
Abstract
The functional integrity of the endothelium is essential for vascular health. In addition to maintaining a delicate balance between vasodilation and vasoconstriction, the endothelium has numerous other complex roles involved in the maintenance of vascular homeostasis. Chronic exposure to cardiovascular risk factors and oxidative stress results in an imbalance in these functions, creating an environment that favors reduced vasodilation and a proinflammatory and prothrombic state. The involvement of endothelial dysfunction in all stages of the cardiovascular continuum makes it an important target for treatment. One of the major endothelial-derived factors involved in the maintenance of endothelial function is nitric oxide (NO). Angiotensin-converting enzyme (ACE) inhibitors increase NO production both directly and indirectly by preventing production of angiotensin II (which diminishes NO production) and inhibiting the degradation of bradykinin (which stimulates local release of NO). Among the ACE inhibitors, perindopril appears to have the greatest effects on bradykinin and has demonstrated efficacy in a number of markers of endothelial dysfunction including arterial stiffness and progression of atherosclerosis. There is also strong evidence supporting the use of perindopril-based therapy for the treatment of hypertension and for reducing the risk of cardiovascular morbidity and mortality in a wide range of patients across the cardiovascular continuum.
Funding: The journal’s Rapid Service Fee was funded by Servier.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cardiovascular diseases (CVD) remain a major cause of premature death and chronic disability worldwide [1]. They represent the final stage in a continuum of cardiovascular risk factors, subclinical organ damage, and eventually cardiovascular, cerebrovascular, and renal events (Fig. 1) [2, 3].

Adapted from Dzau et al. [2]
The cardiovascular continuum. MI myocardial infarction, CHF congestive heart failure, ESRD end-stage renal disease.
The earliest vascular abnormality in the continuum is endothelial dysfunction. Endothelial cells respond to both mechanical stimuli and hormonal signals from the circulation to regulate vascular structure and function by modulating processes such as vascular smooth muscle cell contraction and proliferation, platelet function, coagulation, and monocyte adhesion [4]. Any factors altering the ability of the endothelium to act as a physical barrier or metabolize, synthesize, and release vasoactive mediators will result in endothelial dysfunction. Endothelial dysfunction is a reliable prognostic indicator of cardiovascular events, independently of traditional cardiovascular risk factors in patients with atherosclerotic coronary disease or at high cardiovascular risk [5]. In addition to the correction of cardiovascular risk factors, re-establishing balanced endothelial function is therefore an important target for treatment.
Two pathways that play a major role in endothelial homeostasis, and consequently endothelial dysfunction, are those of the renin–angiotensin and bradykinin–nitric oxide (NO) systems. This review will examine the role of angiotensin II and bradykinin in the pathophysiology of endothelial dysfunction, and the importance of endothelial dysfunction as a target for the prevention of cardiovascular disease. Evidence for the protective effects of angiotensin-converting enzyme (ACE) inhibitors, via their actions on both the angiotensin II and bradykinin pathways, will be presented, with a particular focus on perindopril as one of the most widely studied representatives of the ACE-inhibitor class. This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Pathophysiology of Endothelial Dysfunction and its Role in the Cardiovascular Continuum
Under disease conditions, including the presence of cardiovascular risk factors, the endothelium undergoes functional and structural alterations that result in it losing its cardioprotective role and becoming proatherosclerotic (Fig. 2). A characteristic feature of endothelial dysfunction is impaired NO bioavailability [6]. This may be a consequence of either reduced NO production by endothelial nitric oxide synthase (eNOS) or increased breakdown of NO by reactive oxygen species (ROS) [7, 8]. Following its production and release by the endothelium, NO diffuses to surrounding tissues where it exerts important cardiovascular protective effects including relaxation of media smooth muscle cells, prevention of leukocyte adhesion and migration into the arterial wall, and prevention of muscle cell proliferation, platelet adhesion and aggregation, and adhesion molecule expression [7, 8]. Loss of NO is one of the earliest events that leads to the onset and progression of oxidative stress in the vasculature.

Adapted from Dzau et al. [2]
Oxidative stress and endothelial dysfunction in cardiovascular diseases. LDL-C low-density lipoprotein cholesterol, BP blood pressure, NO nitric oxide, ACE angiotensin-converting enzyme, AngII angiotensin II, SMC smooth muscle cells.
Angiotensin II and the Endothelium
Angiotensin II plays a central role in endothelial dysfunction [9]. In addition to increasing blood pressure via vasoconstriction, it elicits a number of adverse effects on the vascular wall, the majority of which occur through its actions on the angiotensin II type 1 (AT1) receptor. Elevated levels of endothelial angiotensin II stimulate the generation of ROS responsible for NO breakdown [10] and reduce eNOS-derived NO by promoting eNOS uncoupling through monocyte-dependent S-glutathionylation [11]. Angiotensin II also produces conditions favorable for atherogenesis by upregulating endothelial receptors for oxidized low-density lipoprotein (OxLDL), stimulating OxLDL uptake, and enhancing OxLDL-mediated ROS generation. The resulting oxidative stress promotes vascular smooth muscle cell proliferation and collagen deposition, leading to thickening of the vascular media and narrowing of the vascular lumen. Oxidative stress also damages the endothelium, leading to endothelial cell apoptosis [12]. It increases receptors for vascular endothelial growth factors and matrix metalloproteinases, which may account for increased endothelial permeability and vascular remodeling [13,14,15]; angiotensin II-induced vascular remodeling may also occur via activation of the AT2 receptor [16]. Oxidative stress increases the expression of plasminogen activator inhibitor type 1, thereby favoring thrombosis [17], and cytokines, increasing the adhesiveness of the endothelium and subsequently the recruitment and binding of inflammatory cells to the endothelial surface, leading to inflammation and thrombosis [14, 15]. When not sufficiently counterbalanced by the actions of bradykinin, these molecular effects of angiotensin II result in endothelial dysfunction.
Bradykinin and the Endothelium
The systemic hemodynamic and localized endothelial effects of bradykinin are mediated by bradykinin receptor 1 (B1R) and bradykinin receptor 2 (B2R). B2R is constitutively and abundantly expressed in the endothelium, whereas B1R is expressed at low levels and upregulated in response to stress such as ischemia/reperfusion injury, chronic inflammation, or diabetes [18, 19]. In endothelial cells, activation of B2R by bradykinin induces the release of NO, prostacyclin, endothelium-derived hyperpolarizing factor (EDHF), and tissue plasminogen activator, which exert diverse physiological actions on the cardiovascular system, including regulation of vascular tone and local blood flow to organs, coagulation, fibrinolysis, and water–electrolyte balance [20]. Activation of B1R also induces NO-mediated vasodilatation [21]. Experimental studies have demonstrated a protective role of B2R on cardiovascular function, as a result of its opposing effects on angiotensin II AT1 receptor activation [22]. In direct opposition to angiotensin II, bradykinin causes vasodilatation, exerts an anti-inflammatory effect, decreases ROS, and has anti-fibrinolytic and antithrombotic effects [23].
Bradykinin-induced vasodilation has been shown to be the result of at least two pathways: bradykinin-induced NO and prostaglandin release, and the hyperpolarization of the membrane through the EDHF pathway [24, 25]. The anti-inflammatory effects and reduction in ROS induced by bradykinin have been shown to be mediated through the release of NO, a key regulator of endothelial function. Besides anti-inflammation, the release of NO also results in an anti-apoptotic effect of bradykinin [23].
Vascular fibrinolytic balance, which determines the formation and degradation of thrombi, is maintained by the balance between tissue plasminogen activator (T-PA) and plasminogen activator type 1 (PAI-1) [26]. Increases in PAI-1 and decreases in T-PA are associated with thrombus formation and are a major risk factor for the development of cardiovascular events [27]. Bradykinin has been shown to increase T-PA concentration independently of other molecular pathways [28, 29], and consequently has strong anti-thrombotic properties.
ACE Inhibition and Endothelial Function
Rationale for Use in Endothelial Dysfunction
As endothelial dysfunction promotes atherosclerosis and cardiovascular events in patients with cardiovascular risk factors, improving endothelial function is an important treatment target. Thus, antihypertensive compounds, beyond their ability to reduce blood pressure, should ideally have additional properties that specifically restore endothelial function. A detailed description of the pleiotropic properties of available antihypertensive agents is outside the scope of this article, but diuretics and β-blockers show little evidence of being able to improve endothelial dysfunction in patients with hypertension or coronary artery disease (CAD). Calcium channel blockers have been shown to reverse impaired endothelium-dependent vasodilation, mainly in the microcirculation by improving NO bioavailability [30, 31].
The drugs expected to have the greatest influence on endothelial dysfunction are the renin–angiotensin system inhibitors. Both ACE inhibitors and angiotensin receptor blockers (ARBs) prevent the action of angiotensin II, ACE inhibitors by preventing its formation and ARBs by blocking the actions of angiotensin II at AT1 receptors on bloods vessels and other tissues such as the heart. By targeting angiotensin-converting enzymes, ACE inhibitors not only enhance the availability of NO by inhibiting the conversion of angiotensin I to angiotensin II, but also by increasing the availability and preventing the degradation of bradykinin (Fig. 3). ARBs do not inhibit ACE and therefore do not increase bradykinin levels via this pathway. In this respect, it is noteworthy that ACE inhibitors, but not ARBs, have consistently demonstrated reduced cardiovascular mortality and all-cause death compared with placebo in a broad range of hypertensive patients [32,33,34]. Furthermore, a high proportion of the studies demonstrating the benefits of ACE inhibitors on cardiovascular mortality were performed with perindopril.

ACE inhibitor mode of action in balanced endothelial function
Evidence for Target Organ Protection
Endothelial Function
The possibility that ACE inhibitors could improve endothelial function was initially raised following reports from experimental studies that the renin–angiotensin system, and in particular angiotensin II, plays a major role in inhibiting NO production and activity, mainly by inducing ROS generation [35]. However, the role of angiotensin II is not the only explanation for the efficacy of ACE inhibitors regarding endothelial function, as the literature suggests a superiority of ACE inhibitors over ARBs. As described above, bradykinin also has significant effects on the endothelium. The ability of ACE inhibitors to increase bradykinin tissue concentration by blocking its degradation is an important mechanism for vascular protection, especially in the presence of cardiovascular risk factors or CAD. In the presence of these risk factors, chronic overexpression of tissue ACE disrupts the angiotensin II/bradykinin balance, increasing levels of angiotensin II and decreasing tissue levels of bradykinin, the net result being endothelial dysfunction.
The involvement of the bradykinin pathway in endothelial responses to ACE inhibition is well documented. For example, in the PERTINENT (PERindopril Thrombosis INflammation ENdothelial dysfunction and neurohormonal activation Trial) substudy of EUROPA (EURopean trial On reduction of cardiac events with Perindopril in stable CAD), which included 1200 patients with stable CAD, human umbilical vein endothelial cells (HUVECs) were incubated with serum taken from EUROPA patients both at baseline and after 1 year of treatment with either placebo or perindopril 10 mg [36]. Perindopril upregulated eNOS protein activity by 27% (P < 0.05). Furthermore, these changes were highly correlated with increased plasma bradykinin levels (P < 0.05), which were restored to a normal physiological level similar to that of healthy volunteers. PERTINENT showed that perindopril improved endothelial dysfunction by increasing bradykinin and reducing angiotensin II levels, thereby restoring the angiotensin II/bradykinin balance.
The effect of ACE inhibitors on the angiotensin II/bradykinin balance varies markedly among different agents. ACE has two different catalytic domains, one cleaving angiotensin I, and the other inactivating bradykinin. ACE inhibitors have a greater affinity for the bradykinin than for the angiotensin I binding site, supporting the concept that these compounds are primarily inhibitors of bradykinin degradation rather than inhibitors of angiotensin II production [37]. Within the ACE inhibitor class, perindopril has the highest selectivity for the bradykinin binding site when comparing enalapril, perindopril, quinapril, ramipril, and trandolapril [37]. The combination of a lipophilic profile, high tissue affinity, and a high bradykinin/angiotensin I selectivity ratio provide perindopril with a complete and long-lasting inhibition of bradykinin degradation, and may offer a greater physiological impact compared with other ACE inhibitors [38]. In a head-to-head trial comparing ramipril, quinapril, trandolapril, and enalapril, perindopril provided the greatest reductions in lipopolysaccharide-induced endothelial apoptosis [38]. These results are consistent with data from the PERTINENT study, which reported a 31% reduction in apoptosis in HUVECs from patients treated with perindopril [36]. While apoptosis is a marker of endothelial cell death, circulating endothelial progenitor cells (CEPC) are an important marker of endothelial regeneration [39]. Thus, to completely prevent endothelial dysfunction, optimal treatment should not only inhibit inflammation and apoptosis, but also stimulate CEPC production. In experimental conditions, perindopril has been shown to increase the number of CEPCs in the spontaneously hypertensive rat with hind limb ischemia, while the ARB losartan had no effect on the number of CEPCs [40, 41].
Arterial Stiffness
Arterial stiffness of the large vessels is a marker of subclinical target organ damage and an independent predictor of future cardiovascular events and mortality beyond blood pressure alone [42]. Perindopril has been shown to produce a blood pressure-independent decrease in arterial stiffness, suggesting that it is able to reverse adverse structural remodeling of the arterial wall. In the DAPHNET study (Diabetes Artery Perindopril Hypertension Normalization Excess sTiffness), which was conducted in 57 hypertensive patients with diabetes, increasing the perindopril dosage from 4 to 8 mg resulted in significantly higher carotid distensibility and a significantly lower carotid internal diameter and Young’s elastic modulus [43]. Stepwise regression analysis confirmed that these parameters of carotid stiffness were significantly associated with the dosage of perindopril, and not with the 24-h blood pressure.
Stiffening of large vessel walls with age or disease leads to an increase in central aortic blood pressure. In the CAFÉ (Conduit Artery Function Evaluation) substudy of ASCOT-BPLA (Anglo Scandinavian Cardiac Outcomes Trial, Blood Pressure Lowering Arm), perindopril and amlodipine were superior to the combination of atenolol and bendroflumethiazide in lowering central blood pressure and pulse pressure, despite similar reductions in brachial blood pressure [44], which may account for the better cardiovascular outcomes observed in the perindopril/amlodipine arm.
Atherosclerosis
The improvements in functional and structural vascular alterations observed with perindopril have also been shown to contribute to the prevention of atherosclerosis. This was demonstrated in PERSPECTIVE (PERindopril’S Prospective Effect on Coronary aTherosclerosis by IntraVascular ultrasound Evaluation) [45], a substudy of EUROPA, which evaluated the effect of perindopril on progression of coronary atherosclerosis by coronary angiography and intravascular ultrasound. There was no evidence of CAD progression with either perindopril or placebo [46], but a post hoc analysis of data from 118 patients found that the effect of perindopril on coronary plaque progression/regression was dependent on the degree of calcification [45]. Thus, coronary plaques with no or little calcium (0–25%) regressed on perindopril, but did not change on placebo (− 0.33 ± 1.74 vs. − 0.03 ± 1.66, respectively; P = 0.04). Plaques with moderate calcium content (group 25–50%) did not change. Plaques with high calcium content (group 5–100%) progressed similarly. The important conclusion of the study is therefore that noncalcified plaques may be amenable to regression with perindopril treatment. PERSPECTIVE is the only study to have provided evidence for the ability of an ACE inhibitor to prevent progression of coronary atherosclerosis in vivo.
Ventricular Remodeling
Left ventricular remodeling in which left ventricular volumes increase leading to a rise in wall stress and altered contractile function is recognized as a precursor of cardiovascular events, particularly heart failure, and a strong predictor of mortality. The PREAMI (Perindopril and Remodelling in Elderly with Acute Myocardial Infarction) study demonstrated that progressive left ventricular remodeling can occur in elderly post-myocardial infarction (MI) patients with preserved left ventricular ejection fraction (LVEF), and that it is possible to prevent such remodeling with perindopril 8 mg/day [47]. Left ventricular remodeling occurred in 27.7% of perindopril- vs. 51.2% of placebo-treated patients (P < 0.001).
Cardioprotective Actions of Perindopril Translate into Improved Cardiovascular Outcomes
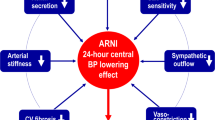
The strong evidence that ACE inhibitors, and in particular perindopril, provide cardiovascular protection beyond their blood pressure-lowering effects, benefits patients at all stages of the cardiovascular continuum. Clinical trials investigating the effects of perindopril on morbidity and mortality have demonstrated efficacy in a wide range of patients at risk of cardiovascular events, from those with uncomplicated hypertension to those with established cardiovascular disease (Fig. 4).

Clinical trials investigating the effects of perindopril on morbidity and mortality have provided evidence of benefits throughout the cardiovascular disease continuum
Hypertension
The ASCOT trial clarified the role of ACE inhibition in the reduction of cardiovascular events in patients with hypertension without cardiovascular disease. In the blood pressure-lowering arm of the trial, patients (n = 19,257) at moderate cardiovascular risk were randomized to amlodipine 5–10 mg (with the addition of perindopril 4–8 mg as required) or the ß-blocker atenolol with the addition of diuretic as required [48]. After a median of 5.5 years, ASCOT was stopped prematurely because patients in the amlodipine/perindopril group showed an 11% reduction in all-cause mortality (P = 0.0247), compared with the beta-blocker/diuretic regimen. The mean blood pressure in the amlodipine/perindopril group was 2.7/1.9 mmHg lower than in the beta-blocker/diuretic group. However, this difference could not entirely account for the differences in outcome. The CAFÉ study (n = 2199) was a prespecified substudy of ASCOT that evaluated the effects of the two ASCOT treatment regimens on central aortic pressure [44]. Despite similar reductions in brachial artery blood pressure, both central aortic blood pressure and central aortic pulse pressure were significantly reduced with amlodipine/perindopril vs. beta-blocker/diuretic (24.3 and 23.0 mmHg, respectively, both P < 0.0001). Moreover, central aortic pulse pressure was significantly associated with an increased risk of cardiovascular events or procedures and the development of renal impairment. These findings suggest that central aortic pressure lowering by perindopril and amlodipine combination therapy disrupts the pathophysiological continuum, thereby slowing progression to the cardiovascular continuum in hypertensive patients. The results of the ASCOT Legacy study, which followed UK participants from the original ASCOT trial for a median of 16 years, confirmed the long-term beneficial effects on mortality of antihypertensive treatment with an amlodipine/perindopril treatment regimen [49]. In the 3975 UK patients in the blood pressure-lowering arm, there were fewer cardiovascular deaths [adjusted hazard ration (HR) 0.79, 0.67–0.93, P = 0.0046] among those assigned to amlodipine/perindopril-based treatment compared with atenolol/diuretic-based treatment (P = 0.022).
Type 2 Diabetes
In patients with diabetes, the ADVANCE (Action in Diabetes and Vascular disease: PreterAx and DiamicroN-MR Controlled Evaluation) study evaluated the clinical benefits of background blood pressure lowering with a fixed combination of perindopril and indapamide on top of standard management in a cohort of 11,140 patients [41, 50]. Over a mean follow-up of 4.3 years, treatment with perindopril/indapamide fixed combination reduced the incidence of the composite primary endpoint of macrovascular (non-fatal stroke, non-fatal MI, and cardiovascular death) and microvascular (new or worsening nephropathy and retinopathy) events by 9% vs. placebo (P < 0.05). Cardiovascular and all-cause mortality were also reduced by 18% and 14%, respectively (both P < 0.05). The beneficial effects of fixed combination of perindopril and indapamide on the primary endpoint were similar in the hypertensive and non-hypertensive patients in ADVANCE [50].
Coronary Artery Disease
CAD is a consequence of endothelial dysfunction and atherosclerosis. The findings of the EUROPA trial provided clear evidence for the efficacy of perindopril in secondary prevention in patients with stable CAD. Patients with documented evidence of CAD, but without uncontrolled hypertension or heart failure (n = 12,218), were randomly allocated to either perindopril or placebo on top of other standard preventative therapies (beta-blockers, lipid-lowering agents, and antiplatelet agents). After a mean follow-up of 4.2 years, there was a significant 20% reduction in the primary endpoint (cardiovascular mortality, non-fatal MI, or resuscitated cardiac arrest) in favor of perindopril (P < 0.001) [51, 52]. As described previously, the EUROPA findings may reflect the restorative action of perindopril on endothelial function. In the PERTINENT substudy of EUROPA, perindopril was found to reduce the rate of endothelial cell apoptosis by 31% (P < 0.05) [36, 38], an effect that could contribute, at least in part, to explaining the results of the main EUROPA study.
Stroke
The PROGRESS (Perindopril pROtection aGainst REcurrent Stroke Study) was the first large-scale study to investigate the role of ACE inhibition in secondary prevention of stroke in patients with a history of stroke or transient ischemic attack. Patients (n = 6105) were randomized to active treatment (perindopril 4 mg/day plus the addition of indapamide at the discretion of the physician) or placebo. There were no blood pressure entry criteria, although subjects with uncontrolled hypertension could receive antihypertensive agents other than an ACE inhibitor. After a mean follow-up of 3.9 years, the relative risk of stroke was reduced by 28% in the perindopril-based regimen vs. placebo (P = 0.0001). Perindopril was also associated with a 27% reduction in major cardiovascular events (non-fatal MI or death from cardiovascular disease) and a 38% reduction in non-fatal MI [53, 54].
Myocardial Infarction
The PREAMI trial was designed to evaluate the hypothesis that ACE inhibition in older patients with acute MI and preserved LVEF would attenuate left ventricular remodeling and reduce the occurrence of heart failure and death [47]. A total of 1252 patients 65 years or older with a LVEF ≥ 40% and recent acute MI were randomized to perindopril 8 mg/day or placebo for 12 months. Ventricular remodeling occurred in 126 (28%) patients in the perindopril group and in 226 (51%) patients in the placebo group. At the end of the study, the composite primary endpoint of death, hospitalization for heart failure, and left ventricular remodeling showed a relative risk reduction of 38% in favor of perindopril (P < 0.001) [47].
Heart Failure
Heart failure represents one of the final stages of the cardiovascular continuum, but successful management can still improve prognosis. The PEP-CHF (Perindopril in Elderly People with Chronic Heart Failure) trial was designed to improve understanding of the effects of ACE inhibition in diastolic heart failure. Patients aged > 70 years diagnosed with diastolic heart failure (n = 870) and preserved LVEF received either perindopril 2 mg/day or placebo, on top of standard management [55]. After 1 year, the event rate was lower than expected and reduced the statistical power of the study, but perindopril still showed a strong tendency to reduce the primary endpoint (all-cause mortality, unplanned heart failure-related hospitalization with a minimum follow-up of 1 year) with a relative risk reduction of 31% (P = 0.055).
Implications for Clinical Practice
Endothelial dysfunction is characterized by a shift in the balance of the endothelial functions that maintain vascular homeostasis to one that favors reduced vasodilation, and a proinflammatory and prothrombic state. It is observed from the earliest stages of the cardiovascular disease continuum and its effects are cumulative, worsening endothelial dysfunction associated with progressive vascular damage. An important priority in cardiovascular disease prevention is therefore to correct or reduce the actions of the impaired endothelium that are driving the cardiovascular continuum. This will involve reducing or eliminating the triggering risk factors for endothelial dysfunction as well as attempting to restore balance to the disrupted endothelial homeostasis.
Certain cardiovascular drugs have pleiotropic effects that improve endothelial function independently of their primary indication. For example, it is well known that statins exert beneficial effects independent of cholesterol lowering, decreasing inflammation and thrombogenesis and stabilizing the atherosclerotic plaque. Among the antihypertensive agents, the ACE inhibitors appear to have the strongest effects on endothelial dysfunction. In addition to correcting elevated blood pressure, a triggering risk factor for the condition, they are also able to increase levels of NO by inhibiting the production of angiotensin II and in particular by inhibiting the degradation of bradykinin. Among the ACE inhibitors, perindopril exerts the greatest selectivity for the bradykinin binding site. It also has good lipid solubility for enhanced tissue penetration and a long half-life resulting in excellent ACE inhibition throughout a 24-h cycle after a single daily dose. These properties may account, at least in part, for the documented superiority of this agent over other renin–angiotensin system inhibitors in the prevention and treatment of all stages of the cardiovascular continuum from hypertension to the most severe forms of cardiovascular disease.
Recently updated European guidelines for the management of arterial hypertension emphasize that initial therapy should comprise a combination of two drugs, usually an ACE inhibitor or ARB in combination with a calcium channel blocker or thiazide-type diuretic, preferably as a single-pill combination, based on evidence from high-quality randomized controlled trials or meta-analyses [56]. This strategy has been implemented to ensure that target blood pressure levels are achieved more rapidly in a greater number of patients with improved treatment adherence. It is well established that regardless of antihypertensive therapy, much greater treatment effects can be achieved by adding a drug from another class than by doubling the dose of the same drug [57]. It is also known that combining drugs with complementary mechanisms of action can minimize adverse events and improve tolerability relative to the use of higher doses of one component. By attenuating the deleterious effects of endothelial dysfunction in addition to lowering blood pressure, the early initiation of agents such as ACE inhibitors and calcium channel blockers could interrupt and slow the development of endothelial dysfunction and progression to clinical disease at all stages of the cardiovascular continuum.
Conclusions
The involvement of endothelial dysfunction in all stages of the cardiovascular continuum makes it an important target for treatment. NO is essential for endothelial function and its production is increased both directly and indirectly via inhibition of angiotensin II production and reduced degradation of bradykinin by the ACE inhibitors. Among the ACE inhibitors, perindopril appears to have the greatest effects on bradykinin and has demonstrated efficacy on a number of markers of endothelial dysfunction including arterial stiffness and progression of atherosclerosis. Evidence from a number of large, randomized controlled trials supports the use of perindopril-based therapy for the treatment of hypertension and for reducing the risk of cardiovascular morbidity and mortality in a wide range of patients across the cardiovascular continuum.
References
Roth GA, Johnson C, Abaiobir A, et al. Global, regional, and national burden of cardiovascular diseases for 10 causes, 1990 to 2015. J Am Coll Cardiol. 2017;70(1):1–25.
Dzau VJ, Antman EM, Black HR, et al. The cardiovascular disease continuum validated: clinical evidence of improved patient outcomes part I: pathophysiology and clinical trial evidence (risk factors through stable coronary artery disease). Circulation. 2006;114:2850–70.
O’Rourke MF, Safar ME, Dzau V. The cardiovascular continuum extended: aging effects on the aorta and microvasculature. Vasc Med. 2010;15(6):461–8.
Galley HF, Webster NR. Physiology of the endothelium. Br J Anaesth. 2004;93:105–13.
Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation. 2005;111:363–8.
Tomasian D, Keaney JF, Vita JA. Antioxidants and the bioactivity of endothelium-derived nitric oxide. Cardiovasc Res. 2000;47(3):426–35.
Lüscher TF, Barton M. Biology of the endothelium. Clin Cardiol. 1997;20:II3–10.
Taddei S, Ghiadoni L, Virdis A, Versari D, Salvetti A. Mechanisms of endothelial dysfunction: clinical significance and preventive non-pharmacological therapeutic strategies. Curr Pharm Des. 2003;9:2385–402.
Dzau VJ. Theodore Cooper Lecture. Tissue angiotensin and pathobiology of vascular disease: a unifying hypothesis. Hypertension. 2001;37(4):1047–52.
Griendling KK, Lassègue B, Murphy TJ, Alexander RW. Angiotensin II receptor pharmacology. Adv Pharmacol. 1994;28:269–306.
Kossmann S, Hu H, Steven S, et al. Inflammatory monocytes determine endothelial nitric-oxide synthase uncoupling and nitro-oxidative stress induced by angiotensin II. J Biol Chem. 2014;289:27540–50.
Li DT, Zhang YC, Philips MI, Sawamura T, Mehta JL. Upregulation of endothelial receptor for oxidized low-density lipoprotein (LOX-1) in cultured human coronary artery endothelial cells by angiotensin II type 1 receptor activation. Circ Res. 1999;84:1043–9.
Watanabe T, Barker TA, Berk BC. Angiotensin II and the endothelium: diverse signals and effects. Hypertension. 2005;45:163–9.
Tamarat R, Silvestre JS, Durie M, Levy BI. Angiotensin II angiogenic effect in vivo involves vascular endothelial growth factor- and inflammation-related pathways. Lab Investig. 2002;82:747–56.
Arenas IA, Xu Y, Lopez-Jaramillo P, Davidge ST. Angiotensin II-induced MMP-2 release from endothelial cells is mediated by TNF-alpha. Am J Physiol Cell Physiol. 2004;286:C779–84.
Levy BI, Benessiano J, Henrion D, et al. Chronic blockade of AT2-subtype receptors prevents the effect of angiotensin II on the rat vascular structure. J Clin Investig. 1996;98:418–25.
Ridker PM, Gaboury CL, Conlin PR, Seely EW, Williams GH, Vaughan DE. Stimulation of plasminogen activator inhibitor in vivo by infusion of angiotensin II. Evidence of a potential interaction between the renin–angiotensin system and fibrinolytic function. Circulation. 1993;87:1969–73.
Kayashima Y, Smithies O, Kakoki M. The kallikrein–kinin system and oxidative stress. Curr Opin Nephrol Hypertens. 2012;21(1):92–6.
Kuhr F, Lowry J, Zhang Y, et al. Differential regulation of inducible and endothelial nitric oxide synthase by kinin B1 and B2 receptors. Neuropeptides. 2010;44(2):145–54.
Su JB. Kinins and cardiovascular diseases. Curr Pharm Des. 2006;12:3423–35.
Su JB, Hoüel R, Héloire F, et al. Stimulation of bradykinin B(1) receptors induces vasodilation in conductance and resistance coronary vessels in conscious dogs: comparison with B(2) receptor stimulation. Circulation. 2000;101:1848–53.
Su JB. Different cross-talk sites between the renin–angiotensin and the kallikrein–kinin systems. J Renin Angiotensin Aldosterone Syst. 2014;15:319–28.
Taddei S, Bortolotto L. Unraveling the pivotal role of bradykinin in ACE inhibitor activity. Am J Cardiovasc Drugs. 2016;16:309–21.
Baudin B, Berard M, Carrier JL, et al. Vascular origin deter-mines angiotensin I-converting enzyme expression in endothelial cells. Endothelium. 1997;5(1):73–84.
Taddei S, Versari D, Cipriano A, et al. Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpo-larizing factor in essential hypertensive patients. J Am Coll Cardiol. 2006;48(3):508–15.
Bentley JP, Asselbergs FW, Coffey CS, et al. Cardiovascular risk associated with interactions among polymorphisms in genes from the renin–angiotensin, bradykinin, and fibrinolytic systems. PLoS One. 2010;5(9):e12757.
Wiman B, Andersson T, Hallqvist J, et al. Plasma levels of tissue plasminogen activator/plasminogen activator inhibitor-1 complex and von Willebrand factor are significant risk markers for recurrent myocardial infarction in the Stockholm Heart Epidemiology Program (SHEEP) study. Arterioscler Thromb Vasc Biol. 2000;20(8):2019–23.
Rahman AM, Murrow JR, Ozkor MA, et al. Endothelium-derived hyperpolarizing factor mediates bradykinin-stimulated tissue plasminogen activator release in humans. J Vasc Res. 2014;51(3):200–8.
Taddei S, Virdis A, Ghiadoni L, et al. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation. 1998;97(22):2222–9.
Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care. 2009;32(Suppl 2):S314–21.
Virdis A, Ghiadoni L, Taddei S. Effects of antihypertensive treatment on endothelial function. Curr Hypertens Rep. 2011;13:276–81.
Blood Pressure Lowering Treatment Trialists’ Collaboration. Blood pressure-dependent and independent effects of agents that inhibit the renin–angiotensin system. J Hypertens. 2007;25(5):951–8.
van Vark LC, Bertrand M, Akkerhuis KM, et al. Angiotensin-converting enzyme inhibitors reduce mortality in hypertension: a meta-analysis of randomized clinical trials of renin–angiotensin–aldosterone system inhibitors involving 158,998 patients. Eur Heart J. 2012;33(16):2088–97.
Strauss MH, Hall AS. Angiotensin receptor blockers do not reduce risk of myocardial infarction, cardiovascular death, or total mortality: further evidence for the ARB-MI paradox. Circulation. 2017;135(22):2088–90.
Virdis A, Neves MF, Amiri F, Touyz RM, Schiffrin EL. Role of NAD(P)H oxidase on vascular alterations in angiotensin II-infused mice. J Hypertens. 2004;22:535–42.
Ceconi C, Fox KM, Remme WJ, EUROPA Investigators, PERTINENT Investigators and the Statistical Committee, et al. ACE inhibition with perindopril and endothelial function. Results of a substudy of the EUROPA study: PERTINENT. Cardiovasc Res. 2007;73:237–46.
Ceconi C, Francolini G, Olivares A, Comini L, Bachetti T, Ferrari R. Angiotensin-converting enzyme (ACE) inhibitors have different selectivity for bradykinin binding sites of human somatic ACE. Eur J Pharmacol. 2007;577:1–6.
Ceconi C, Francolini G, Bastianon D, et al. Differences in the effect of angiotensin-converting enzyme inhibitors on the rate of endothelial cell apoptosis in vitro and in vivo studies. Cardiovasc Drugs Ther. 2007;21(6):423–9.
Goon PK, Boos CJ, Lip GY. Circulating endothelial cells: markers of vascular dysfunction. Clin Lab. 2005;51:531–8.
Ferrari R, Guardigli G, Ceconi C. Secondary prevention of CAD with ACE inhibitors: a struggle between life and death of the endothelium. Cardiovasc Drug Ther. 2010;24:331–9.
You D, Cochain C, Loinard C, et al. Hypertension impairs postnatal vasculogenesis: role of antihypertensive agents. Hypertension. 2008;51:1537–44.
Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all-cause mortality with arterial stiffness: a systematic review and meta-analysis. J Am Coll Cardiol. 2010;55:1318–27.
Tropeano AI, Boutouyrie P, Pannier B, et al. Brachial pressure-independent reduction in carotid stiffness after long-term angiotensin-converting enzyme inhibition in diabetic hypertensives. Hypertension. 2006;48(1):80–6.
Williams B, Lacy PS, Thom SM, CAFE Investigators; Anglo-Scandinavian Cardiac Outcomes Trial Investigators; CAFE Steering Committee and Writing Committee, et al. Differential impact of blood pressure-lowering drugs on central aortic pressure and clinical outcomes: principal results of the Conduit Artery Function Evaluation (CAFE) study. Circulation. 2006;113:1213–25.
Bruining N, deWinter S, Roelandt J, et al. Coronary calcium significantly affects quantitative analysis of coronary ultrasound: importance for atherosclerosis progression/regression studies. Coron Artery Dis. 2009;20:409–14.
Rodriguez-Granillo GA, Vos J, Bruining N, Investigators of the EUROPA Study, et al. Long-term effect of perindopril on coronary atherosclerosis progression (from the perindopril’s prospective effect on coronary atherosclerosis by angiography and intravascular ultrasound evaluation [PERSPECTIVE] study). Am J Cardiol. 2007;100(2):159–63.
Ferrari R. Effects of angiotensin-converting enzyme inhibition with perindopril on left ventricular remodeling and clinical outcome: results of the randomized Perindopril and Remodeling in Elderly with Acute Myocardial Infarction (PREAMI) Study. Arch Intern Med. 2006;166:659–66.
Poulter NR, Wedel H, Dahlöf B, ASCOT Investigators, et al. Role of blood pressure and other variables in the differential cardiovascular event rates noted in the Anglo-Scandinavian Cardiac Outcomes Trial- Blood Pressure Lowering Arm (ASCOT-BPLA). Lancet. 2005;366:907–13.
Gupta A, Mackay J, Whitehouse A, et al. Long-term mortality after blood pressure-lowering and lipid-lowering treatment in patients with hypertension in the Anglo-Scandinavian Cardiac Outcomes Trial (ASCOT) Legacy study: 16-year follow-up results of a randomised factorial trial. Lancet. 2018;392(10153):1127–37.
Patel A; ADVANCE Collaborative Group. Effects of a fixed combination of perindopril and indapamide on macrovascular and microvascular outcomes in patients with type 2 diabetes mellitus (the ADVANCE trial): a randomised controlled trial. Lancet. 2007;370:829–40.
Fox KM. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double-blind, placebo-controlled, multicentre trial (the EUROPA study). Lancet. 2003;362:782–8.
Scheen AJ. Etude Clinique du mois. L’étude EUROPA: protection cardio-vasculaire avec le périndopril chez les patients avec coronaropathie stable. Rev Med Liège. 2003;58:713–6.
PROGRESS Collaborative Group. Randomised trial of a perindopril-based blood-pressure-lowering regimen among 6,105 individuals with previous stroke or transient ischaemic attack. Lancet. 2001;358:1033–41.
Scheen AJ. Etude Clinique du mois. Protection secondaire de l’accident vasculaire cérébral par le périndopril: l’étude PROGRESS. Rev Med Liège. 2001;56:792–5.
Cleland JG, Tendera M, Adamus J, Freemantle N, Polonski L, Taylor J, PEP-CHF Investigators. The perindopril in elderly people with chronic heart failure (PEP-CHF) study. Eur Heart J. 2006;27:2338–45.
Williams B, Mancia G, Spiering W, Authors/Task Force Members, et al. 2018 ESC/ESH Guidelines for the management of arterial hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension: The Task Force for the management of arterial hypertension of the European Society of Cardiology and the European Society of Hypertension. J Hypertens. 2018;36(10):1953–2041.
Wald DS, Law M, Morris JK, Bestwick JP, Wald NJ. Combination therapy versus monotherapy in reducing blood pressure: meta-analysis on 11,000 participants from 42 trials. Am J Med. 2009;122(3):290–300.
Acknowledgements
Funding
No funding or sponsorship was received for this study. The journal’s Rapid Service Fee was funded by Servier.
Medical Writing Assistance
Writing assistance was provided by freelance medical writer Jenny Grice and funded by Servier.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Disclosures
Arnaud Ancion, Julien Tridetti, Mai-Linh Nguyen Trung, Cécile Oury, and Patrizio Lancellotti have nothing to disclose.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any studies with human participants or animals performed by any of the authors.
Data Availability
Data sharing is not applicable to this article as no datasets were generated or analyzed during the current study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Enhanced Digital Features
To view enhanced digital features for this article go to https://doi.org/10.6084/m9.figshare.9896510.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution-NonCommercial 4.0 International License (http://creativecommons.org/licenses/by-nc/4.0/), which permits any non-commercial use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ancion, A., Tridetti, J., Nguyen Trung, ML. et al. A Review of the Role of Bradykinin and Nitric Oxide in the Cardioprotective Action of Angiotensin-Converting Enzyme Inhibitors: Focus on Perindopril. Cardiol Ther 8, 179–191 (2019). https://doi.org/10.1007/s40119-019-00150-w
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-019-00150-w