
Abstract
COVID-19 is still widespread worldwide and up to now there is no established antiviral able to control the disease. Main protease is responsible for the viral replication and transcription; thus, its inhibition is a promising route to control virus proliferation. The present study aims to examine detail interactions between main protease and recently reported ninety-seven inhibitors with available X-ray crystallography to define factors enhance inhibition activity; thirty-two of most potent inhibitors were examined to identify sites and types of interaction. The study showed formation of covalent bond, H-bond and hydrophobic interaction with key residues in the active side. Covalent bond is observed in seventeen complexes, all of them by attack of the 145Cys thiol group on Michael acceptor, aldehyde or its hydrate, α-ketoamide, double bond or acetamide methyl group; the latter type requires H-bonding between acetamide carbonyl oxygen and at least one of 143Gly, 144Ser or 145Cys. Potent inhibitors, disulfiram and ebselen docked in the same binding site. Accordingly, factors identify inhibition include forming covalent bond and existing terminal hydrophobic groups and amidic or peptidomimetic structure. Binding affinity was found correlated with topological diameter up to 24 bond, molecular size, branching, polar surface area up to 199 Å2 and hydrophilicity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
As the critical situation worldwide increases because of the Coronavirus (COVID-19) and the absence of approved or even promising drug till now, there is urgent demand to find antiviral agent able to control the fast virus spread. The newly emerged Coronavirus (COVID-19) is formerly identified as nCoV-19 then SARS-CoV-2 [1, 2]. It belongs to a large virus family called Coronaviridae which contains also the previous severe acute respiratory syndrome Coronavirus (SARS-CoV) and Middle East Respiratory Syndrome (MERS-CoV) virus [3]. Coronaviruses are enveloped viruses with a single positively polar RNA strand (~30 kb) which is large genome relative to other RNA viruses [4]. Coronavirus (COVID-19) disease was first emerged in Wuhan city, China in December 2019 then spread rapidly over Worldwide. On February 27, 2020, in China, a total confirmed cases of Coronavirus disease were 2835 and the death cases were 81 [5]. Since then, the disease spread out rapidly to cover almost the entire world and the situation even get worse where according to the latest WHO Report on February 9, 2021 [6], there were 88,000 new deaths reported last week while the total confirmed cases and total death Worldwide reached 105.4 and 2.3 million cases, respectively.
Coronavirus (COVID-19) 3C-like protease (Mpro also called 3CLpro) is the main virus protein considered critical for the viral replication and transcription; therefore, targeting it controls the virus multiplication and proliferation which makes that enzyme attractive candidate as a drug target [7]. The first X-ray crystal structure of COVID-19 main protease complexed with inhibitor was with N3, released in Protein Data bank on 5-2-2020 (6LU7 2.16 Å) then at resolution 1.7 Å (7BQY) on 26-3-2020 [8]. Since then, a number of studies focused on testing many natural products and FDA-approved drugs for predicting the possible binding modes with COVID-19 main protease by docking and homology techniques [9,10,11,12,13]. However, by May 2020, the number of X-ray crystal structures of COVID-19 main protease bound to various inhibitors was exploded where 97 crystal structures each with different inhibitor were collected; some of them were even experimentally proved to be potent inhibitors and have strong antiviral activity. Therefore, in alternative route, the present work examines the already synthesized inhibitors and experimentally bound to COVID-19 main protease for their binding efficiency according to their binding free energy (ΔG), binding affinity constant (pKd) and inhibitor efficiency (IE). The binding site and interaction as shown by their existing X-ray crystallography as well as searching for the factors that increase these interactions and enhance the binding efficiency were undertaking in details. In addition, the effects of inhibitor hydrophobicity and topological properties on binding efficiency have been examined. All the examined 97 enzyme–inhibitor complexes are recently included in the PDB and most of them have not published yet. Therefore, understanding their binding sites and interactions as well as factors reinforce these interactions facilitates and helps finding out promising drugs among the enormous candidates of natural products and repurpose drugs; some of these compounds were also previously proved to be efficient inhibitors experimentally [8]. Computational chemistry and QSAR study can be strong aid to identify compounds’ reactivity [14]. In addition, compounds’ binding site and binding modes were compared with those of known inhibitors, disulfiram and ebselen.
Material and Methods
Preparation of Protease–Inhibitor Complexes
X-ray crystal structures of 97 inhibitors complexed with main protease of severe acute respiratory syndrome Coronavirus 2 (COVID-19) released in Protein Data Bank up to 20-5-2020 were obtained (www.rcsb.org/). In each complex, water and solvents, e.g., dimethyl sulfoxide were removed using PyMol visualization software (version 4.2.0). Hydrogen atoms were added to complexes for correct ionization and tautomeric states of amino acid residues. Each protein structure was saved (PDB) with and without the ligand.
Preparation and Computations of Ligands
Ligand structures were downloaded from Protein Data Bank then converted to sdf files by PyMol software. Energy was minimized by MM2 calculation and hydrophobic parameter (logP) and topological properties were computed using Chem 3D implemented in ChemOffice 2018. Computed topological parameters are Molecular Topological Index, Cluster Index, Topological Diameter (TD), Radius, Polar Surface Area (PSA), Shape Attribute, Sum of Degrees, Sum of Valence Degrees, Wiener Index, Balaban Index, Total Connectivity and Total Valence Connectivity.
Protein–Ligand Interactions
Protein–ligand interactions were visualized and inspected using protein–ligand interaction profiler (PLIP) [15] server and Discovery Studio-19 software. The examined interactions by PLIP were H-bonding, hydrophobic interaction, covalent bond salt bridge, aromatic ring center, charge center and π-stacking (parallel and perpendicular). The protein–inhibitor binding was assessed by calculating binding affinity constant (pKd), binding free energy (ΔG) and ligand efficiency (LE) using deep convolutional neural networks (DCNNs) in Kdeep predictor server [16]. The inhibitor efficiency was calculated from the formula IE = (ΔG)/N where N is number of non-hydrogen atoms of the ligand.
Docking
Docking of disulfiram and ebselen into COVID-19 main protease was performed using COVID-19 docking server (https://ncov.schanglab.org.cn/). Inhibitor files were loaded in mol2 format as recommended by the site.
Statistical Analysis
Stepwise multiple regression analysis was performed using SPSS software version 25. The validity of models was evaluated by the correlation coefficient (R), standard error of the estimate (SE), the number of data point (N), the least significant difference (p) and the 95% confidence intervals (in parentheses) for each regression coefficient.
Results and Discussion
Main Protease–Inhibitor Interactions
The binding free energy, binding affinity constant and ligand efficiency of 97 complexes released in PDB by 20-5-2020 were computed and arranged in descending order according to their binding strength (ΔG and pKd); complexes with the highest 32 values (≤−5.39 kcal/mole and ≥ 4.00, respectively) are found in Tables 1 and S1 while a complete list are tabulated in Table S2. Types of detected enzyme–inhibitor interactions as presented in Tables 1 and S1 are H-bonding, hydrophobic interaction, covalent bond, salt bridge (electrostatic) and π-stacking interactions; interaction distance as calculated by PLIP.
H-bonding and hydrophobic interactions are found in most complexes. Most compounds showed H-bonding between their amide oxygen or nitrogen and active side residues, e.g., 145Cys, 143Gly, 144Ser, 163His, 25Thr, 26Thr, 41His, 142Asn and 166Glu. Salt bridge attraction is observed either between inhibitor electron-rich group, e.g., hydrate oxygen atom (entry 5), carboxylate (entry 27) and SO2 (entry 30) and basic amino acid, e.g., 41His and 90Lys or between inhibitor nitrogen and acidic amino acids, e.g., 166Glu, 240Glu and 295Asp.
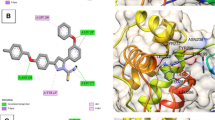
Covalent bond exerts stronger interaction and may lead to irreversible inhibition as found with N3 inhibitor in 7BQY complex [8]. There are fourteen other compounds showed covalent bond (6LZE, 6Y2F, 6YZ6, 5RGO, 5RGM, 5RG2, 5RG3, 5RFQ, 5REM, 5REJ, 5RG0, 5RFY, 5RFO and 5RFV. In all of them, the 145 cysteine sulfur attacks an inhibitor carbon atom to form C–S bond with bond length 1.412 (6YZ6)–1.823 (5RGM) Å indicating the impotence of this amino acid in the binding process of many protease inhibitors by forming either hydrogen or covalent bond and thus affects the 3C-like protease activity since 145Cys has a crucial role in the enzyme catalytic activity and virus replication [9, 13]. On the other hand, the type of reaction and site of attack of forming covalent bond vary. Tables 1 and S1 show that there are five types of reactions responsible for forming the C–S covalent bond. First, Michael addition reaction as in 7BQY (entry 1) complex where the cysteine thiol attacks the β-carbon of carbonyl to form irreversible C–S bond (1.77 Å). The enone moiety is mounted by H-bonds between the carbonyl oxygen and both 143Gly and 145Cys; in addition, the benzyl-O and amine-NH on both sides of enone are H-bonded with 143Gly and 164His, respectively. Figure 1a shows the binding site and the proximity of these amino acids to their bonded ligand atoms; the formed covalent bond between 145Cys and β-carbon to the carbonyl group, which shows in the enolized form, is also presented.

Binding site and key amino acid residue (black) of COVID-19 main protease. Protein is shown in cartoon presentation while inhibitors and 145Cys residue are presented as sticks; 143Gly, 132Asn, 41His and 164His are shown in line model. Carbon, hydrogen, oxygen, nitrogen and sulfur atoms are in grey, white, red, blue and yellow colors, respectively; H-bonds are presented in green lines. Formed covalent bond between 145Cys sulfur and a enone β-carbon (7BQY), b aldehyde carbon (6LZE) and c aldehyde hydrate carbon (6YZ6) are presented
Other listed H-bonds fix the terminal pyrrole and Oxazol rings to decrease the conformational interconversion. These binding efficiency explains the experimentally observed rapid and irreversible enzyme inhibition (kobs/[I] 11,300 M−1 s−1) and strong antiviral activity with EC50 4.67 μM of N3 inhibitor [8].
Second, aldehyde group (CHO, e.g., 6LZE, entry 2) or its hydrate form (CH(OH)2, e.g., 6YZ6, entry 5) are usually present in equilibrium upon water addition in aqueous media; the equilibrium point depends on the media and compound's structure. In 6LZE, nucleophilic addition of the cysteine thiol on the carbonyl group takes place, the resulted hydroxyl group is stabilized by H-bonding of hydroxyl oxygen with NH of 145Cys and 143Gly (Table 1 and Fig. 1b). In 6YZ6, covalent bond is also formed between the 145Cys sulfur and the hydrate carbon; likewise, a hydroxyl oxygen is H-bonded to NH of 145Cys and 143Gly while the hydroxyl OH is H-bonded to 142Asn-C=O residue; besides, the inhibitor-N* accepts a hydrogen in bonding with 41His-imidazol-NH while donates a hydrogen (NH*) to 164His-C=O as listed in Table 1 (entry 5) and illustrated in Fig. 1c). Both inhibitors showed high binding affinity toward COVID-19 main protease with ΔG is −9.37 and 7.18 kcal/mole and pKd is 6.94 and 5.31, respectively. 6LZE showed also experimentally potent enzyme inhibition with IC50 0.053 μM as crystalized and described previously while 6YZ6 crystallization is also recently reported.
Third, the 145Cys thiol group attacks α-ketoamide group as in 6Y2F complex to result a thiohemiketal (Table 1 entry 3). Peptidomimetic α-ketoamides showed broad-spectrum inhibition against main proteases of Coronaviruses and viral replication [17]. The formed hydroxyl group is stabilized by H-bonding of 41His-imidazol-NH with oxygen of the formed hydroxyl group while the carbonyl oxygen of amide group is attached to the NH of 143Gly, 144Ser and 145Cys as illustrated in Fig. 2a where all are in the enzyme active site. The rest of the molecule is also fixed by the other listed H-bonds (Table 1). Inhibitor O6K showed also high COVID-19 main protease inhibition with IC50 0.67 μM [18].

Formed covalent bond between 145Cys sulfur and a α-ketoamid carbon (6Y2F), b double bond secondary carbon (5RG2) and c acetyl methyl carbon (5RFO) are presented. COVID-19 main protease backbone is shown in cartoon presentation while inhibitors and 145Cys residue are presented as sticks; 41His, 143Gly, 144Ser and 26Thr residues are shown in line model. Carbon, hydrogen, oxygen, nitrogen and sulfur atoms are in grey, white, red, blue and yellow colors, respectively; H-bonds are presented in green lines
Fourth, reaction is the electrophilic addition on a carbon–carbon double bond with regioselectivity according to the common Markovnikov's rule as in complexes 5RG2 and 5RG3 crystalized by Fearon et al., (unpublished) forming a C–S bond with length 1.64 and 1.79 Å, respectively (Table S1 entries 17 and 26 respectively). Addition of thiols to unactivated olefins through electrophilic mechanism with Markovnikov's selectivity is known [19] as an opposite to free radical mechanism with anti-Markovnikov addition [20]. Figure 2b shows the formed covalent bond and the rehyperdization of terminal carbon to be sp3 (CH3) in addition to proximity of atoms participating in H-bonging, i.e., 143Gly-NH with Inhibitor CH2–N. It can be noticed in both inhibitors, the double bond is terminal and has a N–H group in β-position that forms hydrogen bonding with either 143Gly (5RG2) or 24Thr (5RG3) to fix the double bond for the addition reaction.
Acetamide, e.g., 5RGO, 5RGM, 5RFQ, 5REM, 5REJ, 5REU, 5RG0, 5RFY, 5RFO and 5RFV complexes (Fearon et al., unpublished) to form α-mercaptoacetamide (C–S 1.80–1.82 Å). The ten complexes show also H-bonding between the acetamide carbonyl oxygen and NH of one or more of 143Gly, 144Ser or 145Cys as presented in Fig. 2c for 5RFO (entry 9); the complex shows also H-bond between the acetamide-N and 41His-imidazol-NH which is also observed in other complexes (entries 11, 19, 23 and 25). This H-bonding seems crucial for effecting the reaction since there are other five complexes (6YZ6, 5RE7, 5R7Z, 5RG2 and 5RG3 have the acetamide moiety but did not undergo the reaction; the five compounds lack a H-bond with the acetamide carbonyl oxygen. In addition, in the first one (6YZ6), the 145Cys thiol prefers to attack the hydrate carbon while in latter two complexes (5RG2 and 5RG3), prefers addition to the double bond because of the stabilization provided by H-bonding discussed above.
Topological Study
To assess the topological factors affecting the COVID-19 main protease–inhibitor interactions; various topological parameters were computed for the collected 97 inhibitors; results are presented in supplementary material (Table S2). Binding affinity constant (pKd) of the most effective thirty-two inhibitors was correlated with each topological parameters and found significantly correlated (p 0.01) with each of Topological Diameter (R 0.804), Radius (R 0.780), Molecular Topological Index (R 0.792), Wiener Index (R 0.788), Cluster Index (R 0.786), Shape Attribute (R 0.786), Sum of Degrees (R 0.786), Sum of Valence Degrees (R 0.745), Balaban Index (R 0.721) and Polar Surface Area (R 0.626). Topological Diameter (TD) is the longest dimension of a molecule. Radius represents how far the farthest atom from the center of the molecular. The cluster index is the number of paths of a given length in the distance matrix. Balaban Index is the sum of topological distances from a given atom to any other atoms in a molecule. Molecular Topological Index, Shape Attribute and Wiener Index are measure of the branching and size of a molecule. Polar Surface Area is the sum of surface of all polar atoms including their attached hydrogen atoms. Sum of Degrees is the sum of the number of heavy atoms bonded to each atom in the molecule. Sum of Valence Degrees is the sum of valence degrees of every atom in the molecule where the valence degree of an atom is the sum of the bonds' orders of bonded atoms including hydrogen [21]. Having positive correlations indicates that the binding affinity can be enhanced by increasing inhibitor molecular size, diameter, branching, surface area of the molecule as well as bond orders and number of polar atoms within the molecule. Correlations between binding affinity (pKd) and each of molecular diameter and Molecular Topological Index can be represented by the following equations.
The binding affinity is also correlated (p 0.01) with the compound hydrophobicity as expressed by log P (R 0.609). Multiple regression analysis including all topological, hydrophobic and interaction parameters retained the topological diameter, number of H-bonds (HB) and logP as presented by the following correlation.
The present results showed that four of the most five potent inhibitors (Table 1) are N3, FHR, O6K and PRD in 7BQY, 6LZE, 6Y2F and 6YZ6 complexes, respectively. All of them have high functionality peptidomimetic structure, responsible for forming several H-bonds, with terminal hydrophobic groups, e.g., t-butyl, isopropyl, cyclopropylmethyl, benzyl or heterocyclic groups that increase the compound hydrophobicity and cause hydrophobic interactions with the amino acid residues; in addition, all of them form one of the early discussed covalent bonds as well as, among other inhibitors, have the highest values of all mentioned topological parameters. Other complexes in Tables 1 and S1 have recently been produced by Fearon et al., (unpublished results); most of them have central urea or amide group and on both sides two hydrophobic groups, e.g., branched alkyl, phenyl or heterocyclic groups. As illustrated in Tables 1 and S1, the central group or the heterocyclic heteroatoms participate in H-bonding while the side groups provide mainly hydrophobic interactions; meanwhile, the presence of acetyl group could afford a covalent bond as discussed above.
Interestingly, all inhibitors in Tables 1 and S1 showed the same binding pocket that falls in the enzyme active site. It is known that the active site in all Coronavirus main proteases is preserved [9, 10, 22,23,24,25]. The key amino acid residues in the active site that participate in H-bonding with most inhibitors are 145Cys, 143Gly, 144Ser, 163His, 164His, 25Thr, 26Thr, 41His, 142Asn and 166Glu; they are shown in Fig. 1 in proximity with N3 inhibitor. N3 inhibitor has the highest binding affinity and highest values of all correlated topological parameters; therefore, increasing these parameters still could enhance the binding affinity, e.g., increasing Topological Diameter (24 bond) and PSA (199 Å2).
Docking of Disulfiram and Ebselen
In the recent efforts to find effective anti-COVID-19 agents, drug screening in silicon and an enzyme inhibitor study reported disufiram and ebselen as potent antiviral promising drugs against COVID-19 [1]. Docking results showed that disulfiram forms a H-bond between the enolized amide carbonyl of 189Gln and disulfiram sulfur (3.36 Å) as presented in Fig. 3a. In addition, hydrophobic interaction was detected between disulfiram and each of 49Met, 141Leu, 165Met, 166Glu, 187Asp and 189Gln (Fig. 3b). Ebselen docking showed the formation of three H-bond between its carbonyl oxygen and each of 143Gly NH (2.16 Å), 144Ser NH (2.35 Å) and 145Cys NH (2.25 Å) as illustrated in Fig. 3c along with the hydrophobic interaction. These results indicate that all the examined inhibitors bind with COVID-19 main protease in the same binding pocket and the above-mentioned key amino acids play a crucial rule in all interactions.

a H-bond between the enolized amide carbonyl of 189Gln and disulfiram sulfur (3.36 Å); b, c are the binding pocket and hydrophobic interactions of the enzyme with disufiram and ebselen, respectively
Conclusion
The inhibition affinity (pKd and ΔG) of the recently X-ray-crystalized COVID-19 main protease with 97 inhibitors was evaluated. Enzyme–inhibitor interactions of the strongest thirty-two inhibitors showed that the key amino acid residues in the active side for binding were 145Cys, 143Gly, 144Ser, 163His, 164His, 25Thr, 26Thr, 41His, 142Asn and 166Glu. Interactions involves H-bonding, covalent bonding and hydrophobic interactions. Inhibitor structure requirements to achieve these interactions include the presence of terminal hydrophobic groups, e.g., t-butyl, cyclopropylmethyl, benzyl or heterocyclic groups and high functionality amidic or peptidomimetic structure. Covalent bond formation takes place on Michael acceptor, α-ketoamide, double bond or acetamide methyl group with H-bonding between the acetamide oxygen and at least one of 143Gly, 144Ser or 145Cys residue. In addition, increasing topological diameter up to 24 bond, molecular size, branching, polar surface area up to 199 Å2 and hydrophilicity enhances inhibitor reactivity.
References
Dong L, Hu S, Gao J (2020) Discovering drugs to treat coronavirus disease 2019 (COVID-19). Drug Discov Therap 14:58–60
Li G, De Clercq E (2020) Therapeutic options for the 2019 novel coronavirus (2019-nCoV). Nat Rev Drug Discov 19:149–150. https://doi.org/10.1038/d41573-020-00016-0
Kandeel M, Ibrahim A, Fayez M, Al-Nazawi M (2020) From SARS and MERS CoVs to SARS-CoV-2: moving toward more biased codon usage in viral structural and nonstructural genes. J Med Virol. https://doi.org/10.1002/jmv.25754
Chen C-N, Lin CPC, Huang K-K, Chen W-C, Hsieh H-P, Po-H L, Hsu JT-A (2005) Inhibition of SARS-CoV 3C-like protease activity by theaflavin-3,3′-digallate (TF3). Evid Based Compl Alternat Med 2:209–215. https://doi.org/10.1093/ecam/neh081
Wang M, Cao R, Zhang L, Yang X, Liu J, Xu M, Shi Z, Hu Z, Zhong W, Xiao G (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res 30:269–271. https://doi.org/10.1038/s41422-020-0282-0
COVID-19 Weekly Epidemiological Update 10:00 am CET 27 December 2020. https://www.who.int/emergencies/diseases/novel-coronavirus-2019/situation-reports
Jin Z, Du X, Xu Y et al (2020) Structure-based drug design, virtual screening and high-throughput screening rapidly identify antiviral leads targeting COVID-19. BioRxiv preprint. https://doi.org/10.1101/2020.02.26.964882
Jin Z, Du X, Xu Y et al (2020) Structure of Mpro from COVID-19 virus and discovery of its inhibitors. Nature. https://doi.org/10.1038/s41586-020-2223-y
Mirza MU, Froeyen M (2020) Structural elucidation of SARS-CoV-2 vital proteins: computational methods reveal potential drug candidates against main protease, Nsp12 polymerase and Nsp13 helicase. J Pharm Anal. https://doi.org/10.1016/j.jpha.2020.04.008
Mothay D, Ramesh KV (2020) Binding site analysis of potential protease inhibitors of COVID-19 using AutoDock. VirusDis. https://doi.org/10.1007/s13337-020-00585-z
Shaghaghi N (2020) Molecular docking study of novel COVID-19 protease with low risk terpenoides compounds of plants. ChemRxiv. https://doi.org/10.26434/chemrxiv.11935722.v1
Kanhed AM, Patel DV, Teli DM, Patel NR, Chhabria MT, Yadav MR (2020) Identification of potential Mpro inhibitors for the treatment of COVID-19 by using systematic virtual screening approach. Mol Div. https://doi.org/10.1007/s11030-020-10130-1
ul Qamar MT, Alqahtani SM, Alamri M, Chen L-L. (2020) Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J Pharm Anal. Doi:https://doi.org/10.1016/j.jpha.2020.03.009
Ali HM, Ali IH (2017) A DFT and QSAR study of the role of hydroxyl group, charge and unpaired-electron distribution in anthocyanidin radical stabilization and antioxidant activity. Med Chem Res 26:2666–2674
Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder M (2015) PLIP: fully automated protein-ligand interaction profiler. Nucl Acids Res 43(W1): W443–W447. doi: https://doi.org/10.1093/nar/gkv315; https://projects.biotec.tu-dresden.de/plip-web/plip/index
Jiménez J, Skalic M, Martinez-Rosell G, de Fabritiis G (2018) Protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J Chem Inf Model 58: 287–296. doi: https://doi.org/10.1021/acs.jcim.7b00650. https://www.playmolecule.org/Kdeep/
Zhang L, Lin D, Kusov Y (2020) α-Ketoamides as broad-spectrum inhibitors of coronavirus and enterovirus replication: structure-based design, synthesis, and activity assessment. J Med Chem 63:4562–4578. https://doi.org/10.1021/acs.jmedchem.9b01828
Zhang L, Lin D, Sun X (2020) Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 368:409–412
Borisov YuA, Dyusengaliev AK, Dyusengaliev KI, Serikov TP (2008) The electrophilic addition of thiols to olefins: a theoretical and experimental study. Russ J Phys Chem A 82:2092–2096. https://doi.org/10.1134/S0036024408120212
Sinha AK, Equbal D (2019) Thiol-ene reaction: synthetic aspects and mechanistic studies of an anti-Markovnikov-selective hydrothiolation of olefins. Asian J Org Chem 8:32–47. https://doi.org/10.1002/ajoc.201800639
Chem3D 18.0 User Guide (2018) PerkinElmer Informatics, Inc
Dai W, Zhang B, Jiang X-M (2020) Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science. https://doi.org/10.1126/science.abb4489
Gunther S, Reinke PYA, Fernandez-Garcia Y (2021) X-ray screening identifies active site and allosteric inhibitors of SARS-CoV-2 main protease. Science 372:642–646. https://doi.org/10.1126/science.abf7945
Yang H, Xie W, Xue X (2005) Design of wide-spectrum inhibitors targeting coronavirus main proteases. PLoS Biol 3: e324. doi:https://doi.org/10.1371/journal.pbio.0030324
Kumar V, Roy K (2020) Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3C-like protease (3CLpro) enzyme inhibitors against SARSCoV diseases. SAR QSAR Environ Res 31:511–526. https://doi.org/10.1080/1062936X.2020.1776388
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Significance’ statement: The present work identifies the binding site, types of interactions, reactions forming a covalent bond and factors affecting binding efficiency of recently X-ray crystallized complexes (97 complexes) and potent inhibitors of COVID-19 main protease, disulfiram and ebselen. The work aims to better understanding of COVID-19 main protease-inhibitor interactions as well as factors reinforce these interactions to facilitate and help finding out promising drugs among the enormous candidates of natural products and repurpose drugs.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Ali, H.M., Soliman, A.G. & Elfiky, H.G.A.G. SAR and QSAR of COVID-19 Main Protease–Inhibitor Interactions of Recently X-ray Crystalized Complexes. Proc. Natl. Acad. Sci., India, Sect. B Biol. Sci. 92, 281–291 (2022). https://doi.org/10.1007/s40011-021-01338-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40011-021-01338-8