations, single nucleotide variation, de novo genomic DNA assembly, copy number variation and chromosome disorders related to heart disease. The most prominent challenge for single-cell genomic DNA sequencing is to obtain the amplification products from a whole and intact genome with high fidelity. The steps usually include selecting a specific population/animal species with specific traits/diseases, isolating the genomic DNA from specific single cells, establishing genome DNA library, making quality control, imputation, association testing, replication (choose the population/animal with the same traits for further validation), and post-GWAS analysis.
Single-Cell RNA Sequencing (scRNA-seq)-Data Exploration at the RNA Level
Due to different research purposes, scRNA-seq can be used for a specific type of cell, and the number of cells can be either large or small. A larger number of cells can provide more genomes, but increased cell density can decrease the capacity to determine mRNA features, such as the detection of expression isoforms or splicing variants. The steps for scRNA-seq are to separate cells, isolate RNA, synthesize cDNA, establish a cDNA library and sequencing, and perform analysis. Because a huge amount of RNA is lost due to RNA degradation during RNA isolation and cDNA synthesis, only 10% to 20% of the whole transcripts can be converted into cDNA, which results in an information loss in RNA sequencing [5]. Uneven amplification during reverse transcription can result in inaccurate results. The introduction of unique molecular identifiers (UMIs) is critically important and can reflect gene expression directly. Illumina HiSeq or NovaSeq are the most currently utilized platforms for scRNA-seq. The most common methods to obtain a single cell to a sequencing library are Drop-seq (a droplet-based sequencing method), SMART-seq (a method that can be paired with index sorting or microfluidics platforms), BD Rhapsody (allows for targeted detection of hundreds of known targets) [5].
Single-Cell Epigenetic Sequencing-Data Exploration at the Post-Translational Level
DNA methylation plays an indispensable role in numerous biological processes. Proper DNA methylation is critical in embryonic development, and abnormal DNA methylation can lead to multiple diseases, such as cancer and CVDs. Bisulfite genomic sequencing is a technically mature method that can be used to determine the DNA methylation in cells from a bulk tissue. Single-cell epigenetics can also be utilized to detect chromosomal conformations by single-cell Hi-C or histone modifications by chromatin immunoprecipitation (ChIP). By the combination of ChIP and sequencing (ChIP-seq), the technique named single-cell chromatin immune-precipitation sequencing (scChIP-seq) is extremely useful in detecting histone modifications and evaluating the transcriptional regulatory elements. In order to solve the low amount of DNA input in scChIP-seq, the microfluidics system can be used. Moreover, chromatin isolated from every single cell must be marked with a different DNA barcode before running ChIP within microfluidics. In the late stage, the total of indexed chromatin isolated from 100 cells is mixed and digested together, then the sequencing will be conducted after the ChIP reactions are finished. There are many single-cell methods that can be used for epigenetics, such as scWGBS and scBS-seq (the target of DNA methylation is 5 Mc), scAba-seq (its target is DNA methylation 5 hmC), scMAB-seq (the target of DNA methylation is 5fC and 5caC), CLEVER-seq (the target of DNA methylation is 5fC), and approaches like scHi-C, scChIP-seq, scDNase-seq, scATAC-seq, scNOMe-seq, all of which have the chromatin accessibility and are extremely useful in exploration of epigenetics in CVDs [6].
Single-Cell Proteomics: Data Exploration at the Protein Level
Western Blotting and enzyme-linked immunosorbent assay are two traditional methods for detecting protein levels. Single-cell proteomics faces a lot of challenges due to its complexity, since a plenty of measurements need to be tested, such as protein expression levels, protein translocation, posttranslational modifications, protein activity, interactions with other proteins, etc. All of these parameters cannot be measured in a single-cell analysis in the same time, posing great challenges to single-cell proteomics.
Single-Cell Metabolomics: Data Exploration at the Metabolite Level
Single-cell metabolomics combined with mass spectrometry facilitates the simultaneous detection of a huge number of metabolites from single cells, which is critical in understanding the underlying mechanisms under the physiological and pathological environments. The metabolites in a single cell are dynamic and can change rapidly. Thus, single-cell metabolomics can provide important insight into the phenotypes of the cells. Although single-cell metabolomics is an emerging area, it has been increasingly used in research on cancer, neuroscience, and CVDs.
Some challenges and limitations exist in single-cell metabolomics: (1) It is difficult to detect the metabolic flux since it is a dynamic process and the metabolome shifts quickly; (2) there is a limited coverage of metabolites. Since there is a wide diversity of metabolite architectures, characteristics, and concentrations, the material with an extremely low volume is unavailable for analysis; (3) the sensitivity of metabolites detection; (4) the accuracy of detecting metabolites [7].
The Current Application of Single-Cell Genomics in CVDs
Single-cell genomics plays fundamental roles in research of CVDs. The current research by application of single-cell genomics in CVDs is summarized in Table 2. Here is the detailed utilization of single-cell genomics in CVDs.
Heart Development
The single-cell relevant analysis has been applied in heart development in recent years. The heart is a sophisticated organ made up of a variety of cells and tissues. Heart development and morphogenesis are hypothesized to be influenced by diverse gene expression patterns. Guang li et al. have applied scRNA-seq in the early developmental stage of mice CMs (embryonic day 10.5) to determine the critical genes in genome-wide analyses of gene expression at the single-cell level. The specific CMs were collected from different anatomic zones, including the left ventricle, left septum, right septum, right ventricle, and the atrial ventricular canal from the early stage developing hearts. The authors found that cell-cycle genes were key factors in transcriptional levels, and CMs in G2/M phase from a specific cardiac chamber presented downregulated genes associated with sarcomeric and cytoskeletal markers matched to different cell-cycle stages. Additionally, specific signaling pathways were discovered to affect the growth of adjacent cell types. The findings from this study indicated that different genes and signaling pathways on cell-cycle activity can induce different cardiac chamber growth during development, and pave the way for a better understanding of the pathogenesis of congenital heart disease [8].
Another group of investigators from Peking University investigated four major cell types [(CMs, cardiac fibroblasts, endothelial cells (ECs), valvar interstitial cells (VICs)] in the heart by application of scRNA-seq. The investigators showed gene-expression profiles from around 4000 cardiac cells from human embryos and found that atrial and ventricular CMs exhibited different characteristics during early cardiac development. Moreover, the gene expression changed gradually in both CMs and fibroblasts. As heart development continues, VICs could be involved in the remodeling stage, while ECs showed a location-specific trait. Finally, the investigators compared the gene-expression patterns of mice and humans and discovered a number of distinctive characteristics in human heart development [9].
By scRNA-seq with over 1200 isolated murine cardiac cells (CMs, ECs, and fibroblast-enriched cells) from embryonic day 9.5 to postnatal day 21, Christine E. Seidman et al. found that different CM populations have distinct dynamic spatiotemporal gene-expression files, including Sh3bgr, Cox6a2, Trim55, Trdn, Bex1, etc. Moreover, Kcnk3, Wnt2, and Tnni3 are three prominent genes differentially expressed in CMs from atria when compared embryonic day 9.5, embryonic day 14.5, and postnatal day 0. Moreover, mice with heterozygous cardiac mutations in Nkx2.5 were identified to cause heart malformations [10].
Myocardial Infarction (MI)
ECs are essential for helping tissues adapt to damage. MI can lead to significant changes in EC activities, which can promote ECs undergoing a mesenchymal transition. Lukas S. Tombor et al. applied scRNA-seq to investigate kinetics and specific biological responses of ECs following MI. They found that the ECs undergo a transient change with numerous mesenchymal gene expressions in the proliferation and inflammation due to metabolism changes post-MI, especially around 3 to 7 days post-MI, all these genes can return to the baseline 14 days post-MI. Moreover, scRNA-seq confirmed increased hypoxic and inflammatory characteristics of ECs throughout early and late phases following damage, as well as the temporary mesenchymal transformation in Cdh5-CreERT2;mT/mG mice. These findings implied that during the initial days following MI, ECs experience a simple transition into the mesenchyme state with a metabolic adaptation, but this process is not a long-term requirement, since this mesenchymal transition may encourage the migration and clonal proliferation of ECs to restore vascular network. Moreover, metabolisms of the activated ECs showed significantly different gene profiles in pentose phosphate pathways (PPP), fatty acid (FA) signaling, tricarboxylic acid (TCA) cycle, glutamine metabolism, and glycolysis pathway comparing mesenchymal activation and none-mesenchymal activation ECs. In addition, compared with the none-mesenchymal activation endothelial cells, the mesenchymal activation showed that increased genes were enriched in the extracellular matrix organization, platelet-derived growth factor binding, regulated exocytosis, collagen biosynthetic process, collagen metabolic process, collagen fibril organization, protein-lysine 6 oxidase activity, regulation of the apoptotic process, platelet degranulation, vascular endothelial growth factor-activated receptor activity [11].
Fibroblasts play a central role in cardiac remodeling after MI. Current research has revealed that fibroblasts react differently to cardiac damage after MI. However, there is a limited characterization of fibroblast heterogeneity due to limited genetic fibroblast markers. Thus, defining the heterogeneity of cardiac fibroblast during ventricular remodeling post-MI is critically important. By application of swine and patient samples via scRNA-seq, bulk RNA sequencing, and assay for transposase-accessible chromatin sequencing, Adrián Ruiz-Villalba et al. discovered a distinct cardiac fibroblast subgroup that appeared in mice following MI. These cardiac fibroblasts had a pro-fibrotic feature with a high expression of Cthrc1 (collagen triple helix repeat containing 1) and were found to reside in the scar. Furthermore, these cardiac fibroblasts were regulated and responded to cardiac damage by non-canonical transforming growth factor-beta signaling and a variety of transcription factors, such as SOX9. In the absence of CTHRC1, the key feature was a significant ventricular rupture. Moreover, these subtypes of cardiac fibroblasts were also determined in the swine and patients with MI [12].
Monocyte macrophages are essential for MI since timely activation are critical in inflammation resolution and initiation of cardiac repair post-MI. The role of histone lactylation in MI is unknown. Naixin Wang et al. discovered that the genes responsible in cardiac repair were activated early before cardiac recruitment in both bone marrow and circulating monocytes by application of scRNA-seq. Moreover, histone H3K18 lactylation was determined in monocyte macrophages at the early stage post-MI. Furthermore, Lrg1, Vegf-a, and IL-10 were proved to be target genes for histone H3K18 lactylation post-MI, indicating that histone lactylation facilitates the early activation of monocytes in reparative transcriptional response to activate the cardiac repair post-MI [13].
Cardiac Ischemia–Reperfusion (I/R) Injury
Epicardial fat (EF) is close to the heart and shares the microvasculature with cardiac muscle, which is an appropriate source for mesenchymal stem cells (MSCs). Little is known about the potential for cardiac regeneration of epicardial adipose tissue-derived stem cells (EATDS). An interesting study from Finosh G. Thankam et al. utilized scRNA-seq to identify the EATDS subpopulations under simulated I/R injury from hyperlipidemic microswine. The results showed 18 unique cell clusters existed in the EATDS, and upregulated genes were enriched in multiple cardioprotective mechanisms including myocardial homeostasis, epigenetic regulation, prevention of fibroblast differentiation, anti-inflammatory responses, differentiation to myocardial lineage, cell integrity and cell cycle, reduction of ER-stress, and enhancing energy metabolism. These unique phenotypes of the heterogeneous EATDS population provide an important translational potential for myocardial regeneration therapy for MI [14].
Monika Gladka et al. applied scRNA-seq to investigate the healthy and diseased hearts of adult mice with I/R injury. When analyzing gene-expression patterns in all major cardiac cell types (CMs, fibroblast, ECs, macrophages), it was found that different cells had a different expression of mitochondrial and genomic transcripts. CMs exhibited a larger proportion of mitochondrial transcripts (58 to 86% of all transcripts). Thus, the mitochondrial transcripts were excluded from the clustering analysis due to the high abundance. The clustering analysis showed different novel markers for different types of cardiac cells. Moreover, cytoskeleton-associated protein 4 was found to be a novel marker for activated fibroblasts, which positively correlated with myofibroblast markers in mouse and human heart tissues, and inhibition of cytoskeleton-associated protein 4 in activated fibroblasts could enhance the gene expressions of activation of fibroblasts, demonstrating that cytoskeleton-associated protein 4 participates in the modulation of activation of fibroblasts in the ischemia-induced heart [15].
Secreted factors are critical post-cardiac I/R injury as well. By application of all types of CMs from mice post cardiac I/R injury, Bas Molenaar et al. have found that multiple unstudied secreted factors were elevated by stressed CMs following IR injury, which were also detected to be increased in circulation. Moreover, beta-2 microglobulin (B2M) can stimulate healing response by activation of fibroblasts after cardiac IR injury [16].
Atherosclerosis
Macrophages participate a major role in atherosclerosis. It can be divided into three major types: resident-like macrophages, inflammatory macrophages, and TREM2hi macrophages. Resident-like macrophages are predominantly located in adventitia tissue and majorly participated in endocytosis and proliferation signaling pathways. Inflammatory macrophages are mainly located in the intima and plaque shoulder, and are involved in inflammation response. TREM2hi macrophages are majorly resided in the intima and necrotic core, and take part in cholesterol metabolism and oxidative phosphorylation during atherosclerosis [17]. By application of scRNA-seq, Kyeongdae Kim et al. identified three major macrophage subsets with an expression of Lyve1 in atherosclerotic plaques from the Ldlr−/− mice. The macrophage subset with the highest Lyve1 expression in aortic CD45 + cells exhibited increased anti-inflammatory markers (such as mannose receptor, CD206), and genes associated with the endocytosis pathway [18]. Moreover, other investigators discovered that a number of resident-like macrophages within atherosclerotic plaques not only express Lyve1 but also other biomarkers like factor XIIIa (F13a1) and growth arrest-specific 6 (Gas6) [19]. Many other single-cell analyses demonstrated that inflammatory macrophages are present in the atherosclerotic aorta [20]. Cochain et al. have demonstrated that these macrophages are very much involved in inflammation and early growth response. Moreover, plenty of transcripts involved in pro-inflammation are enhanced, such as Il1, Tlr2, Cxcl2, Ccl3, and Ccl4 [21].
Smooth muscle cells (SMCs) play key roles in atherosclerosis. SMCs can undergo dedifferentiation, migration, and differentiation into other cell types by means of phenotypic switching. However, it is still unclear how SMCs affect the pathogenesis of atherosclerosis. Huize Pan et al. combined SMC destiny mapping with scRNA-seq of mouse and human atherosclerotic plaques to show the pathways of SMC trans-differentiation during atherosclerosis. The investigators discovered that SMCs underwent an intermediate cell state within human and mouse carotid and coronary atherosclerotic plaques during atherosclerosis. Stem cells, ECs, and monocytes (SEM cells) were SMC-derived intermediate cells that were multipotent and capable of differentiating into macrophage- and fibrochondrocyte-like cells as well as returning to the SMC phenotype. SMC to SEM cell transition was shown to be controlled by retinoic acid (RA) signaling, and RA signaling was dysregulated in symptomatic human atherosclerosis. All-trans RA, a cancer treatment for acute promyelocytic leukemia, activates RA signaling, reduces atherosclerotic load, enhances fibrous cap integrity, and prevents SMC transition to SEM cells [22].
Immune cells establish a highly specific microenvironment in plaque development. This microenvironment is to adapt to various stress and risk factors, but the intrinsic effects of these immune cells on atherosclerosis are still little understood. Pro-atherogenic CD4 + T helper (Th)-1 cells, anti-atherogenic Th-2 cells, and Th-17 cells exert their functions in the formation of atherosclerotic plaque. Moreover, cytotoxic CD8 + T cells have heterogeneous subtypes and are important in the pathogenesis of atherosclerosis. However, their functions and subsets are still unclear. Holger Winkelsa et al. first applied female ApoE-deficient (ApoE −/−) mice at an age of 8 weeks to consume either a standard chow diet (CD) or a cholesterol-rich Western diet (WD) for 12 weeks. Aortic tissues were then collected, and leukocytes were isolated and performed with scRNA-seq. Distinct leukocyte clusters were identified in CD and WD-fed mice. By application of scRNA-seq and mass cytometry (CyTOF) on leukocytes from digested aortae, around 10–30 immune cell subsets, various subtypes of B cells, T cells, myeloid cells, and natural killer cells were confirmed to exist in the aortae. Four B-cell subsets and three macrophage subsets were discovered in the atherosclerotic aorta as well. The differentiation hematopoietic lineage markers and signaling pathways were totally different in these 11 identified cell clusters [23].
Atrial Fibrillation (AF)
GWAS is an extremely useful tool in detecting numerous loci related to CVDs. In order to detect the risk variants and genes in AF, Alan Selewa et al. created an experimental and analytical strategy by combining single-cell epigenomics with GWAS. By applying heart tissues from left and right ventricles, interventricular septum, and apical cardiac tissue from three adult male AF donors, single-nucleus RNA sequencing (snRNA-seq) and scATAC-seq showed that around 49,359 cells and 26,741 cells can be used for testing, respectively. It was shown that eight major cell types existed in the cardiac tissues, and CMs, cardiac fibroblast cells, and ECs were the major types of cells in the cardiac tissue and accounted for 70% of the cells. Lymphoid cells, myeloid cells, pericytes, and SMCs are important cells as well in the cardiac tissues. In addition, scATAC-seq data determined several specific cell types that were critical regulators. By using the data from open chromatin regions (OCRs), the investigators discovered 122 potential causative genetic variations associated with AF. Moreover, 45 risk genes were shown to be remarkably important and enriched in multiple biological processes, such as cardiac ventricle formation, cardioblast proliferation, cardiac neural crest cell differentiation, regulation of relaxation of muscle, and cell proliferation; and enriched in the molecular functions, such as calcium-activated potassium channel activity, sodium channel regulatory activity, alpha-actin binding transmembrane receptor protein, tyrosine kinase activity, and hormone binding. These notable genes are essential in heart development and electrophysiology of CMs [24].
Cardiac Hypertrophy
Hypertrophic cardiomyopathy (HCM) is a hereditary heart condition that the septum of the heart that exhibits the most pronounced hypertrophy, which leads to heart failure (HF) or life-threatening arrhythmias. Although it has been confirmed that sarcomeric gene mutations are the most frequent genetic cause of HCM, the underlying mechanisms of heterogeneous remodeling are still completely unknown. The emergence of scRNA-seq is extremely useful in discovering functional networks between genes, transcripts, and translated proteins for diseases. Martijn Wehrens et al. collected cardiac tissues originating from septal myectomy from HCM patients and performed scRNA-seq. The authors excluded the reads which can map to the mitochondrial genome, and compared their scRNA-seq data with other RNA-seq data from another two research groups. By exclusion of the batch effects, the authors discovered that five different CM clusters and a specific cluster of CMs existed in the HCM hearts. This type of CM was enriched in the cardiac stress gene expression, such as MYH7, NPPA, and XIRP1. Moreover, 83 and 48 genes were positively and negatively correlated with natriuretic peptides A and B (ANP and BNP), both are remarkable biomarkers for cardiac hypertrophy and HF. It is noteworthy that a calcium channel named RYR2, which is critical in calcium-induced calcium release and cardiac contraction, showed a decreased level in the cells with an increased expression of ANP, suggesting a potential mechanism underlying cardiac hypertrophy and HF. Moreover, genes positively correlated with ANP exhibited a higher expression in hypertrophied CMs compared to normal CMs. Xin Actin Binding Repeat Containing 2 (XIRP2) is located at the costamere and intercalated disks, which are important for cardiac function. It is a critical gene expressed in both cardiac and skeletal muscle and can interact with actin and α-actinin. Multiple genes were found to be positively correlated with XIRP2, such as CMYA5, ZNF106, MAP4, TTN, SYNM, MYOM1, etc.; other genes, such as ACTC1 and MYBPC3 exhibited a negative correlation with XIRP2. These findings are significantly important to replenish the mechanisms of HCM [25].
By application of snRNA-seq, Mark Chaffin et al. have discovered that a subset of human hearts with cardiomyopathy showed a unique population of activated fibroblasts, which was entirely absent from non-failing heart samples. Knockout of genes associated with fibroblast transition led to a decreased myofibroblast cell-state transition upon TGFβ1 stimulation [26].
Heart Failure (HF)
One of the leading causes of death in the world is heart failure. Our understanding of cell structure and related gene expression has been hugely improved by the technologies of single-cell genomics. By application of scRNA-seq and snRNA-seq with the heart tissues of 18 patients with DCM and 27 healthy donors, investigators found that there were significantly different gene-expression profiles in healthy and failing human hearts. The cell-specific scRNA-seq data revealed that genes associated with aging were closely relevant to HF. Particularly, CMs, fibroblasts, and myeloid cells went through substantial diversification. Pericytes and ECs exhibited global transcriptional modifications without alterations in cell complexity. These discoveries provided a comprehensive understanding of the cellular environment for the human heart with HF, and uncover cell type-specific transcriptomes and disease-associated biological processes [27].
Li Wang et al. by application of scRNA-seq of human heart tissues from normal, failing, and recovered hearts, revealed that atrial and ventricular cells presented an inter- and intracompartmental CM heterogeneity. Moreover, cellular components and intercellular networks demonstrated that contractility and metabolism in the CMs are the most essential aspects associated with changes in cardiac functions [28]. Another study from Harvard University, utilizing snRNA-seq with 18 healthy control and 61 heart samples from failing, non-ischemic hearts with variants in DCM and arrhythmogenic cardiomyopathy (ACM) genes or idiopathic disease, Daniel Reichart et al. discovered that DCM and ACM ventricular cell atlas had a distinct left and right ventricular responses, intercellular interactions, genotype-associated pathways, and distinct gene expression at a single-cell resolution. These distinct and unique cellular and molecular architectures in human HF provided multiple candidate therapeutic targets [29].
Technology Challenges in Single-Cell Genomics
As the single-cell technologies gradually mature, there are still four major technical challenges existing: (1) The isolation of single cells with good qualities is the most essential step. Cell death and cell debris during cell isolation may result in inaccurate results; (2) For an accurate clinical biopsy, needle aspiration is applied for cancer cell acquisition. However, due to the invasiveness of the biopsy, aspiration is rarely used for heart diseases, and many heart tissue samples are acquired during heart surgeries, which limits the application of single-cell technologies; (3) To obtain sufficient genomic materials from a single cell is critical to make genomic amplifications and downstream analysis; (4) Even though cells are from the same lineage, variations of the final results can be generated due to different microenvironments, disease states, and different temporal and spatial distributions; (5) Loss of genomic material can result in dropouts during the sequencing. Moreover, it is hard to distinguish sequencing errors from mutations [30]; (6) The depth of the scRNA-seq transcriptomes determines the accuracy of the results as well; (7) Appropriate analyzing methods are crucial in interpreting the data correctly. Proper identification and explanation of the biases and mistakes during these initial steps are also significantly important. To best improve the quality of single-cell genomics sequencing, distinguishing the signal from the data and technological noise is essential, and these factors need to be carefully considered [31]; (8) The RNA transcriptome, protein expression, and metabolic profiles might be different in vivo and ex vivo environments, which can result in the bias of the data interpretation as well.
The Future of Single-Cell Technologies
Commercialized single-cell genomics and other technologies have been widely assessable recently. Novel data generated by single-cell technologies are increasingly released as well. The combination of single-cell sequencing with spatial techniques has been widely applied to explore underlying mechanisms of different diseases, and evaluate the molecular genes and signaling changes due to different drug interventions. As the cost is decreasing, it will be utilized more widely in the research for different diseases. Here are some major future directions for the development of single-cell technologies: (1) In order to improve the accuracy and sensitivity of single-cell technologies, standard methods and protocols are highly required, which can be used to remove the doublets, cell debris, and dead cells, and low-quality transcripts during the testing; (2) Imputation software programs can be used to eliminate the variations due to different batches. However, a comparative and integrated analysis for dealing with single-cell data is still in high demand to be generated to exclude the variations across different laboratories, animal species, batches, and single-cell platforms. The development of these comprehensive methods is the future direction for single-cell technologies to reduce undesirable variations while making the analysis during the diverse scRNA-seq datasets; (3) The bioinformatics analysis post-sequencing is another key element for data accuracy of single-cell technologies, which is the major obstacle in current situations. It requires a number of skilled professionals and wide collaborations among researchers with different backgrounds; (4) Moreover, to develop user-friendly databases, file formats, and interfaces for data analysis in single-cell technologies is an important future direction in improving data interpretating and sharing [5].
Conclusions
Single-cell technologies have brought new insights for the research of CVDs. They provide comprehensive genomic data to explain the underlying mechanisms, which can facilitate the novel treatments for CVDs.
References
Cardiovascular Disease Statistics. AHA. 2008.
WHO Cardiovascular Diseases. 2007.
Davis N. What is single cell genomics? December 16, 2015 Updated June 23, 2021.
Simeon M, Dangwal S, Sachinidis A, Doss MX. Application of the pluripotent stem cells and genomics in cardiovascular research-what we have learnt and not learnt until now. Cells. 2021;10.
Williams JW, Winkels H, Durant CP, Zaitsev K, Ghosheh Y, Ley K. Single cell RNA sequencing in atherosclerosis research. Circ Res. 2020;126:1112–26.
Yusuke YASC, Takahiro O. Chapter 22, Single Cell Genomics. HANDBOOK OF SINGLE-CELL TECHNOLOGIES. 2021:521–538.
Ingela Lanekoff VVS, Cátia Ma. Single-cell metabolomics: where are we and where are we going? Curr Opin Biotechnol. 2022;75.
Li G, Tian L, Goodyer W, Kort EJ, Buikema JW, Xu A, Wu JC, Jovinge S and Wu SM. Single cell expression analysis reveals anatomical and cell cycle-dependent transcriptional shifts during heart development. Development. 2019;146.
Cui Y, Zheng Y, Liu X, Yan L, Fan X, Yong J, Hu Y, Dong J, Li Q, Wu X, Gao S, Li J, Wen L, Qiao J, Tang F. Single-cell transcriptome analysis maps the developmental track of the human heart. Cell Rep. 2019;26(1934–1950): e5.
DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE. Single-cell resolution of temporal gene expression during heart development. Dev Cell. 2016;39:480–90.
Tombor LS, John D, Glaser SF, Luxan G, Forte E, Furtado M, Rosenthal N, Baumgarten N, Schulz MH, Wittig J, Rogg EM, Manavski Y, Fischer A, Muhly-Reinholz M, Klee K, Looso M, Selignow C, Acker T, Bibli SI, Fleming I, Patrick R, Harvey RP, Abplanalp WT, Dimmeler S. Single cell sequencing reveals endothelial plasticity with transient mesenchymal activation after myocardial infarction. Nat Commun. 2021;12:681.
Ruiz-Villalba A, Romero JP, Hernandez SC, Vilas-Zornoza A, Fortelny N, Castro-Labrador L, San Martin-Uriz P, Lorenzo-Vivas E, Garcia-Olloqui P, Palacio M, Gavira JJ, Bastarrika G, Janssens S, Wu M, Iglesias E, Abizanda G, de Morentin XM, Lasaga M, Planell N, Bock C, Alignani D, Medal G, Prudovsky I, Jin YR, Ryzhov S, Yin H, Pelacho B, Gomez-Cabrero D, Lindner V, Lara-Astiaso D, Prosper F. Single-cell RNA sequencing analysis reveals a crucial role for CTHRC1 (Collagen Triple Helix Repeat Containing 1) cardiac fibroblasts after myocardial infarction. Circulation. 2020;142:1831–47.
Wang N, Wang W, Wang X, Mang G, Chen J, Yan X, Tong Z, Yang Q, Wang M, Chen L, Sun P, Yang Y, Cui J, Yang M, Zhang Y, Wang D, Wu J, Zhang M, Yu B. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res. 2022;131:893–908.
Thankam FG, Agrawal DK. Single cell genomics identifies unique cardioprotective phenotype of stem cells derived from epicardial adipose tissue under ischemia. Stem Cell Rev Rep. 2022;18:294–335.
Gladka MM, Molenaar B, de Ruiter H, van der Elst S, Tsui H, Versteeg D, Lacraz GPA, Huibers MMH, van Oudenaarden A, van Rooij E. Single-cell sequencing of the healthy and diseased heart reveals cytoskeleton-associated protein 4 as a new modulator of fibroblasts activation. Circulation. 2018;138:166–80.
Molenaar B, Timmer LT, Droog M, Perini I, Versteeg D, Kooijman L, Monshouwer-Kloots J, de Ruiter H, Gladka MM, van Rooij E. Single-cell transcriptomics following ischemic injury identifies a role for B2M in cardiac repair. Commun Biol. 2021;4:146.
Willemsen L, de Winther MP. Macrophage subsets in atherosclerosis as defined by single-cell technologies. J Pathol. 2020;250:705–14.
Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, Jang MY, Seok Jang H, Yun TJ, Lee SH, Yoon WK, Prat A, Seidah NG, Choi J, Lee SP, Yoon SH, Nam JW, Seong JK, Oh GT, Randolph GJ, Artyomov MN, Cheong C, Choi JH. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. 2018;123:1127–42.
Beckers CML, Simpson KR, Griffin KJ, Brown JM, Cheah LT, Smith KA, Vacher J, Cordell PA, Kearney MT, Grant PJ, Pease RJ. Cre/lox studies identify resident macrophages as the major source of circulating coagulation factor XIII-A. Arterioscler Thromb Vasc Biol. 2017;37:1494–502.
Winkels H, Ehinger E, Vassallo M, Buscher K, Dinh HQ, Kobiyama K, Hamers AAJ, Cochain C, Vafadarnejad E, Saliba AE, Zernecke A, Pramod AB, Ghosh AK, Anto Michel N, Hoppe N, Hilgendorf I, Zirlik A, Hedrick CC, Ley K, Wolf D. Atlas of the immune cell repertoire in mouse atherosclerosis defined by single-cell RNA-sequencing and mass cytometry. Circ Res. 2018;122:1675–88.
Albrecht C, Preusch MR, Hofmann G, Morris-Rosenfeld S, Blessing E, Rosenfeld ME, Katus HA, Bea F. Egr-1 deficiency in bone marrow-derived cells reduces atherosclerotic lesion formation in a hyperlipidaemic mouse model. Cardiovasc Res. 2010;86:321–9.
Pan H, Xue C, Auerbach BJ, Fan J, Bashore AC, Cui J, Yang DY, Trignano SB, Liu W, Shi J, Ihuegbu CO, Bush EC, Worley J, Vlahos L, Laise P, Solomon RA, Connolly ES, Califano A, Sims PA, Zhang H, Li M, Reilly MP. Single-cell genomics reveals a novel cell state during smooth muscle cell phenotypic switching and potential therapeutic targets for atherosclerosis in mouse and human. Circulation. 2020;142:2060–75.
Winkels H, Ehinger E, Ghosheh Y, Wolf D, Ley K. Atherosclerosis in the single-cell era. Curr Opin Lipidol. 2018;29:389–96.
Alan SKL, Michael W, Linsin S, Chenwei T, Heather E, Ivan M, Anindita B, Xin H, Sebastian P. Single-cell genomics improves the discovery of risk variants and genes of cardiac traits. medRxiv. 2022.
Martijn WAEdL, Maya WC, Joep ECE, Cornelis JB, Bas M, Petra H van der Kraak, Diederik W D Kuster, Jolanda van der Velden, Michelle Michels, Aryan Vink, Eva van Rooij. Single-cell transcriptomics provides insights into hypertrophic cardiomyopathy. Cell Rep. 2022;39.
Chaffin M, Papangeli I, Simonson B, Akkad AD, Hill MC, Arduini A, Fleming SJ, Melanson M, Hayat S, Kost-Alimova M, Atwa O, Ye J, Bedi KC Jr, Nahrendorf M, Kaushik VK, Stegmann CM, Margulies KB, Tucker NR, Ellinor PT. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature. 2022;608:174–80.
Andrew LKIS, Junedh A, Prabhakar SA, Konstantin Z, Lulu L, Geetika B, Andrea B, Gabriella S, Cameran J, Emily T, Stacey LR, Maxim NA, Kory JL. Single-cell transcriptomics reveals cell-type specific diversification in human heart failure. Nat Cardiovasc Res. 2022;1:263–80.
Wang L, Yu P, Zhou B, Song J, Li Z, Zhang M, Guo G, Wang Y, Chen X, Han L, Hu S. Single-cell reconstruction of the adult human heart during heart failure and recovery reveals the cellular landscape underlying cardiac function. Nat Cell Biol. 2020;22:108–19.
Reichart D, Lindberg EL, Maatz H, Miranda AMA, Viveiros A, Shvetsov N, Gartner A, Nadelmann ER, Lee M, Kanemaru K, Ruiz-Orera J, Strohmenger V, DeLaughter DM, Patone G, Zhang H, Woehler A, Lippert C, Kim Y, Adami E, Gorham JM, Barnett SN, Brown K, Buchan RJ, Chowdhury RA, Constantinou C, Cranley J, Felkin LE, Fox H, Ghauri A, Gummert J, Kanda M, Li R, Mach L, McDonough B, Samari S, Shahriaran F, Yapp C, Stanasiuk C, Theotokis PI, Theis FJ, van den Bogaerdt A, Wakimoto H, Ware JS, Worth CL, Barton PJR, Lee YA, Teichmann SA, Milting H, Noseda M, Oudit GY, Heinig M, Seidman JG, Hubner N, Seidman CE. Pathogenic variants damage cell composition and single cell transcription in cardiomyopathies. Science. 2022;377:1984.
Linnarsson S, Teichmann SA. Single-cell genomics: coming of age. Genome Biol. 2016;17:97.
Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17:175–88.
Acknowledgements
Funding
No funding or sponsorship was received for this study or publication of this article.
Author Contributions
Xuejing Yu, Xianggui Yang, and Jinjin Cao contributed to the conception and literature searches. Xuejing Yu contributed to the writing. All authors reviewed the manuscript and provided a final approval for this version to be submitted and published.
Disclosures
Xuejing Yu, Xianggui Yang, and Jinjin Cao have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Yu, X., Yang, X. & Cao, J. Application of Single-Cell Genomics in Cardiovascular Research. Cardiol Ther 12, 101–125 (2023). https://doi.org/10.1007/s40119-023-00303-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-023-00303-y