
Abstract
Structural and dynamical properties of water confined in carbon nanotubes are investigated by classical molecular dynamics simulations. It is found that density and self-diffusion coefficients of water depend on the diameter of confinant nanotube and exhibit anomalous behavior in narrow tubes. It is shown that water forms several different orderings inside the nanotube channel and these arrangements impact on the energetics of hydrogen bonds. Considering the relationship between the geometry of hydrogen bonds and their energetics, a discussion on the dynamics of enclosed water is presented.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Water is a complex fluid, necessary for life and present in most everyday human activity [1]. Most of its importance may be attributed to its anomalous properties, not found in any other substance. When confined in nanopores or nanochannels its behavior become even more intriguing: it experiences a liquid–solid transition at room temperature [2] and presents large size-dependent viscosity [3] and thermal conductivity [4], among other properties. Besides, hundreds of studies have focused on the remarkable fast diffusion properties of water in nanochannels, with applications in fields as diverse as desalinisation, filtering, gas separation, ion transport and others [5].
The notable physical characteristics of confined water are naturally related to the dynamics of the hydrogen bonds (HB) formed between the water molecules in the nanochannels. The energetics of an HB is much influenced by the relative orientation between each pair of molecules, in a way that any modification in this character has a profound impact on the dynamics of the bond [6]. Recently, Pascal and collaborators [2] showed that the free energy variation of water when it fills the CNT cavity from a bulk reservoir is due to (1) entropical gain in sub-nanometer tubes or (2) enthalpic loss in larger CNTs.
The presence of these competing phenomena expresses how the arrangement of the confined water molecules is important to describe their dynamics. With this aspect in mind, the purpose of this paper is to describe and analyse the geometrical characteristics of the bonding between water molecules in CNT and link these features with the directional character of the hydrogen bonds.
Methodology
For the present study, a classical molecular dynamics approach has been used as implemented in GROMACS 4.5.3 package [7]. It was chosen (n,n) armchair CNT’s, were (the diameter and length are shown in Table 1). The potential describing intermolecular interactions was given by a sum of Lennard-Jones (LJ) and Coulombian contributions. Here, kcal/mol and Å are the LJ parameters for carbon–oxygen interaction, which are similar to the used by Farimani and Aluru [8]. Partial charges on O and H atoms are given respectively by e and e, which corresponds to SPC/E water model [9]. The geometry of water molecules was kept by using SETTLE algorithm [7]. Additionally, long range Coulombian interactions were treated by particle mesh Ewald approximation with a 10 Å real space cutoff and 1.2 Å grid [8]. In all simulations, CNT’s were electrically neutral and formed by rigid walls. This approach is in agreement with previous works, in particular with Won et al. [10]. They have shown that although partial charges are present in carbon atoms at the openings of carbon nanotubes, in the regions far from the ends (such as in an infinite nanotube) the value of electric charges on carbon atoms is nearly zero. Besides, rigid walls are found to be a good approximation, as the degrees of freedom of the walls does not change the overall properties of the fluid according to previous results [11, 12].
The equations of motion were solved by verlet algorithm, in which it was considered a timestep of 1 fs and data saved at each 100 fs. Temperature was set initially in 300 K and controlled by Nosé-Hoover thermostat, within the NVT ensemble (contant number of particlesN, volumeV and temperatureT). In every configuration full periodic boundary conditions (PBC) were considered. The unit cell vectors were chosen in order to guarantee the correct periodic repetition in axial direction (z axis) and to be large in the other directions (x- and y-directions) to avoid inter-tube interaction, such that only a single and infinite nanotube was considered (see “second stage calculation” described bellow).
The study was carried out in two separate stages: first, each CNT was connected to previously thermalised water reservoirs, as shown in Fig. 1. The water was then released and fills the CNT cavity during 10 ns. After that period, the reservoirs and the graphene slabs were removed and new PBC were set, such that the CNT’s could be represented as infinitely long. The second stage was then comprised by a new thermalisation (1 ns) followed by 10 ns of data accumulation of these infinite CNT’s. All results described in this paper are referred only to second stage data.

Definition of simulation cell
Results and discussions
After the first stage is concluded, the CNT’s cavity becomes totally filled with water. The dynamics of filling has been studied in a previous work [13]. In Fig. 2 it is shown the dependence of the number density of water on the nanotube diameter, where it is considered for the calculation of density the inner volume of CNT,
with as CNT diameter and its length.
It can be seen that the water density is strongly dependent on the CNT diameter in narrower nanotubes, what can be attributed to the different arrangements observed for water molecules inside these nanotubes. In (6,6) CNT water forms a single chain of molecules and, as diameter increases, tubular structures are observed, which is in agreement with previous reports [14–19]. These differences on the structural ordering is a direct consequence of the changing in the effective volume of nanotube, in the sense that not all the inner volume is available to water due to the repulsion (scaling to ) between these molecules and CNT wall. This quantity may be expressed similarly as done in Eq. 1, but considering an effective diameter instead of geometrical diameter . Here, the effective diameter can be defined as
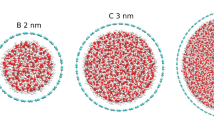
where is the CNT diameter and is the LJ parameter for C and O interaction. In (6,6) CNT, Å. Considering that the van der Waals diameter of a water molecule is about 2.75 Å, a single line of molecules is the only possible stable configuration. From (7,7) up to (12,12) CNT’s, otherwise, the increase in available room due to the larger also increases the number of molecules in the cross section of the nanotube, now arranged in poligonal rings. The (9,9) CNT exhibits a particularly high water density when compared to similar diameter CNT’s, suggesting a highly compact packing of water into this nanotube. For (16,16) CNT, Å. This is about 7 times larger than the van der Waals diameter of water, so a disordered disposition—just as observed in liquid water—is expected. This behaviour extends to (20,20) and must be similar to larger tubes.

Density of water into CNT relative to bulk water density. It is also shown the snapshots of final configuration of three CNT’s

RDF of water in (6,6), (12,12) and (20,20) CNT’s. Theinset is the (6,6) CNT RDF
The different ordering of water molecules into CNT’s can be better observed by the analysis of radial distribution functions (RDF). In Fig. 3 it is shown the RDF taken in xy-plane with respect to the CNT axis. It is shown, for clarity, three representative situations: (6,6), (12,12) and (20,20) CNT’s. For (6,6) CNT, the RDF shows a very sharp peak at the origin, decaying fast to zero as increases, what corroborates the single-chain picture of water molecules. For (12,12) CNT, otherwise, two peaks in RDF are observed at approximately 1.80 and 5.00 Å, which suggest a highly ordered arrangement of molecules. An visual inspection exhibits the formation of two concentrical tubular water layers. Finally, in (20,20), it is observed a typical liquid behaviour, although a peak in RDF appears at 10 Å and can be attributed to the layer formed at the surface of confined water (see snapshots in Fig. 2). Considering all the studied diameters, the results point out to a sequence single-file tubular bulk-like arrangement of inclosed water as the CNT diameter increases. The transition between the tubular to bulk-like phases occurs when the CNT diameter is approximately 13 Å ((10,10) CNT).
The different structures formed by water into CNT determines their dynamics. In Fig. 4 it is shown the axial self-diffusion coefficients () of water for the studied cases, defined as
It is observed that the coefficient increases monotonically for CNT with diameter larger than 13 Å, but an anomalous behaviour is observed in narrower tubes. For (7,7), (8,8) and (9,9) it is noted a sequence of highly and poorly diffusive phases. At this point it is clear that the transition from single-file to bulk-like is not smooth, where the geometric arrangement of water plays a decisive role in the formation of HB’s.

Self-diffusion coefficients of water into CNT’s

Potential energy per HB in CNT confined water
In order to understand this phenomenon it is presented in Fig. 5 the potential energy per HB of water molecules (), calculated as
where , and are, respectively, the Lennard-Jones energy, coulombian energy and the number of hydrogen bonds for the whole system, excluding carbon atoms. The hydrogen bonds are defined according to geometrical criterion, i. e., an HB is considered to exist if the O–HO distance is lower than 3.5 Å and the O–HO angle is lower than 30.
It is observed that in CNT’s with Å ((6,6) to (9,9)) the energy per HB is lower (more negative) than the corresponding for bulk water. An abrupt transition seems to occur in larger tubes, resulting in a maximum (less negative) for (12,12) and a subsequent decrease, with an asymptotic behaviour to the bulk water. These results suggest that water in conditions of extreme confinement forms very tight HB, which are favored by the highly ordered arrangement. As the CNT diameter increases, it is observed a decrease (and further stabilisation to the bulk value) in the energy per bond, exhibiting the liquid-like character of water in CNT with diameter larger than 20 Å.
A remarkably anomalous behaviour is observed in water confined in these narrow tubes. It can be noted that (7,7) and (9,9) present quite similar self-diffusion coefficients. Besides, their energies per HB are also very close, being the lowest observed among the simulated samples. It was previously reported that water crystallizes into these nanotubes [2], as observed in ice. As the interaction between water molecules into these CNT are stronger compared to the other simulated systems, it is noticeable that water forms a quite compact structure, what explains the high density observed in (9,9) CNT, as shown in Fig. 1. Into (7,7) CNT, otherwise, the low density is explained in geometrical terms: the small diameter limits the entrance of water molecules in order to fill the cavity. Further molecules cannot enter into the nanotube as the effective inner volume is fully occupied by the previous molecules. The water filled (8,8) CNT deviates from the previous conduct. The rise in density suggests a higher occupation of the inner cavity, but at the cost of stability: the energy per HB is higher than in (7,7) and (9,9), so that every molecule, although the weaker bonds to its neighbors, cannot move farther due to frequent collisions with other molecules. As a result, self-diffusion coefficient almost vanishes.
Conclusions
This molecular dynamics study showed that water has physical and dynamical properties largely modified when confined into small diameter carbon nanotubes. Density decreases when the CNT channel is narrow and a transition single-file tubular bulk-like arrangement of water molecules is observed as CNT diameter increases from 6 Å up to 20 Å. Many dynamical properties varies anomalously as the diameter of the confining CNT changes: the self-diffusion coefficient of water generally increases with CNT diameter, but this is not observed in (8,8) and (9,9) CNT. A similar behaviour is also observed in the energy per HB in (9,9). These features reveals the complexity of the character of the HB, where even small deviations in angle and bond distances lead to large energetic variations, and show how this is relevant when the system goes to nanometric scale.
References
Chaplin, M.F.: Water: its importance to life. Biochem. Mol. Biol. Educ. 29, 54–59 (2001)
Pascal, T.A., Goddard, W.A., Jung, Y.: Entropy and the driving force for the filling of carbon nanotubes with water. Proc. Natl. Acad. Sci. USA 108(29), 11794–11798 (2011)
Zhang, H., Ye, H., Zheng, Y., Zhang, Z.: Prediction of the viscosity of water confined in carbon nanotubes. Microfluid. Nanofluid. 10, 403–410 (2011)
Thomas, J.A., Lutzi, R.M., McGaughey, A.J.H.: Thermal conductivity and phonon transport in empty and water-filled carbon nanotubes. Phys. Rev. B 81, 045413 (2010)
Zhu, Y., Zhou, J., Lu, X., Guo, X., Lu, L.: Molecular simulations on nanoconfined water molecule behaviors for nanoporous material applications. Microfluid. Nanofluid. 15, 191–205 (2013)
Millen, D.J.: The directional character of the hydrogen bond in gas-phase dimers. J. Mol. Struct. 237, 1–18 (1990)
Hess, B., Kutzner, C., van der Spoel, D., Lindahl, E.: GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 4, 435 (2008)
Farimani, A.B., Aluru, N.R.: Spatial diffusion of water in carbon nanotubes: from fickian to ballistic motion. J. Chem. Phys. B 15, 12145–12149 (2011)
Berendsen, H.J.C., Grigera, J.R., Straatsma, T.P.: The missing term in effective pair potentials. J. Phys. Chem. 91, 6269–6271 (1987)
Won, C.Y., Joseph, S., Aluru, N.R.: Effect of quantum partial charges on the structure and dynamics of water in single-walled carbon nanotubes. J. Chem. Phys. 125, 11471–11479 (2006)
Kotsalis, E.M., Walther, J.H., Koumoutsakos, P.: Multiphase water flow inside carbon nanotubes. Int. J. Multiph. Flow 30, 995–1010 (2004)
Hanasaki, I., Nakatani, A.: Flow structure of water in carbon nanotubes: Poiseuille type or plug-like? J. Chem. Phys. 124, 144708–144716 (2006)
Kipper, A.C., da Silva, L.B.: Water filling of carbon nanotubes membranes: porosity and temperature effects. Chem. Phys. Lett. 552, 84–87 (2012)
Zheng, Y., Ye, H., Zhang, Z., Zhgang, H.: Water diffusion inside carbon nanotubes: mutual effects of surface and confinement. Phys. Chem. Chem. Phys. 14, 964–971 (2012)
Thomas, J.A., McGaughey, A.J.H.: Water flow in carbon nanotubes: transition to subcontinuum transport. Phys. Rev. Lett. 102, 184502–184505 (2009)
Striolo, A.: The mechanism of water diffusion in narrow carbon nanotubes. Nano Lett. 6, 633–639 (2006)
Wang, J., Zhu, Y., Zhou, J.: Lu XH: diameter and helicity effects on static properties of water molecules confined in carbon nanotubes. Phys. Chem. Chem. Phys. 6, 829–835 (2004)
Mashl, R.J., Joseph, S., Aluru, N.R., Jakobsson, E.: Anomalously immobilized water: a new water phase induced by confinement in nanotubes. Nano Lett. 3, 589 (2003)
Gordillo, M.C.: MartíJ: Hydrogen bond structure of liquid water confined in carbon nanotubes. Chem. Phys. Lett. 329, 341–345 (2000)
Acknowledgments
Author acknowledges Brazilian agencies CAPES, CNPq and FAPERGS for funding, CPAD/UFSM and CENAPAD/UNICAMP for computer time.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under license to BioMed Central Ltd. Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
da Silva, L.B. Structural and dynamical properties of water confined in carbon nanotubes. J Nanostruct Chem 4, 104 (2014). https://doi.org/10.1007/s40097-014-0104-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s40097-014-0104-3