
Abstract
Familial hypercholesterolemia (FH) is an inherited disorder of lipid metabolism characterized by premature cardiovascular disease. It is one of the most common metabolic disorders affecting humans. There are two clinical manifestations: the milder heterozygous form and more severe homozygous form. Despite posing a significant health risk, FH is inadequately diagnosed and managed. As the clinical outcome is related to the degree and duration of exposure to elevated low-density lipoprotein cholesterol (LDL-C) levels, early treatment is vital. Diagnosis can usually be made using a combination of clinical characteristics such as family history, lipid levels, and genetic testing. Mutations in the gene encoding the LDL receptor (LDLR), apolipoprotein B, the pro-protein convertase subtilisin/kexin 9 (PCSK9), and LDLR adaptor protein are the commonest abnormalities. Early identification and treatment of patients, as well as screening of relatives, helps significantly reduce the risk of premature disease. Although statins remain the first-line therapy in most cases, monotherapy is usually inadequate to control elevated LDL-C levels. Additional therapy with ezetimibe and bile acid sequestrants may be required. Newer classes of pharmacotherapy currently under investigation include lomitapide, mipomersen, and monoclonal antibodies to PCSK9. Lipoprotein apheresis may be required when multiple pharmacotherapies are inadequate, especially in the homozygous form. Effective early detection and treatment of the index individual and initiation of cascade screening will help reduce the complications associated with FH. In this article, we review the disease of FH, complexity of diagnosis and management, and the challenges faced in preventing the significant morbidity and mortality associated with it.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Familial hypercholesterolemia (FH) is an inherited autosomal dominant disorder of premature atherosclerosis [1]. It was first described in the late 1930s by Carl Mϋller, a Norwegian clinician in a landmark paper by putting forward the idea that hypercholesterolemia and tendinous xanthomas were linked to cardiovascular (CV) disease through a single gene inheritance [2]. The clinical phenotype was further characterized in 1964 into the mild heterozygous (HeFH) and more severe homozygous (HoFH) forms with dominant inheritance [3]. While Dr Mϋller had initially postulated that causal and prophylactic intervention may be beneficial, it was almost 50 years before this became a possibility.
Historically, untreated HeFH begins to manifest its clinical consequences in the fourth decade in men and fifth decade in women. Patients with HoFH, however, may suffer significant CV events as early as in the first decade of life. By early adulthood, these patients without treatment have 100 times greater mortality risk from CV disease resulting from coronary atherosclerosis or supra-vascular aortic valve calcification as compared to those without FH [1]. In general, those with HoFH do not survive past 30 years without therapeutic interventions [1]. Ever since the work of Goldstein and Brown in 1983, it had been generally accepted that FH has a population prevalence of 1:500 in HeFH and 1:1,000,000 in HoFH [4]. These figures, however, vastly underestimate the true prevalence. Recent work by the European Atherosclerosis Society (EAS) noted widening inconsistency of diagnosis, with 71 % of patients with FH diagnosed in Netherlands, 43 % in Norway, and only 6 % in Spain [5]. In addition, these figures were only obtainable for 22 countries out of approximately 200 in the world. Despite posing a significant health and economic burden, the true prevalence is yet to be accurately determined. Data suggest that HoFH may actually affect 1 in 160,000–300,000 people [6]. The Copenhagen General Population Study and EAS estimate an HeFH prevalence of 1:200 instead of the historical 1:500, which would mean that between 14 and 34 million individuals may in fact be affected worldwide [5]. This represents a staggering number whose risk may be easily reduced with lipid-lowering therapy, yet are currently being overlooked.
In this article, we review the disease of FH, complexity of diagnosis and management, and the challenges faced in preventing the significant morbidity and mortality associated with it. This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Pathophysiology
Abnormal elevations in plasma low-density lipoprotein cholesterol (LDL-C) in FH are largely due to functional genetic mutations in the LDL receptor (LDLR), and less frequently apolipoprotein B (ApoB) or gain-of-function mutations of pro-protein convertase subtilisin/kexin 9 (PCSK9). Autosomal recessive FH is associated with LDLR-adaptor protein (LDLRAP) mutation [1]. These mutations mainly result in four molecular defects: failed internalization of bound LDL, reduced LDL binding, lack of receptor expression, and failure of LDLR to reach the plasma membrane [7]. Hepatocyte and peripheral cell-regulated endocytosis of LDL-C occurs via ApoB, a protein that transports lipids through the lymphatic and circulatory system. Genetic mutation of ApoB leads to decreased LDL affinity for the LDLR and reduced hepatic LDL plasma clearance. FH is also associated with raised levels of lipoprotein (a) [Lp(a)] by an unknown mechanism [8]. Lp(a) levels are higher in HoFH than HeFH. Defective high-density lipoprotein (HDL)-driven cholesterol efflux may also be associated with low levels of HDL cholesterol in HoFH [8]. If FH is inadequately treated or untreated, it leads to cholesterol retention in the arterial wall with foam cell formation within the intima, accelerated occlusive atherosclerosis, and early CV disease [5, 9].
HeFH is characterized by only one normal allele and may be due to loss-of-function LDLR mutation, gain-of-function mutation in PCSK9, or mutations in ApoB affecting the LDLR-binding domain [5]. To date, more than 1200 LDLR mutations are documented in this gene [9]. HoFH, on the other hand, is relatively rare with patients having two mutant copies of the gene, with impaired LDLR function being the most common and associated with devastating clinical consequences. Other variations of the clinical HoFH phenotype include compound heterozygotes with mutations at two different sites of the same gene or double heterozygotes, for example having one copy of ApoB gene defect and one copy of an LDL-R defect.
Clinical Presentation and Diagnosis
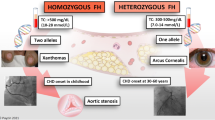
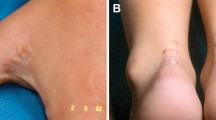
In the primary care setting, the diagnosis of FH may be easily missed. Patients are commonly only identified after experiencing a CV event at an unexpected age or as a result of a family member being diagnosed. HoFH is likely when LDL-C levels are greater than 13 mmol/L (500 mg/dL) in adults and 11 mmol/L (420 mg/dL) in children [10]. Triglyceride (TG) levels may remain normal [11]. LDLR-negative HoFH patients usually have a worse prognosis, succumbing to complications by the second decade [6]. In addition to cholesterol levels, diagnosis may be supported by features seen during physical examination such as tendon xanthomas on the dorsal aspect of the metacarpophalangeal joints or at the calcaneal tendon, and arcus senilis. The EAS diagnostic criterion for HoFH is outlined in Table 1 [12].
For early effective prevention of CV disease, HeFH should be suspected in asymptomatic individuals with elevated plasma total cholesterol (TC) or LDL-C concentrations, relevant clinical history, physical signs, or a family history of premature coronary disease. TC levels greater than 6.7 mmol/L (260 mg/dL) and 7.5 mmol/L (290 mg/dL)—or untreated LDL-C levels greater than 4 mmol/L (155 mg/dL) and 4.9 mmol/L (190 mg/dL)—warrant further investigation in children and adults, respectively, after exclusion of secondary causes of hypercholesterolemia such as diabetes, hypothyroidism, and obesity [5, 13].
Although clinical and biochemical findings provide valuable diagnostic information, specialized genetic testing is often required. With the advent of DNA-based mutation screening methods, direct detection of mutations in the LDLR, ApoB, PCSK9, and LDLRAP genes are now widely utilized. However, it is reported that up to 40 % of patients with a clinical diagnosis may in fact not have a genetic diagnosis of their hyperlipidemia [14, 15]. This may be due to causal mutations yet to be discovered, insensitivity of current testing, or even misdiagnosis using the biochemical and clinical criteria. The National Institute for Health and Care Excellence (NICE) guidelines in the United Kingdom (UK) recommend referral to an FH specialist post-diagnosis for initiation of cascade testing [10]. Cascade testing allows identification of people at risk by the process of family tracing, using LDL-C levels and/or a DNA test if the mutation has already been identified in the index individual/proband. DNA-positive relatives identified through cascade screening may not have elevated LDL-C levels and not fulfill the clinical diagnostic criteria [5, 15, 16]. This raises a complex dilemma of treating without elevation of LDL-C levels. On balance, due to the lifetime exposure and risk, lipid-lowering therapy should be considered and ultimately decided with the patient themselves.
To date, however, there is no single internationally accepted criterion for the diagnosis of FH. The three most commonly used and validated diagnostic tools are the Simon Broome Register in the UK, the Dutch Lipid Clinic Network criteria, and the United States (US) Make Early Diagnosis to Prevent Early Death (MEDPED) [17–19]. The Simon Broome and the Dutch criteria take into account the family history, clinical history, physical signs, LDL-C concentration, and molecular genetic testing results to classify the likelihood of FH. The main difference between the two being that the Simon Broome criteria recognizes DNA evidence of a mutation as evidence of definite FH, while the Dutch require one other criteria to be met in addition to the molecular diagnosis for definite FH. All three systems also use different age cutoffs for defining premature coronary heart disease (CHD). The US MEDPED criterion uses age-specific thresholds of TC concentration to diagnose FH with TC cutoff levels being lower in the first-, second- and third-degree relatives than the general population. The main disadvantages of its use are that clinical characteristics and FH-associated gene mutation are not considered.
Treatment
The goal of treatment in FH is to reduce the risk of atherosclerotic heart disease. All patients with FH, whether heterozygous or homozygous, should undergo a comprehensive program of lifestyle modification. This has three primary objectives: dietary changes, exercise and behavioral therapy [13]. Dietary changes include reduction in saturated fats, transfats, and cholesterol. Referrals should be made to a nutritionist and smoking cessation encouraged. Risk factors such as hypertension, diabetes, and smoking should be addressed. Although these measures are of benefit, they are unlikely to lower the LDL-C levels sufficiently and direct intervention is invariably needed to reduce the levels.
Treatment for HeFH
To date, no randomized controlled trials have been conducted assessing the benefit of lipid-modification treatment on CHD events among patients with HeFH. As such, much of the pharmacotherapy currently used is based on an extrapolation of data among non-FH patients or from a few observational studies conducted principally using hydroxymethylglutaryl co-enyzme A (HMG CoA) reductase inhibitors (statins) in patients with FH [20, 21]. Statins remain the only class of lipid-lowering therapy to reduce total and coronary mortality post-myocardial infarction. It is widely accepted that maximal potent statin dose should be initiated as first-line therapy in adults post-diagnosis of HeFH [5]. If started prophylactically in early adulthood, statin use has been shown to lower the risk of CHD by up to 80 % [20]. NICE recommends a target of 50 % reduction in LDL-C concentration [22]. In accordance with the European Society of Cardiology, the EAS has outlined new LDL targets [5]:
-
children <3.5 mmol/L (<135 mg/dL);
-
adults <2.5 mmol/L (<100 mg/dL);
-
adults with CHD or diabetes <1.8 mmol/L (<70 mg/dL).
Although the 2013 American College of Cardiology/American Heart Association Guidelines do not recommend a target LDL level, high-intensity statin therapy is recommended in asymptomatic arteriosclerotic CV disease (ASCVD) with LDL-C levels greater than 190 mg/dL where tolerated [23]. Due to its net benefit in terms of reduction in ASCVD risk versus potential adverse effects, statin therapy is recommended for those at increased risk.
Despite maximal dose statin therapy, LDL-C levels may yet remain elevated. The addition of ezetimibe (a cholesterol absorption inhibitor) to statins or as monotherapy reduces CVD events with NICE and EAS both recommending its co-administration, which may help reduce LDL-C levels by 60 to 70 % in total [18, 21]. Results from the recent Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT; ClinicalTrials.gov #NCT00202878) presented at the American Heart Association 2014 Scientific Sessions have demonstrated modest benefit with combined use of ezetimibe and simvastatin in stable patients post-acute coronary syndrome (ACS) [72]. Significant reduction in the primary end point—a composite of CV death, major coronary events, and stroke—by 6.4 % compared to monotherapy with statins was noted (P = 0.016). The absolute risk reduction was 2 %. Ezetimibe may also be used as monotherapy in patients unable to tolerate statins.
Bile acid sequestrants, such as cholestyramine, colestipol, or colesevelam, may be added as a third agent in very high-risk patients with CHD, type 2 diabetes, or LDL-C levels greater than 1.8 mmol/L (>70 mg/dL) [18]. Colesevelam is preferred due to its lower gastrointestinal side effect profile than cholestyramine and colestipol, both of which are also associated with poor patient compliance and significant multi-drug interactions [24].
Despite a lack of evidence of the clinical benefit of niacin co-administration, high-dose therapy has until recently been recommended, especially in the USA and Canada where its use had doubled [23, 25, 26]. Its availability in Europe has started to decline due to two recently published neutral CVD outcome studies. The AIM-HIGH (ClinicalTrials.gov #NCT00120289) study was prematurely stopped at 3 years due to a lack of clinical benefit of niacin therapy when compared to placebo in patients with established CV disease who were already being treated with statins and ezetimibe [27]. One of the criticisms of the study was that it was not powered to determine a difference in CV events. The recent randomized, placebo-controlled HPS2-THRIVE (ClinicalTrials.gov #NCT00461630) trial attempted to address this by recruiting 25,673 patients with atherosclerotic vascular disease and also found no benefit of the addition of niacin therapy to statin-based LDL-C-lowering therapy on major vascular events [28]. Quite worryingly, serious adverse effects were noted involving the gastrointestinal, musculoskeletal, and cutaneous systems. In a subgroup analysis, there was a trend toward improved outcomes in patients with a high baseline LDL (≥58 mg/dL). Routine administration of niacin should be curtailed, although it may still have a role in very select statin-intolerant patients at high risk of CV events who are unable to reduce LDL-C levels optimally despite multiple therapies. This decision should ultimately be made by specialized clinicians with expertise in dyslipidemia.
Although the mechanism of action of fibric acid is not fully understood, its effect is thought to be due to β-oxidation of fatty acids in peroxisomes and mitochondria. Fibrates reduce plasma TG and cholesterol levels, while elevating HDL-C levels [29]. Due to the increased risk of myopathy, rhabdomyolysis, and liver impairment when co-administered with statins, fibrate use should be restricted to patients with raised TG levels (>4.5 mmol/L or 170mg/dL) and low HDL levels only [30]. A recent meta-analysis, however, showed that neither niacin nor fibrate treatment reduced all-cause mortality, CHD mortality, myocardial infarction, or stroke in patients already treated with statins [31].
Patients with very high CV risk, whose LDL-C levels remain elevated despite combination therapies, may be candidates for weekly or bi-weekly adjunctive lipoprotein apheresis, especially if there is evidence of progression of disease. LDL-C and Lp(a) levels may be reduced by 50–75 % and, while effective, availability of this service, high costs, and the inconvenience and invasive nature of this treatment (use of peripheral veins and, occasionally, requirement of a fistula) limit its widespread use [5, 32, 33].
Treatment for HoFH
All patients with HoFH should be initiated on lipid-lowering therapy as early as possible, with LDL-C targets the same as in HeFH [6]. Statins remain the cornerstone of treatment with the observed benefit due to inhibition of hepatic lipoprotein synthesis, up-regulation of LDLR, or an increase in trans-intestinal cholesterol excretion [34]. While no randomized controlled trials have been conducted looking at the end point of CV mortality in HoFH, statins in the non-FH population are known to reduce the incidence of major vascular events in primary prevention, with intensive regimens producing greater reduction than less intensive regimens [35, 36]. In HoFH, observational studies show a dose-dependent effect on LDL-C reduction and maximum tolerable doses should generally be prescribed [37, 38]. The degree of reduction is however less than that seen in HeFH. Although individual response may be variable, LDLR-defective patients may achieve LDL-C reduction of 25 %, while LDLR-negative patients achieve around 15 %, thus suggesting that some receptor function is needed for clinical benefit [6, 39, 40]. Despite this, statin monotherapy does not reduce LDL-C levels sufficiently and co-administration of ezetimibe is often necessary, yielding a further 15–20 % reduction [41].
Medical therapy with statins alone or in combination with other lipid-lowering agents such as ezetimibe, bile acid sequestrants, niacin, or probucol rarely provide an adequate solution, and the majority of patients ultimately require LDL apheresis. In the late 1970s unselective plasmapheresis was used in patients with HoFH in an attempt to control hypercholesterolemia, slow coronary atherosclerosis, and prolong survival with limited success [42, 43]. Subsequently, work began to focus on removing LDL more selectively [44, 45]. Due to its dramatic benefits, extracorporeal removal of LDL-C by lipoprotein apheresis is now the treatment of choice in HoFH. Despite being expensive, time-consuming, and not readily available, the substantial benefits of single treatment reducing LDL-C levels by up to 70 % have been recommended by the EAS and make it cost-effective overall [12].
Liver transplantation was first described in 1983 and has now emerged as the most effective treatment, markedly improving LDL-C levels long term [46, 47]. Surgery provides a liver with functional LDLRs, thereby correcting the molecular defect. It is indicated in patients who despite maximal medical therapy and apheresis fail to reduce LDL-C levels sufficiently. Although successful, it poses significant challenges including procedure-related morbidity and mortality, lack of available donors, and the need for long-term immunosuppression [48]. Although effective, transplantation is not a realistic option for many patients. Due to the limitations of existing therapies, novel lipid-lowering agents are being developed and provide a new avenue for research for the management of FH.
Newer Therapies for FH
Lomitapide
Microsomal TG transfer protein (MTP) localized on the endoplasmic reticulum is an intracellular lipid-transfer protein essential for the assembly and secretion of ApoB-containing chylomicrons and very low-density lipoproteins (VLDL) in the intestines and liver [49]. The concept that pharmacological inhibition of MTP by lomitapide may reduce LDL-C stemmed from the discovery that loss-of-function mutation in the gene encoding MTP results in abetalipoproteinemia, a rare disorder marked by hypocholesterolemia with absent ApoB-containing lipoproteins [50]. Lomitapide binds to MTP in the liver and intestines, inhibiting lipid transfer. It was first approved by the US Food and Drug Administration (FDA) in December 2012 and the European Medicines Agency (EMA) as additional therapy to other lipid-lowering regimes including LDL apheresis in those aged 18 years and over with HoFH.
In a single-arm, open-label phase 3 study of 29 individuals with HoFH, lomitapide was given as an adjunct to other therapies including LDL apheresis [51]. In the intention-to-treat analysis of all 29 patients, LDL-C reduced by 40 % and ApoB by 39 % by week 26. In 23 patients who completed the efficacy phase, lomitapide at maximal tolerated dose reduced plasma LDL-C levels by 50 % and ApoB by 49 % from baseline, although there was considerable variability in the response to the therapy [49]. A potential and as yet uncertain consequence is the increase in hepatic fat during therapy, so serum transaminase levels should be monitored as ApoB-containing lipoproteins accumulate in the liver.
Mipomersen
Mipomersen is an antisense oligonucleotide targeting the messenger ribonucleic acid of ApoB with eventual reduction in the secretion of VLDL [6]. It was approved by the FDA in January 2013 based on a randomized, double-blinded, placebo-controlled study of 51 patients with HoFH [52]. Mipomersen was seen to reduce LDL-C levels by 25 %, ApoB by 27 %, and Lp(a) by 31 % when compared to placebo (stable low-fat diet, pre-existing maximal tolerated lipid-lowering drugs, and those not receiving apheresis). However, doubts remain due to its significant side effects as the EMA rejected its use in March 2013 [53]. EMA noted that a high proportion of patients stopped the drug within 2 years due to liver dysfunction and there were uncertainties of its effect on long-term CV outcome. Numerical imbalance in overall CV events, major adverse cardiac events, and CV-associated hospitalization compared to placebo were of considerable concern [6]. The commonest reason for discontinuation of mipomersen is injection site reactions, such as erythema, pain, pruritus, and local swelling, being seen in around 78 % of patients [54]. Further evaluation of the long-term efficacy, outcome, and side effects is urgently required.
PCSK9 Inhibitors
LDLR is a cell-surface receptor playing a vital role in regulating the circulating cholesterol levels. It allows LDL binding with subsequent endocytosis and lysosomal degradation, following which it is recycled back to the plasma membrane [55]. PCSK9 is a serine protease secreted in the liver and acts by inhibiting this LDLR recycling with consequential reduction in LDL clearance. PCSK9 binds to LDLR resulting in co-internalization and degradation of the receptor within the lysosome. This inverse relationship between plasma PCSK9 levels and LDLR results in reduced LDL-C metabolism and eventual hypercholesterolemia [56]. Gain-of-function mutation in the PCSK9 gene was first discovered in 2003, linking it to a phenotype identical to classical FH [57]. What caused widespread interest was the discovery that loss-of-function mutation resulted in low plasma levels of LDL-C and ApoB and thus protection against CHD events [58]. Since then, several large epidemiological studies have confirmed this association. The ARIC study in 2006 showed that loss-of-function mutations in the PCSK9 gene were associated with lower LDL-C levels in African Americans (by 28 %) and white individuals (by 15 %), with consequential lower risk of coronary events [hazard ratio (HR) 0.11, 95 % confidence interval (CI) 0.02–0.81, P = 0.03 for African Americans; HR 0.50, 95 % CI 0.32–0.79, P = 0.003 for white individuals] [59]. A 37 mg/dL reduction in LDL-C levels resulting from these mutations was associated with an 88 % reduction in incident CHD in African Americans and a 21 mg/dL reduction with a 47 % reduction in white individuals. Analysis of Copenhagen Heart Studies combined further supported the notion that PCSK9 mutations were associated with LDL-C reduction and CHD events [60]. Combined data from three Copenhagen studies (prospective, cross sectional, and case control) showed an LDL-C reduction of 13 % and risk reduction of 30 % for ischemic heart disease (IHD) in the mutation carrier group. Meta-analysis which included seven general population studies noted 12 % lower LDL-C levels in carriers compared to non-carriers [mean LDL difference in fixed effects model −0.43 mmol/L (95 % CI −0.48 to −0.38 mmol/L)]. Combined analysis showed 23 % risk reduction in IHD [odds ratio (OR) 0.77 (95 % CI 0.65 to 0.92)] in patients with PCSK9 mutation compared to non-carriers [60].
Therapeutic inhibition of PCSK9 was first hypothesized by Berge and colleagues in 2006 [61]. They reported that specific mutations in the genome could be associated with hypocholesterolemia and possibly increase the response to statin, with additional LDL-C lowering. This has led to the development of several new treatments targeting the PCSK9 pathway [6]. Phase 3 trials on monoclonal antibody therapies are underway and have yielded promising results so far. In the MENDEL-2 (ClinicalTrials.gov #NCT01763827) trial, biweekly and monthly evolocumab treatment was compared to placebo and ezetimibe [62]. Biweekly 140 mg evolocumab treatment reduced LDL-C levels from baseline by an average of 57 % (95 % CI −59.5 to −54.6 %), while ezetimibe reduced levels by an average of 17.8 % (95 % CI −21.0 to −14.5 %). Monthly treatment with 420 mg evolocumab reduced levels by 56.1 % (95 % CI −58.3 to −53.9 %), with ezetimibe reducing by an average 18.6 % (95 % CI −21.6 to −15.5 %). The GAUSS-2 (ClinicalTrials.gov #NCT01763905) trial demonstrated similar benefit in patients intolerant to statins [63]. Mean percent reductions in LDL-C from baseline with biweekly evolocumab was 56.1 % (95 % CI −59.9 to −52.4 %), and with ezetimibe 18.1 % (95 % CI −23.1 to − 13.1 %). Monthly treatment showed similar results to the MENDEL-2 trial, with 52.6 % LDL-C reduction with evolocumab (95 % CI −55.7 to −49.5 %) and 15.1 % with ezetimibe (95 % CI −19.3 to −10.9 %) [62].
The randomized double-blinded placebo-controlled LAPLACE-2 (ClinicalTrials.gov #NCT01763866) study found that treatment with evolocumab reduced LDL-C levels by 66–75 % and 63–75 % versus placebo in patients already receiving moderate and high-intensity statin therapy, respectively [64]. Similar results have also been seen in the DESCARTES-2 (ClinicalTrials.gov #NCT01516879) trial [65]. In patients with HoFH, evolocumab therapy caused a significant reduction in LDL-C by 30.9 % (95 % CI −43.9 to −18.0 %, P < 0.0001) in the recently published TESLA Part B (ClinicalTrials.gov #NCT01588496) trial in patients with LDLR mutations in both alleles of which at least one was defective [66]. Benefit has also been noted in patients with HeFH. The RUTHERFORD-2 (ClinicalTrials.gov #NCT01763918) trial randomly assigned 331 patients with HeFH to evolocumab or placebo every 2 or 4 weeks [67]. Being assigned to the monoclonal antibody resulted in 56–63 % reduction in LDL-C after 12 weeks in patients already receiving a statin and 60 % of those who were taking ezetimibe. These trials have shown promising results, especially in patients with high cholesterol who cannot tolerate statins or where other lipid-lowering therapy does not reduce the LDL-C levels enough. The long-term safety of these compounds and very low DL-C remain unclear and data from larger phase 3 or 4 trials are keenly awaited.
Gene Therapy
The primary defect in 85 % of FH cases is mutation or deletion of the LDLR-encoding gene responsible for removing LDL-C via endocytosis and intracellular degradation [11]. The availability of functional LDLRs post-liver transplantation in HoFH has provided backing to the idea that gene replacement may constitute an important strategy for treatment. In animal models, viral vector-based gene transfer has been seen to result in overexpression of LDLR with long-term stabilization in hypercholesterolemia [68]. A pilot study in 1995 in the USA used recombinant retroviruses in five patients with HoFH with inconsistent results [69]. Since then, stepwise technological advances and refinements have been made. Gene therapy using vectors based on serotype 8 of the adeno-associated virus (AAV8) is one of the other methods under development [70]. Although it remains at an investigational stage, gene therapy does provide an exciting new avenue for future research, especially for patients with HoFH who are LDL-R defective.
Challenges Faced by the Clinician
Despite the significant progress in our understanding, FH remains one of the most poorly diagnosed and managed diseases in the world. Early detection and management is not only cost-effective, but also preserves life. However, achieving this remains a considerable challenge. The discovery of elevated blood cholesterol may be opportunistic or may be sought during cascade screening of relatives of an affected individual. Furthermore, a diagnosis of FH should be considered in cases of premature CHD or among first-degree relatives or individuals with premature CHD.
FH is overexpressed among those with CHD occurring in about 1:10 people with CHD compared with 1:200–300 of the general population. While the norm is to initiate high-intensity statin therapy following an ACS irrespective of cholesterol levels, it is unlikely that patients with FH reach LDL-C goals of 70 mg/dL. Several factors make the diagnosis of FH difficult. Firstly, clinicians do not think about this and, as cardiology is largely a procedure-driven specialty and with a drive in most health-care systems to reduce bed stay, a detailed family history and an elevated LDL-C are more likely to be overlooked as compared to a hemoglobin or a creatinine value post-percutaneous coronary intervention. Furthermore, there is a broad spectrum of LDL-C levels in FH which make this difficult [71]. There may also be a lack of understanding of FH among cardiologists in particular and overconfidence in the benefits of statin therapy. Most patients are either simply discharged on moderate or high-intensity statin, cholesterol levels ignored, or simply left to the patient’s own general practitioner to investigate further, who is likely to know less than the cardiologist. An integrated care pathway where data flows seamlessly post-ACS to medical personnel directly involved in the patient’s care would be one solution, especially if abnormal results triggered data clinical tasks such as further investigation or referrals. Efforts should also be made to ensure that there is a better uptake of a nurse-led physician-supported cardiac rehabilitation program where every patient goes through the system and is adequately followed up, as abnormal results could be picked up in the first 3 months post-ACS.
Once a diagnosis of FH is made, focus should shift to family members, but this seldom occurs. Most countries do not possess an integrated system or resources to adequately initiate cascade screening, where the focus is about preserving health, rather than treating the consequences of disease. The organizational and legislative environments are variable according to locality. After gaining informed consent, relatives should be invited to specialist lipid clinics with plasma cholesterol level measurement and genetic assessment. Another challenge is the ability to provide appropriate psychological support to those being screened, and especially asymptomatic children. Advice on contraception and pregnancy needs to be provided, especially as statins need to be avoided in a gestating or lactating patient. The real challenge in delivering FH care is to have an effective care model, with an integration of hospital and community-based services, having an appropriate informational technology setup, education and research of not only the patient but also the physician, and establishing an appropriate family support group. This would provide an effective framework for good clinical practice.
References
Austin MA, Hutter CM, Zimmern RL, Humphries SE. Genetic causes of monogenic heterozygous familial hypercholesterolemia: a HuGE prevalence review. Am J Epidemiol. 2004;160:407–20.
Nutrition classics. Archives of internal medicine, volume 64, October 1939: angina pectoris in hereditary xanthomatosis. By Carl Muller. Nutr Rev. 1987;45:113–5.
Khachadurian AK. The inheritance of essential familial hypercholesterolemia. Am J Med. 1964;37:402–7.
Goldstein JKHH, Brown MS. Familial hypercholesterolemia. 2001st ed. New York: McGraw-Hill; 2001.
Nordestgaard BG, Chapman MJ, Humphries SE, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a.
Cuchel M, Bruckert E, Ginsberg HN, et al. Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–57.
Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–8.
Kontush A, Chapman MJ. Functionally defective high-density lipoprotein: a new therapeutic target at the crossroads of dyslipidemia, inflammation, and atherosclerosis. Pharmacol Rev. 2006;58:342–74.
Watts GF, Gidding S, Wierzbicki AS, et al. Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Int J Cardiol. 2014;171:309–25.
In: Identification and management of familial hypercholesterolaemia (FH). London; 2008.
Hovingh GK, Davidson MH, Kastelein JJ, O’Connor AM. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013;34:962–71.
Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97:3956–64.
Wadden TA, Webb VL, Moran CH, Bailer BA. Lifestyle modification for obesity: new developments in diet, physical activity, and behavior therapy. Circulation. 2012;125:1157–70.
Civeira F, Ros E, Jarauta E, et al. Comparison of genetic versus clinical diagnosis in familial hypercholesterolemia. Am J Cardiol 2008;102:1187–93, 93 e1.
Palacios L, Grandoso L, Cuevas N, et al. Molecular characterization of familial hypercholesterolemia in Spain. Atherosclerosis. 2012;221:137–42.
Huijgen R, Hutten BA, Kindt I, Vissers MN, Kastelein JJ. Discriminative ability of LDL-cholesterol to identify patients with familial hypercholesterolemia: a cross-sectional study in 26,406 individuals tested for genetic FH. Circ Cardiovasc Genet. 2012;5:354–9.
Risk of fatal coronary heart disease in familial hypercholesterolaemia. Scientific Steering Committee on behalf of the Simon Broome Register Group. BMJ 1991;303:893–6.
Williams RR, Hunt SC, Schumacher MC, et al. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–6.
Civeira F. Guidelines for the diagnosis and management of heterozygous familial hypercholesterolemia. Atherosclerosis. 2004;173:55–68.
Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423.
Raal FJ, Pilcher GJ, Panz VR, et al. Reduction in mortality in subjects with homozygous familial hypercholesterolemia associated with advances in lipid-lowering therapy. Circulation. 2011;124:2202–7.
Wierzbicki AS, Humphries SE, Minhas R. Familial hypercholesterolaemia: summary of NICE guidance. BMJ. 2008;337:a1095.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S1–45.
Robinson DM, Keating GM. Colesevelam: a review of its use in hypercholesterolemia. Am J Cardiovasc Drugs. 2007;7:453–65.
Jackevicius CA, Tu JV, Ko DT, de Leon N, Krumholz HM. Use of niacin in the United States and Canada. JAMA Intern Med. 2013;173:1379–81.
Reiner Z, Catapano AL, De Backer G, et al. ESC/EAS guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–818.
Boden WE, Probstfield JL, Anderson T, et al. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N Engl J Med. 2011;365:2255–67.
Landray MJ, Haynes R, Hopewell JC, et al. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12.
Watts GF, Dimmitt SB. Fibrates, dyslipoproteinaemia and cardiovascular disease. Curr Opin Lipidol. 1999;10:561–74.
Staels B, Dallongeville J, Auwerx J, Schoonjans K, Leitersdorf E, Fruchart JC. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–93.
Keene D, Price C, Shun-Shin MJ, Francis DP. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: meta-analysis of randomised controlled trials including 117,411 patients. BMJ. 2014;349:g4379.
Stefanutti C, Morozzi C, Di Giacomo S. Italian multicenter study on low-density lipoprotein apheresis Working Group 2009 survey. Ther Apher Dial. 2013;17:169–78.
Fernandez-Fuertes LF, Tapia Martin M, Nieves Pla I, Novoa Mogollon FJ, Diaz Cremades J. Low-density lipoprotein apheresis using double filtration plasmapheresis: 27-month use in a child with homozygous familial hypercholesterolemia. Ther Apher Dial. 2010;14:484–5.
Le May C, Berger JM, Lespine A, et al. Transintestinal cholesterol excretion is an active metabolic process modulated by PCSK9 and statin involving ABCB1. Arterioscler Thromb Vasc Biol. 2013;33:1484–93.
Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–78.
Baigent C, Blackwell L, Emberson J, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. 2010;376:1670–81.
Marais AD, Raal FJ, Stein EA, et al. A dose-titration and comparative study of rosuvastatin and atorvastatin in patients with homozygous familial hypercholesterolaemia. Atherosclerosis. 2008;197:400–6.
Raal FJ, Pilcher GJ, Illingworth DR, et al. Expanded-dose simvastatin is effective in homozygous familial hypercholesterolaemia. Atherosclerosis. 1997;135:249–56.
Schaefer JR, Kurt B, Sattler A, Klaus G, Soufi M. Pharmacogenetic aspects in familial hypercholesterolemia with the special focus on FHMarburg (FH p.W556R). Clin Res Cardiol Suppl 2012;7:2–6.
Postmus I, Trompet S, Deshmukh HA, et al. Pharmacogenetic meta-analysis of genome-wide association studies of LDL cholesterol response to statins. Nat Commun. 2014;5:5068.
Gagne C, Gaudet D, Bruckert E. Efficacy and safety of ezetimibe coadministered with atorvastatin or simvastatin in patients with homozygous familial hypercholesterolemia. Circulation. 2002;105:2469–75.
Thompson GR, Miller JP, Breslow JL. Improved survival of patients with homozygous familial hypercholesterolaemia treated with plasma exchange. Br Med J (Clin Res Ed). 1985;291:1671–3.
de Gennes JL, Touraine R, Maunand B, Truffert J, Laudat P. Homozygous cutaneo-tendinous forms of hypercholesteremic xanthomatosis in an exemplary familial case. Trial of plasmapheresis ans heroic treatment. Bull Soc Med Hop Paris. 1967;118:1377–402.
Lupien PJ, Moorjani S, Awad J. A new approach to the management of familial hypercholesterolaemia: removal of plasma-cholesterol based on the principle of affinity chromatography. Lancet. 1976;1:1261–5.
Stoffel W, Borberg H, Greve V. Application of specific extracorporeal removal of low density lipoprotein in familial hypercholesterolaemia. Lancet. 1981;2:1005–7.
Starzl TE, Bilheimer DW, Bahnson HT, et al. Heart-liver transplantation in a patient with familial hypercholesterolaemia. Lancet. 1984;1:1382–3.
Shirahata Y, Ohkohchi N, Kawagishi N, et al. Living-donor liver transplantation for homozygous familial hypercholesterolemia from a donor with heterozygous hypercholesterolemia. Transpl Int. 2003;16:276–9.
Malatack JJ. Liver transplantation as treatment for familial homozygous hypercholesterolemia: too early or too late. Pediatr Transplant. 2011;15:123–5.
Rader DJ, Kastelein JJ. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–32.
Wetterau JR, Aggerbeck LP, Bouma ME, et al. Absence of microsomal triglyceride transfer protein in individuals with abetalipoproteinemia. Science. 1992;258:999–1001.
Cuchel M, Meagher EA, du Toit Theron H, et al. Efficacy and safety of a microsomal triglyceride transfer protein inhibitor in patients with homozygous familial hypercholesterolaemia: a single-arm, open-label, phase 3 study. Lancet. 2013;381:40–6.
Raal FJ, Santos RD, Blom DJ, et al. Mipomersen, an apolipoprotein B synthesis inhibitor, for lowering of LDL cholesterol concentrations in patients with homozygous familial hypercholesterolaemia: a randomised, double-blind, placebo-controlled trial. Lancet. 2010;375:998–1006.
Kynamro (mipomersen sodium). 2013. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/002429/human_med_001622.jsp&mid=WC0b01ac058001d124. Accessed 16 Oct 2014.
Thomas GS, Cromwell WC, Ali S, Chin W, Flaim JD, Davidson M. Mipomersen, an apolipoprotein B synthesis inhibitor, reduces atherogenic lipoproteins in patients with severe hypercholesterolemia at high cardiovascular risk: a randomized, double-blind, placebo-controlled trial. J Am Coll Cardiol. 2013;62:2178–84.
Akram ON, Bernier A, Petrides F, Wong G, Lambert G. Beyond LDL cholesterol, a new role for PCSK9. Arterioscler Thromb Vasc Biol. 2010;30:1279–81.
Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714–21.
Abifadel M, Varret M, Rabes JP, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Fasano T, Cefalu AB, Di Leo E, et al. A novel loss of function mutation of PCSK9 gene in white subjects with low-plasma low-density lipoprotein cholesterol. Arterioscler Thromb Vasc Biol. 2007;27:677–81.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Benn M, Nordestgaard BG, Grande P, Schnohr P, Tybjaerg-Hansen A. PCSK9 R46L, low-density lipoprotein cholesterol levels, and risk of ischemic heart disease: 3 independent studies and meta-analyses. J Am Coll Cardiol. 2010;55:2833–42.
Berge KE, Ose L, Leren TP. Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler Thromb Vasc Biol. 2006;26:1094–100.
Koren MJ, Lundqvist P, Bolognese M, et al. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–40.
Stroes E, Colquhoun D, Sullivan D, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–8.
Robinson JG, Nedergaard BS, Rogers WJ, et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–82.
Blom DJ, Hala T, Bolognese M, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19.
Raal FJ, Honarpour N, Blom DJ, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2014;385(9965):341–50.
Raal FJ, Stein EA, Dufour R, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2014;385(9965):331–40.
Wilson JM, Chowdhury NR, Grossman M, et al. Temporary amelioration of hyperlipidemia in low density lipoprotein receptor-deficient rabbits transplanted with genetically modified hepatocytes. Proc Natl Acad Sci USA. 1990;87:8437–41.
Grossman M, Rader DJ, Muller DW, et al. A pilot study of ex vivo gene therapy for homozygous familial hypercholesterolaemia. Nat Med. 1995;1:1148–54.
Chen SJ, Sanmiguel J, Lock M, et al. Biodistribution of AAV8 vectors expressing human low-density lipoprotein receptor in a mouse model of homozygous familial hypercholesterolemia. Human Gene Ther Clin Dev. 2013;24:154–60.
Harada-Shiba M, Arai H, Oikawa S, et al. Guidelines for the management of familial hypercholesterolemia. J Atheroscler Thromb. 2012;19:1043–60.
IMPROVE-IT Trial: a comparison of ezetimibe/simvastatin versus simvastatin monotherapy on cardiovascular outcomes after acute coronary syndromes. 2014. Available from: http://my.americanheart.org/professional/Sessions/ScientificSessions/ScienceNews/SS14-Late-Breaking-Clinical-Trials_UCM_468855_Article.jsp. Accessed 3 Mar 2015.
Acknowledgments
No funding or sponsorship was received for this study or publication of this article. All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this manuscript, take responsibility for the integrity of the work as a whole, and have given final approval for the version to be published.
Conflict of interest
Professor Ray has received honoraria (modest) for consulting and is a member of the speaker’s bureaus and advisory committees from Pfizer Inc., AstraZeneca Pharmaceuticals LP, Merck Sharp & Dohme, Abbott Laboratories, Roche Therapeutics Inc., Regeneron Pharmaceuticals, Inc., Aegerion Pharmaceuticals, Inc., The Sanofi-Aventis Group, Novartis Pharmaceuticals Corporation, Novo Nordisk Inc., Eli Lilly and Co., Daiichi-Sankyo, Inc., Amgen Inc., and Bristol-Myers Squibb Co. Dr Najam has not received any honoraria for consulting or other related purposes.
Compliance with ethics guidelines
This article is based on previously conducted studies and does not involve any new studies of human or animal subjects performed by any of the authors.
Open Access
This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0), which permits use, duplication, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Najam, O., Ray, K.K. Familial Hypercholesterolemia: a Review of the Natural History, Diagnosis, and Management. Cardiol Ther 4, 25–38 (2015). https://doi.org/10.1007/s40119-015-0037-z
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40119-015-0037-z