
Abstract
The aim of this work is to study the structural, electrical, NMR, NBO and quantum parameters of the adsorption of CO molecule on pristine, As, B and BAs doped (4, 4) armchair aluminum nitride nanotubes (AlNNTs). The considerable changes in the adsorption energies, gap energies, global hardness, NBO, and NMR parameters show the high sensitivity and significant change of the electronic properties of AlNNTs to doping of As, B, BAs atoms and CO molecule adsorption. The results reveal that the As, B, and BAs atoms replaced in AlNNTs are good candidates for CO adsorption and increase the adsorption of CO molecule on the surface of nanotube. The gap energy of all adsorption models is in the range of 3.51–4.45 eV. The analysis of NMR parameters indicate that the CSI values of Al and N sites around the adsorption position of the AIV model decrease significantly from the original values, while they increase significantly from the original values for the AII and BII models. The NBO charge values of CO molecule in the AIV model are negative while they are positive for the other models. The positive values of NBO charge prove that CO molecule is an electron acceptor and decrease charge density around nanotube surface.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
With the rapid growth of industrial expansion and the increase in automotive emissions, combustion or natural gas manufacturing, the environmental pollution has significantly increased which is very dangerous for human health. Therefore, monitoring environmental characteristics and providing a reliable accurate and low-level detection limit of pollutant gas in the environment are of great importance for the modern society [1, 2]. Among all the pollutants, carbon monoxide (CO) is a more hazardous gas in the atmosphere and is to be monitored [3]. Carbon monoxide is a common industrial hazard resulting from the incomplete burning of natural gas and any other material containing carbon such as gasoline, kerosene, oil, propane, coal, or wood and coke ovens. When the concentration of carbon monoxide increases in the atmosphere, it becomes dangerous for the human body and other life forms [2–5]. Some research groups have investigated the detection and adsorption of CO gas with various methods [6–9]. After the discovery and synthesis of carbon nanotubes (CNTs) [10] and studying all their properties and applications, many researchers have undertaken to investigate other non-carbon based nanotubes that exhibit electronic properties independent of these features. Among these, aluminum nitride nanotubes (AlNNTs) which are neighbors of carbon in the periodic table have been an interesting subject of many studies [11, 12]. After the prediction of AlNNTs by Zhang [13], Tondare et al. [14], other articles reported the synthesis of AlNNTs through different methods [15, 16]. AlNNTs is the largest band-gap semiconductors with the energy of 6.2 eV and have several unique properties such as high thermal conductivity, high electrical resistivity, low thermal expansion coefficient, high dielectric breakdown strength, good mechanical strength, excellent chemical stability, and non-toxicity [17–19]. In recent years, theoretical investigations have shown that AlNNTs is sensitive to NH3, H2, N2, and CO2 gas. Ahmadi et al. showed that the pristine type of these tubes is not sensitive to NH3 and CO molecules, while the O-doped AlNNTs would be a potential candidate for NH3 molecule detection [20–25]. Ahmadi Peyghan et al. calculated structural and electrical parameters of the adsorption of CO gas on the surface of AlNNTs at different parallel electric field strengths and indicated that the electric field effect on the AlNNTs is a suitable method for CO adsorption [26]. In our previous work, we studied the NMR, NQR and structural parameters of AsGa, Ga, C doped on BPNTs and CO adsorption on the surface of pristine and AsGa doped of (4, 4) armchair boron phosphide nanotubes (BPNTs) [27–31]. In this research, we consider eight models (AI–BIV) (see Fig. 1) for the adsorption of CO molecule on pristine, As, B and BAs doped AlNNTs. The structural, quantum parameters, adsorption energies, band gaps of HOMO–LUMO orbitals, NBO and NMR parameters of the (AI–BIV) models have been investigated by using density function theory.

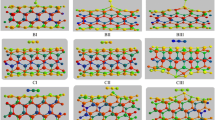
2D views for adsorption CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV): AI vertical adsorption of CO gas on the Al site of pristine AlNNTs via carbon head. AII Vertical adsorption of CO gas on the Al site of B-doped of AlNNTs via carbon head. AIII Vertical adsorption of CO gas on the Al site of As-doped of AlNNTs via carbon head. AIV Vertical adsorption of CO gas on the Al site of BAs-doped of AlNNTs via carbon head. BI Vertical adsorption of CO gas on the N site of pristine AlNNTs via carbon head. BII Vertical adsorption of CO gas on the N site of B-doped of AlNNTs via carbon head. BIII Vertical adsorption of CO gas on the N site of As-doped of AlNNTs via carbon head. BIV Vertical adsorption of CO gas on the N site of BAs-doped of AlNNTs via carbon head
Results and discussion
Structural parameters
The structural parameters: the (Al–N) bond lengths and the (Al–N–Al) bond angles of eight stable (AI–BIV) models (See Fig. 1) are determined from optimized structures and the results are given in Fig. 2. The average (Al–N) bond length is 1.811 Å parallel with the tube axis, and another is 1.818 Å diagonal to the tube axis which is in agreement with other researches [27, 32–35]. With doping of B atom, the bond lengths (Al–N) decrease significantly to 1.48 Å and with doping of As the bond lengths increase significantly to 2.369 Å. The radius of B atom is smaller than Al atoms while the radius of As atom is larger than N atoms. As a result, with doping of B atom the bond lengths (Al–N) decrease significantly from original values whereas with doping of As atom the bond lengths increase significantly from the original values. On the other hand, when the CO molecule is adsorbed on the surface of AlNNTs (the AI–BIV models) the bond length changes slightly from the original values. Comparing the results reveal that doping of (B and As) has a significant effect on the charge transfer from Al and N atoms and yielding asymmetric electronic charge density distribution along B–N and Al–As bonds. The adsorption of CO molecule on the surface of AlNNTs has a slight effect on the charge transfer from Al and N atoms and therefore the bond length slightly changes. The bond angles of neighbor atoms of As-doped in all the models decrease from the original values due to the larger radius of As atom. We found that when CO gas is adsorbed on the Al site of BAs doped on AlNNTs (AIV model) due to strong adsorption of CO molecule the bond length of B–As increases largely to 3.25 Å. The dipole moment results (see Table 1) indicate that the electrical dipole moment of AIV model is more than those of the other models. When a nanotube is placed under interaction with foreign molecules, its atomic charge distribution is easily changed and the center of the positive and negative charges of the nanotube change due to redistribution of the structures of nanotube, consequently leading to the polarization of the nanotube and giving it an induced electric dipole moment. The dipole moment and charge distribution of BIV model are lower than those of other models, whereas the dipole moment and charge distribution of AIV model are more than those of other models.

2D views of bond length and bond angle for CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV)
We calculated the adsorption energies (E ads) of CO molecule/AlNNTs by using Eq. 1 (see Table 1; Fig. 3). As shown in Fig. 3, E ads adsorption of CO molecule on the surface of AlNNTs is in the range of −2.31 to −9.83 kcal/mol. The range of adsorption energy in this research confirms that the adsorption of CO molecule on the surface of AlNNTs is in physisorption form due to the weak van der Waals interaction between the nanotube and CO molecule. Comparing the results shows that the adsorption energy depended on the orientation and location of the CO molecule on the surface of AlNNTs. Therefore, the adsorption energy of AII model is more than those of other models and therefore this model is thermodynamically more stable than the other models. On the other hand, the adsorption energy of BI model is lower than those of the other models and it is more unstable than the other models. It is notable that the adsorption of CO molecule on the Al site of nanotube is stronger than N site. Quantitative results revealed that doping of B and As atoms increase the adsorption of CO molecule on the surface of nanotube.

Diagram of adsorption energy for adsorption CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV)
Quantum molecular descriptors
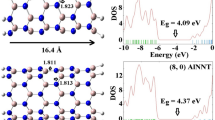
To study the distribution of electron density on the surface of nanotubes, the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO) are calculated for the (AI–BIV) models and the results are shown in Fig. 4. Notations 1 and 2 are used to determine the HOMO and LUMO orbital, respectively. Comparing the results of electron distribution shows that the HOMO orbital density in the AlNNTs is mainly localized on the N atoms, while the density of LUMO orbital is located on the Al ones, resulting in a more strong HOMO/LUMO interaction. By adsorbing CO molecule on the surface of AlNNTs at the (AI–BIV) models the density of HOMO orbital is mostly localized on the surface of nanotubes around the adsorption position. Meanwhile, the density of LUMO orbitals for all models is mostly localized on the CO molecule. Due to acceptor effects of CO molecule, the electron charge density around adsorption sites decreased. It is notable that the density of HOMO orbital on the AIV model is localized on the CO surface, which is in agreement with NBO charge distribution. The quantum parameters including gap energy between HOMO–LUMO orbitals, electronic chemical potential (μ), global hardness (η), electrophilicity index (ω), density of state (DOS plot), global softness (S), electronegativity (χ) and the fractional number of electrons transfer (∆N) of the AlNNTs/CO complex are calculated by Eqs. (4–11). The calculated results are summarized in Table 1 and the plots are shown in Figs. 5, 6 and 7. GaussSum program [32] is used to obtain the plots of density of states (DOS) of all the models. The results of DOS plots for the (AI–BIV) models are shown in Fig. 5. From DOS plots of AI and BI models, it can be concluded that these materials are semiconductors with an E gap of 4.45 and 4.16 eV, respectively. When the B and As atoms are doped on nanotube, the interaction between them is approximately strong and the electronic properties of nanotube are changed significantly, and as a result the E gap of the (AII, AIII,AIV,BII,BIII and BIV) models decrease significantly from original values. The results show that the gap energy of the AIV and BIV models decrease significantly from 4.45 and 4.16 eV to 3.51 and 3.94 eV, respectively. These results indicate that the electronic properties of AlNNTs are very sensitive to the adsorption of CO molecule and doping of BAs. It can be concluded that for CO/AlNNTs complexes, the DOS plots in the gap regions are dramatically affected by the adsorption of CO and doping of As and B atoms.

2D views of HOMO and LUMO orbitals for CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV)

DOS plot for adsorption CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV) and a–b unadsorbed models

Diagram of quantum parameters for adsorption CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV)

Diagram of quantum parameters for adsorption CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV)
The comparison DOS spectrum of (a–d) models of AlNNTs before CO adsorption and (AI–BIV) models of AlNNTs after CO adsorption show that the number of DOS peaks at the HOMO region are five peaks and at the LUMO region are three peaks in range of -10 to 0 eV. By doping B, As and BAs atoms the altitude of all pecks increase significantly from pristine models, and so the photoelectric properties of all doped nanotube increase from original form. In addition, the comparison of DOS spectrum before and after CO adsorption show that the numbers of peaks are constant, the altitude of all pecks change slightly. In other words, the comparison results indicate that the interaction of CO gas with AlNNTs is rather weakly, and that no significant hybridization of the respective orbitals of the two entities takes place; the small interaction is obtained quantitatively in terms of binding energies. On the other hand, we found that the doping of BAs and CO adsorption generated a virtual peak in the region of LUMO orbital.
The HOMO, LUMO and Fermi level (E FL) energy of the (AI–BIV) models are given in Fig. 6. The canonical assumption for Fermi level (E FL) is that in a molecule (at T=0 K) it lies approximately in the middle of the highest occupied molecular orbital (HOMO) and the lowest unoccupied molecular orbital (LUMO). It is worth mentioning that, in fact, the Fermi level energy is the chemical potential of free gas electrons as traditionally defined. Herein, the Fermi level energy of all the models is at the center of the energy gap.
The calculated results of Fig. 7 show that the global hardness of the AIV model is lower than those of other models and therefore this model is more reactive than the other models. The decrease in global hardness and the gap energy proves that the stability of the nanotube/CO complex decreases significantly from original values and the reactivity of the complex increases. The chemical potential values of all the mentioned models are negative which indicate that all structures are stable from a thermodynamic approach (see Table 1). The electrophilicity index (ω) of the AI model is lower than those of the other models, on the other hand, the electrophilicity index of BII and BIII models are slightly more than those of the other models. The electrophilicity indexes are a measure of the electrophilicity power of a molecule. The chemical potential and chemical hardness are key indicators of the overall reactivity of the molecule and are the most fundamental descriptors of charge transfer during a chemical reaction. A molecule with low chemical potential is a good electrophile and an extremely hard molecule has feeble electron acceptability. Consequently, a measure of molecular electrophilicity depends both on the chemical potential and on the chemical hardness. The electronegativity (χ) of nanotube in the AIV model is lower than those of other models are. On the other hand, the electrons transfer (∆N) parameter of the AIV model is more than those of the other models and therefore the charge transfer takes place between the CO and AlNNTs sidewalls.
By using NBO analysis [36] the charge transfer (Δρ) between CO molecule and AlNNTs is calculated from the difference between the natural charge concentration on CO molecule after adsorption and an isolated CO. The results of Table 1 show that the Δρ(NBO) values for AI, AII, AIII, BI, BII, BIII and BIV models have positive values and the results prove that CO molecule is a donor electron. Nevertheless, when CO molecule is adsorbed on the Al site of BAs doped AlNNTs (model AIV), the NBO charge of CO molecule has a negative value due to donor electron effects of BAs doped on nanotube. The results indicate that the CO molecule at the AIV model is an acceptor electron, which is in good agreement with the decrease of electronegativity (χ) and the increase of the electrons transfer ∆N of nanotube.
The NMR parameters of adsorption of CO on AlNNTs
To study the NMR parameters of adsorption of CO molecule on the surface of pristine and As, B atoms doped of AlNNTs, we calculate the CSI and CSA parameters of 13Alnuclei and 15N nuclei by Eqs. (2, 3) and the evaluated NMR parameters are presented in Fig. 8. The results indicate that in the AIV model the CSI values of N51 and Al52 sites decrease significantly from those of pristine models. The negative values of NBO charge distribution for AIV model prove that adsorption of CO gas significantly decreases the charge density around adsorption position and so the CSI tensors at these sites decrease. On the other hand, by doping of B atoms the CSI values at the site of Al21 of AII model increase significantly from those of pristine models due to donor electron effects of B doped. The values of CSI tensors of the (AI, AIII, BI, BII, BIII and BIV) models show slight changes due to CO adsorption.

CSI plot of Al and N site for adsorption CO gas on the surface of pristine and B, As, BAs-doped of (4, 4) armchair model of AlNNTs (models AI–BIV)
Conclusions
The adsorption energy results reveal that the adsorption of CO molecule in vertical direction of Al site of B-doped of AlNNTs is more stable than those of other models from a thermodynamic approach. The adsorption energy of AII model is −9.83 kcal/mol and distance of CO from nanotube is 2.26 Å. The results indicate that when CO molecule is adsorbed on the Al site of BAs doped AlNNTs (model AIV), the NBO charge of CO molecule becomes negative due to donor electron effects of nanotube and BAs doped. These results are in good agreement with the decrease of the NMR parameters of the N51 and Al52 sites of AIV model. On the other hand, the NBO charge of CO molecule at AI, AII, AII, BI, BII, BII, and BIV models are positive and these results prove that CO molecule at these models is a donor electron.
Computational methods
In the current research, to study the interaction and adsorption of carbon monoxide (CO) gas on the surface of AlNNTs, we consider eight models AI, AII, AII, AIV, BI, BII, BIII and BIV (see Fig. 1). The structures of (AI–BIV) representatives models are optimized successfully using GAMESS suite of programs [37] with density functional theory (DFT) and using B3LYP level of theory and 6-31G (d) basis set.
The adsorption energy (E ads) of CO gas on the surface of AlNNTs at the (AI–BIV) models was calculated as follows:
where E AlNNTs-CO was obtained from the scan of the potential energy of the AlNNTs/CO, E AlNNTs is the energy of the optimized AlNNTs structure, and E CO is the energy of optimized CO gas.
Subsequently, the CS tensors were calculated from the optimized structures by using the same level of the theory. Furthermore, the chemical shielding (CS) tensors at the sites of 13Al, 15N nuclei were calculated based on the gauge included atomic orbital (GIAO) approach [38]. The calculated CS tensors in the principal axes system (PAS) (σ 33 > σ 22 > σ 11) were converted to measurable NMR parameters, chemical shielding isotropic (CSI) and chemical shielding anisotropic (CSA) by using Eqs. (2) and (3), respectively [28–31].
Form results of HOMO and LUMO energies, Fermi level energy (E FL), chemical potential (μ), global hardness(η), electrophilicity index (ω), gap energy (E gap), global softness (S), electronegativity (χ) and the fractional number of electrons transfer ∆N of the nanotubes were calculated as follows:
where I (−E HOMO) is the ionization potential and A (−E LUMO) the electron affinity of the molecule [31, 39–42].
References
Prockop, L.D., Chichkova, R.I.: Carbon monoxide intoxication: an updated review. J. Neurol. Sci. 262, 122–130 (2007)
Buckley, N.A., Isbister, G.K., Stokes, B., Juurlink, D.N.: Hyperbaric oxygen for carbon monoxide poisoning. Toxicol. Rev. 24, 75–92 (2005)
Omaye, S.T.: Metabolic modulation of carbon monoxide toxicity. Toxicology 180, 139–150 (2002)
Donnay, A.: A True Tale of A Truly Haunted House. Ghostvillage.com. Retrieved 2008, pp. 12–16 (2004)
Ernst, A., Zibrak, J.D.: Carbon monoxide poisoning. N. Engl. J. Med. 339, 1603–1608 (1998)
Zhuiykov, S., Wlodarski, W., Li, Y.: Nanocrystalline V 2 O 5–TiO 2 thin-films for oxygen sensing prepared by sol–gel process. Sens. Actuators B Chem. 77, 484–490 (2001)
Malavasi, L., Tealdi, C., Montenero, A., Tulliani, J.M., Moggi, P., Guglielmi, M., Flor, G., Lorenzi, A., Martucci, A., Montanaro, L.: Materials development for CO-detection with improved selectivity through catalytic activation. Sens. Actuators, B 118, 121–128 (2006)
Nagarajan, V., Chandiramouli, R.: NiO nanocone as a CO sensor DFT investigation. Struct. Chem. 25, 1765–1771 (2014)
Beheshtian, J., Kamfiroozi, M., Bagheri, Z., Ahmadi, A.: Computational study of CO and NO adsorption on magnesium oxide nanotubes. Physica E 44, 546–549 (2011)
Iijima, S.: Helical microtubules of graphitic carbon. Nature 354, 56–58 (1991)
Balasubramanian, C., Bellucci, S., Castrucci, P., De Crescenzi, M., Bhoraskar, S.V.: Chem. Phys. Lett. 383, 188–191 (2004)
Stan, G., Ciobanu, C., Thayer, T., Wang, G., Creighton, J., Purushotham, K., Bendersky, L., Cook, R.: Elastic moduli of faceted aluminum nitride nanotubes measured by contact resonance atomic force microscopy. Nanotechnology 20, 35706–357014 (2009)
Zhang, D., Zhang, R.: Theoretical prediction on aluminum nitride nanotubes. Chem. Phys. Lett. 371, 426–432 (2003)
Tondare, V., Balasubramanian, C., Shende, S., Joag, D., Godbole, V., Bhoraskar, S., Bhadhade, M.: Field emission from open ended aluminum nitride nanotubes. Appl. Phys. Lett. 80, 4813–4815 (2002)
Yin, B., Bando, Y., Zhu, Y., Li, M., Tang, C., Golberg, D.: Single-crystalline AlN nanotubes with carbon-layer coatings on the outer and inner surfaces via a multiwalled-carbon-nanotube-template-induced route. Adv. Mater. 17, 213–217 (2005)
Stan, G., Ciobanu, C.V., Thayer, T.P., Wang, G.T., Creighton, J.R., Purushotham, K.P., Bendersky, L.A., Cook, R.F.: Elastic moduli of faceted aluminum nitride nanotubes measured by contact resonance atomic force microscopy. Nanotechnology 20, 35706–357014 (2009)
Fan, Y.: Formation of crystalline AlN nanotubes by a roll-up approach. Mater. Lett. 65, 1900–1902 (2011)
Strite, S., Morkoc, H.: GaN, AlN, and InN: a review. J. Vac. Sci. Technol., B 10, 1237–1266 (1992)
Jain, C., Willander, M., Narayan, J., Van Overstraeten, R.: III–nitrides: growth, characterization, and properties. J. Appl. Phys. 87, 965–1006 (2000)
Ruterana, P., Albrecht, M., Neugebauer, J.: Nitride Semiconductors: Handbook on Materials and Devices. Wiley, New York (2006)
Ahmadi, A., Beheshtian, J., Hadipour, N.L.: Interaction of NH3 with aluminum nitride nanotube: electrostatic vs. covalent. Phys. E 43, 1717–1719 (2011)
Ahmadi, A., Kamfiroozi, M., Beheshtian, J., Hadipour, N.L.: The effect of surface curvature of aluminum nitride nanotubes on the adsorption of NH3. Struct. Chem. 22, 1261–1265 (2011)
Jiao, Y., Du, A., Zhu, Z., Rudolph, V., Smith, S.C.: A density functional theory study of CO2 and N2 adsorption on aluminium nitride single walled nanotubes. J. Mater. Chem. 20, 10426–10430 (2010)
Beheshtian, J., Bagheri, Z., Kamfiroozi, M., Ahmadi, A.: A theoretical study of CO adsorption on aluminum nitride nanotubes. Struct. Chem. 23, 653–657 (2012)
Ahmadi, A., Hadipour, N.L., Kamfiroozi, M., Bagheri, Z.: Theoretical study of aluminum nitride nanotubes for chemical sensing of formaldehyde. Sens. Actuators B Chem. 161, 1025–1029 (2012)
Ahmadi, A., Omidvar, A., Hadipour, N.L., Bagheri, Z., Kamfiroozi, M.: Can aluminum nitride nanotubes detect the toxic NH 3 molecules? Physica E 44, 1357–1360 (2012)
Ahmadi-Peyghan, A., Baei, M.T., Hashemian, S., Torabi, P.: Adsorption of CO molecule on AlN nanotubes by parallel electric field. J. Mol. Model. 19, 859–870 (2013)
Rezaei-Sameti, M.: Gallium doped in armchair and zigzag models of boron phosphide nanotubes (BPNTs): a NMR study. Phys. B 407, 3717–3721 (2012)
Rezaei-Sameti, M.: The effect of doping three Al and N atoms on the chemical shielding tensor parameters of the boron phosphide nanotubes: a DFT study. Phys. B 407, 22–26 (2012)
Rezaei-Sameti, M.: The effects of SiC-doped on the NMR parameters of the armchair and zigzag models of aluminum phosphide nanotubes: a DFT study. Physica E 44, 1770–1775 (2012)
Rezaei-Sameti, M., Yaghoobi, S.: Theoretical study of adsorption of CO gas on pristine and AsGa-doped (4, 4) armchair models of BPNTs. Comput. Condens. Matter 3, 21–29 (2015)
O’Boyle, N.M., Tenderholt, A.L., Langner, K.M.: Cclib: a library for package‐independent computational chemistry algorithms. J. Comput. Chem. 29, 839–845 (2008)
Beheshtian, J., Baei, M.T., Ahmadi Peyghan, A., Bagheri, Z.: Electronic sensor for sulfide dioxide based on AlN nanotubes: a computational study. J. Mol. Model. 18, 4745–4750 (2012)
Rastegar, S.F., Hadipour, N.L., Soleymanabadi, H.: Theoretical investigation on the selective detection of SO2 molecule by AlN nanosheets. J. Mol. Model. 20, 1–6 (2014)
de Almeida, J.E., de Brito, M.F., de Castilho, C., Kakanakova-Georgieva, A., Gueorguiev, G.K.: Defects in hexagonal-AlN sheets by first-principles calculations. Eur. Phys. J. B 85, 1–9 (2012)
Reed, A.E., Curtiss, L.A., Weinhold, F.: For a deeper discussion of this approach, see for example. Chem. Rev. 88, 899–926 (1988)
Schmidt, M.W., Baldridge, K.K., Boatz, J.A., Elbert, S.T., Gordon, M.S., Jensen, J.H., Koseki, S., Matsunaga, N., Nguyen, K.A., Su, S.J., Windus, T.L., Dupuis, M., Montgomery, J.A.: General atomic and molecular electronic structure system. J. Comput. Chem. 14, 1347–1363 (1993)
Ditchfield, R., Hehre, W.J., Pople, J.A.: GAUSSIAN 70 Program, QCPE No 236. J. Chem. Phys. 54, 724–728 (1971)
Parr, R.G., Pearson, R.G.: Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 105, 7512–7516 (1983)
Politzer, P., Murray, J.S., Bulat, F.A.: Average local ionization energy: a review. J. Mol. Model. 16, 1731–1742 (2010)
Mulliken, R.S.: A new electroaffinity scale; together with data on valence states and on valence ionization potentials and electron affinities. J. Chem. Phys. 2, 782–793 (1934)
Tabtimsai, C., Keawwangchai, S., Nunthaboot, N., Ruangpornvisuti, V., Wanno, B.: Density functional investigation of hydrogen gas adsorption on Fe-doped pristine and Stone–Wales defected single-walled carbon nanotubes. J. Mol. Model. 18, 3941–3949 (2012)
Acknowledgments
The authors thank the Centre of computational Nano of Malayer Universities for supporting this research.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Rezaei-Sameti, M., Samadi Jamil, E. The adsorption of CO molecule on pristine, As, B, BAs doped (4, 4) armchair AlNNTs: a computational study. J Nanostruct Chem 6, 197–205 (2016). https://doi.org/10.1007/s40097-015-0183-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40097-015-0183-9