
Abstract
Thermally activated persulfate (TAP) was applied for the degradation of Acid Blue 92 (AB92) dye in its aqueous solution. The effects of pH (3–11), temperature (298–333 K), contact time (15–75 min), sodium persulfate (SPS) concentration (0.05–0.5 mM) and initial AB92 concentration (50–400 mg/L) on the degradation of AB92 using TAP were examined. The initial and residual AB92 concentrations were determined spectrophotometrically at the wavelength of 260 nm and the dye mineralization was examined via the total organic carbon analysis. In addition, the chemical oxygen demand was also measured. The activation energy (Ea) of AB92 degradation was calculated as 17.38 kJ mol−1 based on the Arrhenius equation. Maximum degradation efficiency of 86.47% was reached after 75 min of treatment at a pH of 5, AB92 concentration of 200 mg/L, SPS concentration of 0.5 mM and temperature of 333 K. The degradation efficiency declined with the addition of different sodium chloride concentrations and organic radical scavengers. AB92 degradation was reduced from 86.5 to 74%, 65, and 59.1% using ethylenediaminetetraacetic acid, tert-butanol, and ethanol, respectively. A kinetic model was also developed to estimate the pseudo-first-order constants as a function of the main operational parameters (initial dye concentration and TAP concentration). Decolorization rate constants (k) of 0.0009, 0.001, 0.0012, 0.0014, and 0.0018 min−1 were obtained at 303, 308, 313, 328, and 333 K, respectively, using the Langmuir–Hinshelwood kinetic model. The results obtained indicate that the TAP degradation process has great potential for the reduction of azo dyes in aqueous environments.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The textile industry is considered as a prominent dye production sector [1]. The utilization of various types of colors in addition to chemical substances in dyeing processes generates wastewater with unique characteristics such as pH, color, and composition [2]. The disposal of colored wastewater into the aquatic ecosystem significantly hinders the penetration of light into the deep waters [3, 4]. It may also disturb the process of photosynthesis; this can also lead to the obliteration of aquatic plants [4]. In addition, colored dye effluents are significantly hazardous to the environment even at lower concentrations [5]. Moreover, the majority of dyes employed by textile industries are of organic origin; they are produced from phthalocyanine, diazo and anthraquinone salts which contain benzene rings that are highly carcinogenic and toxic in nature [6, 7]. An example of such dye is the C.I. Acid Blue 92(AB92), which is utilized on a regular basis by textile industries. Many researchers have proven that dyes are not completely removed during biological treatment, and they enter into water resources via wastewater effluents originating from treatment plants [8]. These compounds are not eliminated effectively through traditional wastewater removal procedures since they are non-biodegradable.
Several techniques have been employed for the elimination of dyes from polluted waters including coagulation–flocculation [9, 10], chemical treatment [11], oxidation [12, 13], adsorption [14,15,16,17,18,19,20,21,22] and photocatalytic degradation [23,24,25,26,27,28,29,30,31,32,33,34,35]. Adsorption is the most widely used because of its simplicity, low cost and adsorption recovery properties in removing contaminants [36]. Adsorption is usually done with an adsorbent such as activated carbon to eliminate dyes, but this process only transfers pollutants from one phase to another [37].
Several advanced techniques in recent studies have been reported for wastewater treatment containing azo dyes [38,39,40]. Among various implemented methods for dye removal, the advanced oxidation process (AOP) is applied favorably to destroy resistant contaminants. This process fundamentally creates hydroxyl radicals (·OH) and sulfate radicals (SO ·−4 ) [41]. The sulfate radical contains significant oxidation potential (E0 = 2.6 V) in addition to being selective in reactions with organic compounds via electron transfer [42], whereas ·OH is non-selective and reacts with various compounds [43,44,45]. Activated persulfate is applied extensively for environmental remediation because the produced radicals react easily with the organic compounds by complete or partial mineralization [46]. The radical is typically created from potassium peroxymonosulfate (KHSO5, PMS) or sodium persulfate (Na2S2O8, SPS) using thermal, transition metal and UV activation methods [47,48,49]. In addition, they can be applied alone to eliminate pollutants of high concentrations, since they usually possess a strong adsorption capacity [43, 50]. Even though S2O82− is a formidable oxidant, it reacts efficiently with pollutants at slow rates that are deemed impractical. This is due to the fact that S2O82− may be activated into hydroxyl (·OH) and sulfate (SO ·−4 ) radicals which are extremely powerful oxidants that react with pollutants within close diffusion-limited rates [51, 52]. The production of a highly reactive sulfate radical is depicted in Eqs. 1–6 [53, 54]:
SO ·−4 is comparably more stable to oxidize organic contaminants, thus providing the possibility for greater dispersion distance and improved mineralization in water [55, 56]. SO ·−4 has been proven to possess great potential for methylparaben degradation via UV-activated persulfate compared to other activators [57]. Cai et al. [58] proved that the bimetallic Fe–Co/GAC catalyst may be utilized to heterogeneously activate SPS oxidation for Acid Orange 7 degradation, which has also been proven in other studies. Also, very toxic persistent organic pollutants (POPs) can be decontaminated with persulfates [59].
The thermally activated persulfate (TAP) process is widely applied in ISCO procedures to treat hazardous and organic contaminants in water. Thermal activation may basically reduce the reaction time and cause higher drops in SPS usage in comparison to other methods [60]. When temperature increases, S2O82− is disintegrated into sulfate ions (SO42−) as seen in the reaction below [61]:
It is clear that SPS reaction with heat (Eq. 7) causes the generation of SO ·−4 [62].
Thus, in this research, the impact of heat activation on SPS for the elimination of AB92 from its aqueous solution was examined. The impact of different operating parameters such as pH, contact time, sodium persulfate concentration, and initial AB92 concentration on the degradation process was also examined. No prior studies on AB92 dye removal using the TAP process have been observed in the literature. The effect of different radical scavengers including ethylenediaminetetraacetic acid (EDTA), tert-butanol (TBA), and ethanol (EtOH) on the removal of AB92 was examined. The impact of sodium chloride concentrations was also considered.
Materials and methods
Chemicals
AB92 (Acid Blue 92) dye was purchased from Alvan Sabet Corporation, Hamadan, Iran, and used as the pollutant. All reagents [sodium persulfate (SPS) (Na2S2O8, 99%, molecular weight: 238.09 g mol−1), sodium hydroxide (NaOH, 98%), and sulfuric acid (H2SO4, 99.99%)] were of analytical grade and purchased from Merck (Germany). The specifications of AB92 are shown in Table 1. All solutions were prepared using de-ionized water. The pH of the solution was adjusted using HCl or NaOH 0.1 N solutions.
Experimental procedure and analysis
The effects of different parameters such as pH (3, 5, 7, 9, and 11), contact time (10, 20, 40, 60, and 80 min), dye concentration (50, 75, 100, 150, and 200 mg/L), SPS concentration (0.05, 0.07, 0.09, 0.3, and 0.5 mM), and temperature (313, 323, 333, 343 and 353 K) on AB92 degradation were studied.
Initially, the initial AB92 dye concentration, SPS concentration, pH, and temperature were set to be constant at 200 mg/L, 0.07 mM, 3, and 333 K, respectively. The effect of each parameter on the removal percentage of AB92 dye was evaluated. AB92 dye and SPS stock solutions were prepared using double-distilled water. The reactor was filled with 250 mL of the prepared AB92 solution. The initial and residual AB92 dye concentrations were determined using a UV–visible spectrophotometer (Shimadzu Model: CE-1021-UK). The COD concentration was determined using a spectrophotometer (COD VAQIO). The total organic carbon (TOC) was determined through the TOC analyzer (ANATOC Series II). Before adding SPS to the solution, the solution temperature was set using a shaker incubator (Froilabo EC180). To ensure the complete mixing of the mixture during the experiment, the shaker speed was set at 150 rpm.
The results were reported as the average of three replications of each experiment. The apparent activation energy, Ea (J mol−1), required to decompose AB92 at the studied temperatures was calculated using the Arrhenius equation (Eq. 8) [46, 63, 64]:
where A is the frequency factor, R is the ideal gas constant (8.314 J mol−1 K−1), k is the rate constant and T is the absolute temperature (K).
The rate of AB92 decolorization was analyzed using the first-order and Langmuir–Hinshelwood kinetic models. The first-order equation is given as [65]:
where C0([AB92]0) and C ([AB92]) are the concentrations at the initial time, t = 0 and contact time t, respectively, and k is the first-order rate constant [27, 30, 31].
The Langmuir–Hinshelwood kinetic model is given as [64, 66]:
where C0 and Ct are the concentrations of the dye at the initial time (t = 0) and contact time; the rate constant (k) is obtained from the slope of the straight-line plot of Ln(C0/Ct) versus t.
Results and discussion
Effect of pH
Solution pH is one of the most important parameters that influence SO ·4 -based oxidation of organic pollutants in wastewater or natural water. The effect of solution pH (3–11) on the degradation of AB92 was studied at initial dye concentration ([AB92]0) of 100 mg/L, initial persulfate concentration ([SPS]0) of 0.5 mM and temperature of 333 K. Highest removal of AB92 was observed at pH of 5 (Fig. 1). COD percentage removals of 55, 65.5, 54.2, 53, and 59.7% were obtained at pH of 3, 5, 7, 9, and 11, respectively, at a reaction time of 15 min. Maximum COD removal was observed at a pH of 5. According to the reactions below (Eqs. 11–13), in an acidic environment, SPS was decomposed to bisulfate (\({\text{HSO}}_{4}^{ - }\)) and tetra sulfate oxide (\({\text{SO}}_{ 4}\)). Also,\({\text{SO}}_{ 4}\) decomposed to \({\text{O}}_{ 2}\) and \({\text{H}}_{ 2} {\text{SO}}_{ 5}\) in a weak and strong acidic environment, respectively.

Effect of pH on AB92 and COD degradation by TAP. ([AB92]0:100 mg/L, temperature: 333 K, [SPS] 0: 0.5 Mm)
Therefore, it was predicted that dyes at pH of 3 and 4 were less decomposed than at other conditions because of catalytic–acidic decomposition of persulfate. The more neutral and alkaline the pH of the environment, the more was the reaction of the sulfate radical with OH−/H2O resulting in the production of hydroxyl radicals (Eqs. 14 and 15).
Although the hydroxyl radicals showed high oxidation potential (E0 = 2.8 V), they were attracted by non-target species or converted to hydrogen peroxide (E0 = 1.8 V) because of their non-selective reaction [43, 45]. Sulfate radical showed selective reactions too. The difference in reaction mechanisms could differentiate the dye decomposition rate in alkaline and acidic conditions because electron transfer is the main mechanism of sulfate radicals’ reaction [67]. Ji et al. [68] showed that the maximum rate of trichloroethylene (atrazine) removal using persulfate occurred at a pH of 5.
Effect of initial dye concentration
The effect of AB92 concentration (50, 100, 200, 300, and 400 mg/L) on the removal efficiency of AB92 was investigated at the reaction time of 15 min and optimum pH of 5. Figure 2 shows that the efficiency of AB92 decomposition was increased with increasing dye concentration. Therefore, AB92 radical reaction with hydroxyl radicals at low AB92 concentration would be increased, which will result in an increase of AB92 decomposition by the free radical [69]. Previous studies also observed that an increase in the concentration of organic compounds accelerated the rate of S2O82− loss, presumably due to radical chain reactions that propagated S2O82− decomposition [70, 71].

The effect of initial AB92 concentration on AB92 and COD degradation using TAP (Temperature: 333 K, [SPS]0: 0.5 mM, pH 5)
The effect of SPS concentration
Sodium persulfate concentration is regarded as a vital and influential factor for thermally activated SPS oxidation due to its direct impact on the sulfate radical equilibrium. Thus, the impact of SPS concentration on dye removal was investigated. As depicted in Fig. 3, AB92 was eliminated to a high extent by increasing the SPS concentration from 0.05 to 0.5 mM. The sulfate radical showed increased reduction/oxidation properties compared to the persulfate anion and was able to disintegrate a high amount of organic compounds. Radicals’ performance was commensurate to the persulfate concentration at a constant temperature. Based on Eqs. 16 and 17, higher concentrations of SPS caused extra sulfate radicals’ diffusion, thus acting as a scavenger that was effective in decomposing the pollutants [72]. These sulfate radicals may react with each other to form persulfate ion over again [73].

The effect of initial SPS concentration on AB92 and COD degradation using TAP ([AB92]0: 200 mg/L, temperature: 333 K, pH 3)
Furthermore, as depicted in Fig. 3, the average percentage of COD elimination was obtained as 12.21, 33.05, 33.26, 39.5, and 58.2% at initial SPS concentration of 0.05, 0.07, 0.09, 0.3, and 0.5 mM, respectively. The highest COD removal rate was achieved at 0.5 mM. The increased AB92 and COD removal efficiency may be attributed to higher SO ·−4 production with increasing SPS concentrations. The created sulfate radicals disintegrated the AB92 molecules via hydrogen abstraction effecting the double bonds and electron transfer. In addition, based on Eq. 17, the SPS may react directly with AB92 to generate sulfate radicals [74, 75], which entails the gradual decomposition of AB92. Based on Eqs. 18–28, the generated free radicals can cause a series of chain reactions in radical production and attacks on AB92 molecules resulting in AB92 degradation [76, 77].
Yang et al. [48] proved that when the initial persulfate concentration exceeded a particular point, the azo dye, Acid Orange 7 decomposition rate is marginally slowed down.
Effect of temperature
Temperature plays an important role in the decomposition of pollutants using activated persulfate. The effect of temperature on the degradation of AB92 was studied at temperatures of 303, 308, 313, 328, and 333 K at constant [AB92]0 of 200 mg/L, [SPS]0 of 0.5 mM and pH 5. As seen in Fig. 4, AB92 and COD removal efficiency was higher at the temperature of 333 K. The decomposition of persulfate to sulfate radicals could be increased by increasing the temperature (Fig. 4). The efficiency of dye removal increased linearly with increasing temperature. The bond fission of O–O in SPS caused by increased temperature resulted in an increased rate of AB92 decomposition [78, 79]. The high temperature provided enough energy to separate the O–O bond in the SPS leading to the production of the active species such as ·OH and \({\text{SO}}_{4}^{ \cdot - }\) [80], which led to the pollutants’ removal. Although the high temperature was useful in the decomposition of non-organic compounds using active SPS, the attraction and reduction reactions of SPS would proceed simultaneously. Therefore, the optimal temperature has to be determined [67]. A study by Potakis et al. [81] on the bisphenol oxidation using active persulfate showed that increasing temperature from 313 to 343 K accelerated the removal of bisphenyl at the same rate as that seen in the current study.

The effect of temperature on AB92 and COD degradation by TAP. ([AB92]0: 200 mg/L, [SPS]0: 0.5 mM, pH 5)
The reaction time is another parameter of importance in SPS oxidation with heat [82]. After some time, the production of sulfate radicals increased parallel to the rate of pollutants’ removal.
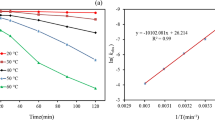
The activation energy (Ea = − Slope × R) was calculated as 17.38 kJ mol−1 by plotting Ln k against 1/T (Fig. 5). In addition, the change in enthalpy \(\left( {\Delta H^{0} } \right)\) and change in entropy \((\Delta S^{0} )\) were calculated as 2.091 kJ mol−1 and − 1.011 kJ mol−1 K−1, respectively, using the Eyring equation [83, 84]:
where kB is the Boltzmann’s constant (1.381 × 10−23 J/K), h is the Planck’s constant (6.626 × 10−34 Js), R is the universal gas constant (8.314 J mol− 1 K−1), and T is the absolute temperature (K). ΔH‡ is the enthalpy of activation (the same as \(\Delta H^{0} )\) and ΔS‡ is the entropy of activation (the same as \(\Delta {\text{S}}^{0} )\).

Arrhenius plot for AB92 degradation
Equation 30 shows the relationship between temperature (T) and free energy (\(\Delta G^{0} )\)
Ghauch and Tuqan [63] proved that bisoprolol decomposition via heated persulfate/H2O requires an activation energy of 119.8 kJ mol−1.
Degradation kinetics
The plot of Ln(C0/Ct) versus contact time, t, at various temperatures is shown in Fig. 6. The rate constant, k, estimated from the slopes of the plots were 0.0009, 0.001, 0.0012, 0.0014, and 0.0018 min−1 at temperatures of 303, 308, 313, 328, and 333 K, respectively. As seen in the study, an increase in the temperature of persulfate activation could lead to an increase in the degradation rate constant. The correlation coefficients, R2, show that the data follow the Langmuir–Hinshelwood equation at all temperatures except 308 K.

The degradation kinetics of AB92 using heat-activated persulfate
TOC removal
To ascertain the TAP process potential for AB92 mineralization, TOC removal was assessed at the initial AB92 concentration of 200 mg/L, SPS concentration of 0.5 mM, pH of 5, and temperature of 333 K. As presented in Fig. 7, AB92 mineralization levels were obtained as 55.38, 65.5, 67, 77.44, and 89.2% at the reaction times of 15, 30, 45, 60, and 75 min, respectively. Even though significant AB92 removal efficiency was acquired through the TAP process, AB92 mineralization may not be complete and may decompose into various products. The results obtained from this study proved that AB92 and its by-products were aptly mineralized via the TAP process. Hence, at a reaction time of 75 min, 89.2% of the TOC was decomposed.

TOC removal during the TAP process. ([AB92]0 = 200 mg/L, temperature = 333 K, pH 5, [SPS]0 = 0.5 mM)
Effect of chloride ion
The impact of chloride on the degradation of AB92 using the TAP process was studied in the range of 50–500 mg/L at different reaction times (Fig. 8). From Fig. 8, it can be seen that the degradation of AB92 was better without the addition of chloride ion ([NaCl]0 = 0 mg/L). The removal efficiency was decreased with the addition of sodium chloride and decreased further as the concentration and reaction time were increased. Chloride (Cl−) is pervasive in environmental systems and often hinders the degradation of contaminants in some cases while expediting degradation in others [70, 85]. Chloride is vital since it is reactive with SO ·−4 , with KSO ·−4 = 2.6 × 108 M−1 s−1 [86] and is a typical co-contaminant for chlorinated pollutants. In relation to chloride, SO ·−4 , and Cl− reaction creates Cl· that subsequently intervenes in a series of reactions (Eqs. 31–41) which create either ClO3− or ·OH [86,87,88,89]:

Effect of NaCl concentration on AB92 degradation. ([AB92]0 = 200 mg/L, pH 5, [SPS]0 = 0.5 mM, pH 5)
The SO ·−4 and Cl− reaction at neutral pH creates ·OH. Thus, Cl− is not regarded as a competing solute, since the result of this reaction is another profound oxidant that can cause a reaction with the contaminants. The Cl− is regarded as a radical scavenger, that is, a computing solute at pH < 5 since the reaction result was not a secondary radical applicable to oxidizing the contaminants. Furthermore, at this pH, the reaction creates ClO3− which is possibly a toxic compound.
Effect of inorganic and organic radical scavengers
The effect of various organic radical scavengers (EDTA, tert-butanol, and ethanol) on AB92 removal with time was examined at a scavenger concentration of 10 mg/L (Fig. 9). As shown in Fig. 9, the degradation efficiency was reduced from 86.5% to 74, 65, and 59.1%, respectively, using ethylenediaminetetraacetic acid (EDTA), tert-butanol (TBA), and ethanol (EtOH). The impact of the organic scavengers (TBA, and EtOH) on AB92 degradation efficiency was lower compared to the inorganic scavenger (EDTA), which implies that EtOH may efficiently hinder the effects of the oxidizing species. Based on Fig. 9, this research showed that the assessed organic species may significantly suppress the studied procedure due to their scavenging impact on reactive radicals’ reaction [85, 90]. Based on Eqs. 41 and 44, EtOH reaction rate with OH· [k = (1.2–2.8) × 109 M−1 s−1] is 50 times greater compared to that of SO ·−4 [k = (1.6–7.7) × 107 M−1 s−1]. Furthermore, TBA is regarded as an influential scavenger for OH· [k = (3.8–7.6) × 108 M−1 s−1]. Based on Eqs. 42 and 45, it can quickly react with OH· [60, 68]

Effect of organic and inorganic scavengers on AB92 degradation. ([AB92]0 = 200 mg/L, [SPS]0 = 0.5 mM, pH 5, temperature = 353 K and [scavenger] = 10 mg/L)
In addition, EDTA is an organic molecule that can react with ·OH and SO ·−4 . Therefore, the addition of an excess amount of EDTA should be avoided to prevent the EDTA from competing with the contaminant for the reactive radical species.
Comparison with other AOPs for the degradation of AB92
As presented in Table 2, numerous researchers have assessed the removal of AB92 from aqueous environments using several modern oxidation procedures. Compared to the prior studies, the TAP process is a beneficial method to eliminate AB92. Other advantages of this method include favorable removal efficiency, cost-effectiveness, non-toxicity, and aqueous solubility.
Conclusion
The applicability of thermally activated sodium persulfate (SPS) for the degradation of Acid Blue 92 (AB92) from its aqueous solution was investigated. The effects of pH (3–11), temperature (298–333 K), contact time (15–75 min), SPS concentration (0.05–0.5 mM), and initial AB92 concentrations (50– 400 mg/L) on the degradation of AB92 were examined. The AB92 disintegration kinetics was studied. The degradation of AB92 by SPS was fitted into the Langmuir–Hinshelwood model equation. The effect of various organic radical scavengers (EDTA, tert-butanol, and ethanol) and chloride ion concentration on AB92 removal was examined. Optimum conditions of pH 5, SPS concentration of 0.5 mM, reaction time of 75 min, and temperature of 333 K and initial AB92 concentration of 200 mg/L were obtained for the AB92 removal using the TAP process, which gave a removal efficiency of 86.74%. AB19 degradation efficiency declined with the addition of radical scavengers and chloride ion. The TAP degradation process was employed efficiently for the treatment of azo dyes in aqueous environments.
References
Paul SA, Chavan SK, Khambe SD (2012) Studies on characterization of textile industrial waste water in Solapur city. Int J Chem Sci 10:635–642
Holkar CR, Jadhav AJ, Pinjari DV, Mahamuni NM, Pandit AB (2016) A critical review on textile wastewater treatments: possible approaches. J Environ Manag 182:351–366. https://doi.org/10.1016/j.jenvman.2016.07.090
Rietschel RL, Fowler JF, Fisher AA (2008) Fisher’s contact dermatitis: in textiles and shoes. BC Decker Inc., Hamilton, pp 339–401
Daneshvar N, Salari D, Khataee AR (2004) Photo catalytic degradation of azo dye acid red 14 in water on ZnO as an alternative catalyst to TiO2. J PhotochemPhotobiol A: Chem 162:317–322. https://doi.org/10.1016/S1010-6030(03)00378-2
Goshadrou A, Moheb A (2011) Continuous fixed bed adsorption of CI Acid Blue 92 by exfoliated graphite: an experimental and modeling study. Desalination 269:170–176. https://doi.org/10.1016/j.desal.2010.10.058
Cheng MY, Yu IC, Wang PK (2002) Degradation of azo dye Procion Red MX-5B by photo catalytic oxidation. Chemosphere 46:905–912. https://doi.org/10.1016/S0045-6535(01)00153-9
Hachem C, Bocquillon F, Zahraa O, Bouchy M (2001) Decolorisation of textile industry wastewater by the photo catalytic degradation process. Dyes Pigments 49:117–125. https://doi.org/10.1016/S0143-7208(01)00014-6
Bhatia D, Sharma NR, Singh J, Kanwar RS (2017) Biological methods for textile dye removal from wastewater: a review. Crit Rev Environ Sci Technol 47:1836–1876. https://doi.org/10.1080/10643389.2017.1393263
Somasundaran P, Runkana V (2005) Investigation of the flocculation of colloidal suspensions by controlling adsorbed layer microstructure and population balance modeling. Chem Eng Res Des 83:905–914. https://doi.org/10.1205/cherd.04345
Obiora-Okafo IA, Onukwuli OD (2018) Characterization and optimization of spectrophotometric colour removal from dye containing wastewater by coagulation–flocculation. Pol J Chem Technol 20:49–59. https://doi.org/10.2478/pjct-2018-0054
Gao BY, Yue QY, Wang Y, Zhou WZ (2007) Color removal from dye-containing wastewater by magnesium chloride. J Environ Manage 82:167–172. https://doi.org/10.1016/j.jenvman.2005.12.019
Ahmadi S, Mohammadi L, Igwegbe CA, Rahdar S, Banach AM (2018) Application of response surface methodology in the degradation of Reactive Blue 19 using H2O2/MgO nanoparticles advanced oxidation process. Int J Ind Chem 213:272. https://doi.org/10.1007/s40090-018-0153-4
Muruganandham M, Swaminathan M (2007) Solar driven decolourisation of Reactive Yellow 14 by advanced oxidation processes in heterogeneous and homogeneous media. Dyes Pigments 72:137–143. https://doi.org/10.1016/j.dyepig.2005.08.009
Kavitha D, Namasivayam C (2008) Capacity of activated carbon in the removal of acid brilliant blue: determination of equilibrium and kinetic model parameters. Chem Eng J 139:453–461. https://doi.org/10.1016/j.cej.2007.08.011
O’Mahony T, Guibal E, Tobin JM (2002) Reactive dye biosorption by Rhizopus arrhizus biomass. Enzyme Microb Technol 31:456–463. https://doi.org/10.1016/S0141-0229(02)00110-2
Igwegbe CA, Banach AM, Ahmadi S (2018) Adsorption of Reactive Blue 19 from aqueous environment on magnesium oxide nanoparticles: kinetic, isotherm and thermodynamic studies. Pharm Chem J 5:111–121
Igwegbe CA, Onukwuli OD, Nwabanne JT (2016) Adsorptive removal of vat yellow 4 on activated Mucuna pruriens (velvet bean) seed shells carbon. Asian J Chem Sci 1:1–16. https://doi.org/10.9734/AJOCS/2016/30210
Igwegbe CA, Onyechi PC, Onukwuli OD, Nwokedi IC (2016) Adsorptive treatment of textile wastewater using activated carbon produced from Mucuna pruriens seed shells. World J Eng Technol 4:21–37. https://doi.org/10.4236/wjet.2016.41003
Igwegbe CA, Onyechi PC, Onukwuli OD (2015) Kinetic, isotherm and thermodynamic modelling on the adsorptive removal of malachite green on Dacryodes edulis seeds. J Sci Eng Res 2:23–39
Mittal A, Mittal J, Malviya A, Gupta VK (2010) Removal and recovery of Chrysoidine Y from aqueous solutions by waste materials. J Colloid Interface Sci 344:497–507. https://doi.org/10.1016/j.jcis.2010.01.007
Asfaram A, Ghaedi M, Agarwal S, Tyagib I, Gupta VK (2015) Removal of basic dye Auramine-O by ZnS: cu nanoparticles loaded on activated carbon: optimization of parameters using response surface methodology with central composite design. RSC Adv 5:18438–18450
Mohammadi N, Khani H, Gupta VK, Amereh E, Agarwal S (2011) Adsorption process of methyl orange dye onto mesoporous carbon material–kinetic and thermodynamic studies. J Colloid Interface Sci 362:457–462. https://doi.org/10.1016/j.jcis.2011.06.067
Saleh A, Gupta VK (2012) Photo-catalyzed degradation of hazardous dye methyl orange by use of a composite catalyst consisting of multi-walled carbon nanotubes and titanium dioxide. J Colloid Interface Sci 371:101–106. https://doi.org/10.1016/j.jcis.2011.12.038
Saravanan R, Gupta VK, Mosquera E, Gracia F (2014) Preparation and characterization of V2O5/ZnO nanocomposite system for photocatalytic application. J Mol Liq 198:409–412. https://doi.org/10.1016/j.molliq.2014.07.030
Saravanan R, Gupta VK, Narayanan V, Stephen A (2013) Comparative study on photocatalytic activity of ZnO prepared by different methods. J Mol Liq 181:133–141. https://doi.org/10.1016/j.molliq.2013.02.023
Gupta VK, Jain R, Nayak A, Agarwa S, Shrivastava M (2011) Removal of the hazardous dye: tartrazine by photodegradation on titanium dioxide surface. Mater Sci Eng: C 31:1062–1067. https://doi.org/10.1016/j.msec.2011.03.006
Saravanan R, Sacari E, Gracia F, Khan MM, Mosquera E, Gupta VK (2016) Conducting PANI stimulated ZnO system for visible light photocatalytic degradation of coloured dyes. J Mole Liq 221:1029–1033. https://doi.org/10.1016/j.molliq.2016.06.074
Saravanan R, Khan MM, Gracia F, Qin J, Gupta VK, Arumainathan S (2016) Ce3+-ion-induced visible-light photocatalytic degradation and electrochemical activity of ZnO/CeO2 nanocomposite. Sci Rep 6:31641
Saravanan R, Karthikeyan S, Gupta VK, SekaranG NV, Stephen A (2013) Enhanced photocatalytic activity of ZnO/CuO nanocomposite for the degradation of textile dye on visible light illumination. Mater Sci Eng C33:91–98. https://doi.org/10.1016/j.msec.2012.08.011
Saravanan R, Gupta VK, Narayanan V, Stephen A (2014) Visible light degradation of textile effluent using novel catalyst ZnO/γ-Mn2O3. J Taiwan Inst Chem Eng 45:1910–1917. https://doi.org/10.1016/j.jtice.2013.12.021
Saravanan R, Khan MM, Gupta VK, Mosquera E, Gracia F, Narayanang V, Stephen A (2015) ZnO/Ag/CdO nanocomposite for visible light-induced photocatalytic degradation of industrial textile effluents. J Colloid Interface Sci 452:126–133. https://doi.org/10.1016/j.jcis.2015.04.035
Saleh TA, Gupta VK (2011) Functionalization of tungsten oxide into MWCNT and its application for sunlight-induced degradation of rhodamine B. J Colloid Interface Sci 362:337–344. https://doi.org/10.1016/j.jcis.2011.06.081
Saravanan R, Khan MM, Gupta VK, Mosquera E, Gracia F, Narayanan V, Stephen A (2015) ZnO/Ag/Mn2O3 nanocomposite for visible light-induced industrial textile effluent degradation, uric acid and ascorbic acid sensing and antimicrobial activity. RSC Adv 5:34645–34651. https://doi.org/10.1039/c5ra02557e
Saravanan R, Joicy S, Gupta VK, NarayananV Stephen A (2013) Visible light induced degradation of methylene blue using CeO2/V2O5 and CeO2/CuO catalysts. Mater Sci Eng: C 33:4725–4731. https://doi.org/10.1016/j.msec.2013.07.034
Saravanan R, Karthikeyan N, GuptaVK Thirumal E, Thangadurai P, Narayanan V, Stephen A (2013) ZnO/Ag nanocomposite: an efficient catalyst for degradation studies of textile effluents under visible light. Mater Sci Eng: C 33:2235–2244. https://doi.org/10.1016/j.msec.2013.01.046
Ahmadi S, Banach A, Mostafapour FK, Balarak D (2017) Study survey of cupric oxide nanoparticles in removal efficiency of ciprofloxacin antibiotic from aqueous solution: adsorption isotherm study. Desalination Water Treat 89:297–303. https://doi.org/10.5004/dwt.2017.21362
Ikehata K, Gamal El-Din M, Snyder SA (2008) Ozonation and advanced oxidation treatment of emerging organic pollutants in water and wastewater. Ozone Sci Eng 30:21–26. https://doi.org/10.1080/01919510701728970
Chakma S, Praneeth S, Moholkar VS (2017) Mechanistic investigations in sono-hybrid (ultrasound/Fe2+/UVC) techniques of persulfate activation for degradation of Azorubine. Ultrason Sonochem 38:652–663. https://doi.org/10.1016/j.ultsonch.2016.08.015
Chakma S, Das L, Moholkar VS (2015) Dye decolorization with hybrid advanced oxidation processes comprising sonolysis/Fenton-like/photo-ferrioxalate systems: a mechanistic investigation. Sep Purif Technol 156:596–607. https://doi.org/10.1016/j.seppur.2015.10.055
Chakma S, Moholkar VS (2016) Mechanistic analysis of hybrid sono-photo-ferrioxalate system for decolorization of azo dye. J Taiwan Inst Chem Eng 60:469–478. https://doi.org/10.1016/j.jtice.2015.11.009
Rahdar S, Igwegbe CA, Rahdar A, Ahmadi S (2018) Efficiency of sono-nano-catalytic process of magnesium oxide nano particle in removal of penicillin G from aqueous solution. Desalination Water Treat 106:330–335. https://doi.org/10.5004/dwt.2018.22102
Fang G-D, Dionysiou DD, Zhou D-M, Wang Y, Zhu X-D, Fan J-X, Cang L, Wang Y-J (2013) Transformation of polychlorinated biphenyls by persulfate at ambient temperature. Chemosphere 90:1573–1580. https://doi.org/10.1016/j.chemosphere.2012.07.047
Guerra-Rodríguez S, Rodríguez E, Singh DN, Rodríguez-Chueca J (2018) Assessment of sulfate radical-based advanced oxidation processes for water and wastewater treatment: a review. Water 10(12):1828. https://doi.org/10.3390/w10121828
Zhao Q, Mao Q, Zhou Y, Wei J, Liu X, Yang J, Luo L, Zhang J, Chen HCh et al (2017) Metal-free carbon materials-catalyzed sulfate radical-based advanced oxidation processes: a review on heterogeneous catalysts and applications. Chemosphere 189:224–238. https://doi.org/10.1016/j.chemosphere.2017.09.042
Liang C, Bruell CJ (2008) Thermally activated persulfate oxidation of trichloroethylene: experimental investigation of reaction orders. Ind Eng Chem Res 47:2912–2918. https://doi.org/10.1021/ie070820l
Wang C-W, Liang C (2014) Oxidative degradation of TMAH solution with UV persulfate activation. Chem Eng J 254:472–478. https://doi.org/10.1016/j.cej.2014.05.116
Liu X, Zhang T, Zhou Y, Fang L, Shao Y (2013) Degradation of atenolol by UV/peroxymonosulfate: kinetics, effect of operational parameters and mechanism. Chemosphere 93:2717–2724. https://doi.org/10.1016/j.chemosphere.2013.08.090
Yang S, Yang X, Shao X, Niu R, Wang L (2011) Activated carbon catalyzed persulfate oxidation of Azo dye acid orange 7 at ambient temperature. J Hazard Mater 186:659–666. https://doi.org/10.1016/j.jhazmat.2010.11.057
Wacławeka S, Lutze HV, Grübele K, Padila VVT, Černíka M, Dionysiouf DD (2017) Chemistry of persulfates in water and wastewater treatment: a review. Chem Eng J 330:44–62. https://doi.org/10.1016/j.cej.2017.07.132
Duan X, Sun H, Tade M, Wang S (2018) Metal-free activation of persulfate by cubic mesoporous carbons for catalytic oxidation via radical and nonradical processes. Catal Today 307:140–146. https://doi.org/10.1016/j.cattod.2017.04.038
Bard AJ, Parsons R, Jordan J (1985) Standard potentials in aqueous solution. CRC Press, Boca Raton
Buxton GV, Greenstock CL, Helman WP, Ross AB (1988) Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O−) in aqueous solution. J Phys Chem Refer Data 17(2):513–886. https://doi.org/10.1063/1.555805
Fang G, Juan G, Dionysiou DD, Liu C, Zhou D (2013) Activation of persulfate by quinones: free radical reactions and implication for the degradation of PCBs. Environ Sci Technol 47(9):4605–4611. https://doi.org/10.1021/es400262n
He X, de la Cruz AA, Dionysiou DD (2013) Destruction of cyanobacterial toxin cylindrospermopsin by hydroxyl radicals and sulfate radicals using UV-254 nm activation of hydrogen peroxide, persulfate and peroxymonosulfate. J Photochem Photobiol A: Chem 251:160–166. https://doi.org/10.1016/j.jphotochem.2012.09.017
Jiang X, Wu Y, Wang P, Li H, Dong W (2013) Degradation of bisphenol A in aqueous solution by persulfate activated with ferrous ion. Environ Sci Pollut Res 20:4947–4953. https://doi.org/10.1007/s11356-013-1468-5
Wilmarth WK, Haim A (1962) Mechanisms of oxidation by peroxydisulfate ion. In: Edwards JO (ed) Peroxide reaction mechanisms. Interscience, New York, pp 175–225
Dhaka S, Kumar R, Khan MA, Paeng KJ, Kurade MB, Kim SJ, Jeon BH (2017) Aqueous phase degradation of methyl paraben using UV-activated persulfate method. Chem Eng J 321:11–19. https://doi.org/10.1016/j.cej.2017.03.085
Cai C, Wang L, Gao H, Hou L, Zhang H (2014) Ultrasound enhanced heterogeneous activation of peroxydisulfate by bimetallic Fe–Co/GAC catalyst for the degradation of Acid Orange 7 in water. J Environ Sci 26:1267–1273. https://doi.org/10.1016/S1001-0742(13)60598-7
Hrabák P, Homolková M, Wacławek S, Černík M (2016) Chemical degradation of PCDD/F in contaminated sediment. Ecol Chem Eng S 23:473–482. https://doi.org/10.1515/eces-2016-0034
Tan C, Gao N, Deng Y, Rong W, Zhou S, Lu N (2013) Degradation of antipyrine by heat activated persulfate. Sep Purif Technol 109:122–128. https://doi.org/10.1016/j.seppur.2013.03.003
Mora VC, Rosso JA, Mártire DO, Gonzalez MC (2011) Phenol depletion by thermally activated peroxydisulfate at 70°C. Chemosphere 84(9):1270–1275. https://doi.org/10.1016/j.chemosphere.2011.04.062
Liang C, Su H (2009) Identification of sulfate and hydroxyl radicals in thermally activated persulfate. Ind Eng Chem Res 48(11):5558–5562. https://doi.org/10.1021/ie9002848
Ghauch A, Tuqan AM (2012) Oxidation of bisoprolol in heated persulfate/H2O systems: kinetics and products. Chem Eng J 183:162–171. https://doi.org/10.1016/j.cej.2011.12.048
Ghauch A, Tuqan AM, Kibbi N, Geryes S (2012) Methylene blue discoloration of heated persulfate in aqueous solution. Chem Eng J 213:259–271. https://doi.org/10.1016/j.cej.2012.09.122
Taghavi M, Tabatabaee M, Ehrampoush MH, Ghaneian MT, Afsharnia M, Alami A, Mardaneh J (2018) Synthesis, characterization and photocatalytic activity of TiO2/ZnO-supported phosphomolybdic acid nanocomposites. J Mol Liq 249:546–553. https://doi.org/10.1016/j.molliq.2017.11.031
Banach AB, Mollazehi A, Ahmadi S (2018) Survey on the removal of ciprofloxacin from aqueous solutions by nano-sono-catalytic process. Desalination Water Treat 136:207–211. https://doi.org/10.5004/dwt.2018.23108
Tan C, Gao N, Deng Y, Li L, Deng J, Zhou S (2015) Kinetic oxidation of antipyrine in heat-activated persulfate. Desalination Water Treat 53:263–271. https://doi.org/10.1080/19443994.2013.848414
Ji Y, Dong C, Kong D, Lu J, Zhou Q (2015) Heat-activated persulfate oxidation of atrazine: implications for remediation of groundwater contaminated by herbicides. Chem Eng J 263:45–54. https://doi.org/10.1016/j.cej.2014.10.097
Hoseini M, Safari GH, Kamani H, Jaafari J, Mahvi AH (2015) Survey on removal of tetracycline antibiotic from aqueous solutions by nano-sonochemical process and evaluation of the influencing parameters. Iran J Health Environ 8:141–151
Liang C, Chen C (2009) pH dependence of persulfate activation by EDTA/Fe(III) for degradation of trichloroethylene. J Contam Hydrol 106:173–182. https://doi.org/10.1016/j.jconhyd.2009.02.008
Liu H, Bruton TA, Li W, Buren JV, Prasse C, Doyle FM, Sedlak DL (2016) Oxidation of benzene by persulfate in the presence of Fe(III)- and Mn(IV)-containing oxides: stoichiometric efficiency and transformation products. Environ Sci Technol 50(2):890–898. https://doi.org/10.1021/acs.est.5b04815
Pokhalekar P, Chakraborty M (2015) Degradation of bisphenol A and 4-tert-octylphenol: a comparison between ultrasonic and photocatalytic technique. Desalination Water Treat 1:1–8. https://doi.org/10.1080/19443994.2015.1041053
Soubh A, Mokhtarani N (2016) Post treatment of composting leachate with combination of ozone and persulfate oxidation process. RSC Adv 6:76113. https://doi.org/10.1039/C6RA09539A
Dogliotti L, Hayon E (1967) Flash photolysis of per[oxydi]sulfate ions in aqueous solutions. The sulfate and ozonide radical anions. J Phys Chem 71(8):2511–2516. https://doi.org/10.1021/j100867a019
Matzek LW, Carter KE (2016) Activated persulfate for organic chemical degradation: a review. Chemosphere 151:178–188. https://doi.org/10.1016/j.chemosphere.2016.02.055
Huang K-C, Couttenye RA, Hoag GE (2002) Kinetics of heat-assisted persulfate oxidation of methyl tert-butyl ether (MTBE). Chemosphere 49:413–420. https://doi.org/10.1016/S0045-6535(02)00330-2
Waldemer RH, Tratnyek PG, Johnson RL, Nurmi JT (2007) Oxidation of chlorinated ethenes by heat-activated persulfate: kinetics and products. Environ Sci Technol 41(3):1010–1015. https://doi.org/10.1021/es062237m
Rahmani AR, Rezaeivahidian H, Almasi M, Shabanlo A, Almasi H (2016) A comparative study on the removal of phenol from aqueous solutions by electro–Fenton, and electro–persulfate processes using iron electrodes. Res Chem Intermed 42:1441–1450. https://doi.org/10.1007/s11164-015-2095-1
Luo Q (2014) Oxidative treatment of aqueous monochlorobenzene with thermally-activated persulfate. Front Environ Sci Eng 8:188–194. https://doi.org/10.1007/s11783-013-0544-x
Gao Y-q, Gao N-y, Deng Y, Yin D-q, Zhang Y-s, Rong W-l et al (2015) Heat-activated persulfate oxidation of sulfamethoxazole in water. Desalination Water Treat 56:2225–2233. https://doi.org/10.1080/19443994.2014.960471
Potakis N, Frontistis Z, Antonopoulou M, Konstantinou I, Mantzavinos D (2017) Oxidation of bisphenol A in water by heat-activated persulfate. J Environ Manage 195:125–132. https://doi.org/10.1016/j.jenvman.2016.05.045
Fan Y, Ji Y, Kong D, Lu J, Zhou Q (2015) Kinetic and mechanistic investigations of the degradation of sulfamethazine in heat-activated persulfate oxidation process. J Hazard Mater 300:39–47. https://doi.org/10.1016/j.jhazmat.2015.06.058
Sanjeev R, Padmavathi DA, Jagannadham V (2018) The ‘yard stick’ to interpret the entropy of activation in chemical kinetics: a physical–organic chemistry exercise. World J Chem Ed 6:78–81. https://doi.org/10.12691/wjce-6-1-12
Glasstone S, Laidler KJ, Eyring H (1941) The theory of rate processes. McGraw-Hill, New York
Bennedsen LR, Muff J, Søgaard EG (2012) Influence of chloride and carbonates on the reactivity of activated persulfate. Chemosphere 86:1092–1097. https://doi.org/10.1016/j.chemosphere.2011.12.011
Padmaja S, Neta P, Huie RE (1993) Rate constants for some reactions of inorganic radicals with inorganic ions temperature and solvent dependence. Int J Chem Kinet 25(6):445–455. https://doi.org/10.1002/kin.550250604
Jayson GG, Parsons BJ, Swallow AJ (1973) Some simple, highly reactive, inorganic chlorine derivatives in aqueous solution. J Chem Soc Faraday Trans Phys Chem Condens Phases 69:1597. https://doi.org/10.1039/f19736901597
Kläning UK, Wolff T (1985) Laser flash photolysis of HCIO, CIO−, HBrO, and BrO− in aqueous solution. Reactions of Cl− and Br− Atoms. Berichte Der BunsengesellschaftFür Physikalische Chem 89(3):243–245
Buxton GV, Subhani MS (1972) Radiation chemistry and photochemistry of oxychlorine ions. Part 2.—Photodecomposition of aqueous solutions of hypochlorite ions. J Chem Soc Faraday Trans 1 Phys Chem Condens Phases 68:958. https://doi.org/10.1039/F19726800958
Wang C, Zhang X, Yuan B, Wang Y, Sun P, Wang D, Wei Y, Liu Y (2014) Multi-heterojunction photocatalysts based on WO3 nanorods: structural design and optimization for enhanced photocatalytic activity under visible light. Chem Eng J 237:29–37. https://doi.org/10.1016/j.cej.2013.10.003
Khataee A, Sheydaei M, Hassani A, Taseidifar M, Karaca S (2015) Sonocatalytic removal of an organic dye using TiO2/montmorillonite nanocomposite. Ultrason Sonochem 22:404–411. https://doi.org/10.1016/j.ultsonch.2014.07.002
Parsa JB, Negahdar SH (2012) Treatment of wastewater containing Acid Blue 92 dye by advancedozone-based oxidation methods. Sep Purif Technol 98:315–320. https://doi.org/10.1016/j.seppur.2012.06.041
Rezaei S, Tahmasbi H, Mogharabi M, Ameri A, Forootanfar H, Khoshayand MR, Faramarzi MA (2015) Laccase-catalyzed decolorization and detoxification of Acid Blue 92: statistical optimization, microtoxicity, kinetics, and energetic. J Environ Health Sci Eng 13:31. https://doi.org/10.1186/s40201-015-0183-1
Acknowledgements
The authors are grateful to the Zabol University of Medical Sciences for their financial support in this study (Project No. 1396.329).
Author information
Authors and Affiliations
Contributions
SA and CAI Conceived and designed the experiments; Analyzed and interpreted the data; Wrote the paper. Contributed reagents, materials, analysis tools or data. SR Performed the experiments; Analyzed and interpreted the data.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Ahmadi, S., Igwegbe, C.A. & Rahdar, S. The application of thermally activated persulfate for degradation of Acid Blue 92 in aqueous solution. Int J Ind Chem 10, 249–260 (2019). https://doi.org/10.1007/s40090-019-0188-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40090-019-0188-1