
Abstract
Purpose
Carbapenemase-producing Enterobacterales (CPE) pose a serious threat for healthcare facilities worldwide, yet the mode of transmission is often unclear. Recently, we recorded an increase of blaOXA-48-harboring isolates at our hospital associated with patients with previous medical treatment in the Ukraine. We used long-read whole genome sequencing (lrWGS) to characterize these isolates including their plasmids.
Methods
Samples were collected as part of clinical routine diagnostic or screening of multi-drug resistance bacteria (MDRB). Antimicrobial susceptibility testing was performed and all MDRB (n = 10) were sequenced by lrWGS for genotyping, identification of antimicrobial resistance (AMR) genes, and characterization of plasmids.
Results
While routine analysis of core genome multilocus sequence typing (cgMLST) did not show any genetic similarities between isolates, we found an unexpected high similarity in the plasmid diversity of different Enterobacterales in patients with previous medical treatment in the Ukraine. This included an IncL/M plasmid carrying blaOXA-48 and additional small non-AMR-coding plasmids.
Conclusion
Our results show that lrWGS can be used in the routine setting to uncover similarities in plasmids and may give further information about potential epidemiologic associations. In the future, analysis of both AMR and non-AMR plasmids may provide an additional layer of information for molecular surveillance of CPE.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction and purpose
Carbapenemase-producing Enterobacterales (CPE) are still rare in our 1,500-bed university hospital in the northwestern part of Germany. However, in 2022, we noticed an increase of blaOXA-48-harboring isolates in screening and clinical samples compared to previous years. To rule out intrahospital transmission of CPE, we reviewed the case history of the affected patients (PatA and PatB, Table 1) and realized that all blaOXA-48-harboring isolates in 2022 were associated with Ukrainian refugees admitted to our hospital for medical emergency treatment. In fact, other publications have described similar findings, suggesting a high prevalence of blaOXA-48-carrying Enterobacterales in the Ukrainian healthcare system at present [1, 2]. We performed long-read whole genome sequencing (lrWGS) of all blaOXA-48-harboring isolates and of all ESBL-producing Enterobacterales (n = 5) from these patients. As part of routine surveillance, we sequence all MDRB isolates using lrWGS at the time of first detection at our hospital; therefore, we were able to compare the isolates associated with the Ukraine and their plasmids with other samples with IncL/M plasmids from our hospital (n = 5). Moreover, we included previously well-characterized plasmids from the literature as comparison.
Methods
In addition to the detection of multi-drug resistance bacteria (MDRB) from clinical samples, anorectal swab samples were collected as part of routine surveillance for multi-drug-resistant Gram negative bacteria. Screening samples were cultivated on chromID® ESBL Agar (biomérieux, Marcy-l’Etoile, France), CHROMagar™ Acinetobacter (Mast Group, Paris, France), and Pseudomonas Cetrimide Agar (Thermo Fisher Scientific, Basingstoke, UK). Species identification was performed by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF/MS) (Bruker, Bremen, Germany). Antimicrobial susceptibility testing was performed using VITEK® 2 (biomérieux, Marcy-l’Etoile, France) and Etest® (biomérieux, Marcy-l’Etoile, France). Antibiotic susceptibility was interpreted according to the 2022 European Committee on Antimicrobial Susceptibility Testing (EUCAST, v.12.0) criteria.
Genomic DNA (gDNA) from bacterial isolates was extracted using the NEB Monarch Genomic Purification Kit (New England Biolabs, Ipswich, MA, USA). All isolates were sequenced on a PacBio® Sequel IIe system (Pacific Biosciences, Menlo Park, CA, USA). Next, raw sequences were assembled de novo and analyzed using the SMRT® Link software suite v.10 or v.11 with default parameters. Isolates were genotyped based on core genome multilocus sequence typing (cgMLST) targets implemented in Ridom SeqSphere+ software version 8.99 (Ridom GmbH, Münster, Germany) [3]. In addition, the MLST sequence types (ST) were identified using the schemes for the respective species. Antimicrobial resistance (AMR) genes and their location were determined using target gene sets for antimicrobial resistance based on the NCBI AMRFinderPlus [4]. To predict the plasmid replicon type, the respective contigs were checked for complete circularization and analyzed with MobSuite [5]. For annotation, we used the online pipeline dfast (including HMM scan against TIGRFAM and RPSBLAST against COG database from NCBI) [6]. We performed BLAST analyses using BRIG Software (BLAST Ring Image Generator) [7]. Same secondary cluster ID by MobSuite was the indicator for us to perform BLASTn analysis via BRIG Software.
Results
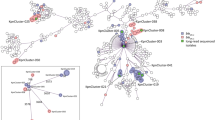
At first glance, cgMLST did not reveal any significant genetic relation among the five Ukrainian isolates. The closest genetic relationship observed was between two ST11 Klebsiella pneumoniae isolates with an allelic distance of 11 and no documented contact during hospitalization. Next, we investigated the plasmid content of the isolates in the same patient based on lrWGS data (shown in Table 1). Interestingly, we identified an identical blaOXA-48 plasmid in two isolates, a K. pneumoniae and a Morganella morganii. The plasmid is classified by MobSuite as an IncL/M plasmid with a length of 65,540 bp. Within this plasmid, a resistance cassette with a blaOXA-48 AMR gene was detected. As the plasmid is classified as conjugative and shows 100% overlap in the Blast analysis (Supplementary_Figure B, plasmids PatA_MM_p2 and PatB_KpnST11_p1) in isolates of two different species, we assume horizontal gene transfer has occurred at some point in the past. This is consistent with the already known high rate of conjugation in IncL/M-blaOXA-48 positive plasmids [8]. A deeper view into the case history of the patients showed that they had escaped the Ukraine because of the Russian invasion and both had previous treatment in the region of Charkiw in 2022. To estimate how prevalent this plasmid is in the bacterial population, we compared the respective plasmids with another five IncL/M plasmids, all of which were collected in our hospital from 2022 to April 2023 (see Supplementary_Figure A). No IncL/M plasmid with the same degree of similarity was identified. However, in a Blast comparison with the NCBI database, we found an identical IncL/M plasmid with the same AMR gene collected at the Charles University in Prague, Plzen, Czech Republic in 2016 (SAMN14227071) (see Supplementary_Figure A). As a next step, we compared all the non-blaOXA-48-containing plasmids of the five isolates associated with the Ukraine, using MobSuite based on a mash-distance analysis [9]. Interestingly, characterization of small cryptic plasmids (SCPs) (< 10 kb) [10] revealed additional links among the isolates. Here we detected four clusters with nine plasmids in the Ukrainian isolates. We found no SCP cluster in the isolates from our hospital. This observation also suggests ongoing plasmid exchange among bacteria in the same health region, as the co-conjugation of SCPs has already been shown in the case of AMR plasmid transmission [10]. So far the function of these small plasmids (Fig. 1) (3.5 kb, 3.6 kb, 4.5 kb and 5.2 kb) is unclear, as they only encode a few proteins. The 3.5 kb plasmid contains three hypothetical proteins and a Rop family plasmid primer RNA-binding protein; the 4.5 kb plasmid encodes for three hypothetical proteins, a Rop family plasmid primer RNA-binding protein, and a type II toxin-antitoxin system RelE/ParE family toxin. The 5.2 kb plasmid harbors four hypothetical proteins and a DNA cytosine methyltransferase and the 3.6 kb plasmid only for hypothetical proteins (Annotation and comparison is shown in Fig. 1, BLAST analysis in Supplementary_Figure B).

Genetic content of the four SCP clusters. Kpn = K. pneumoniae, MM = M. morganii, Ec = E. coli, ST = MLST sequence type. The full names of the compared plasmids are written in the center of the plasmid rings. Names and further information related to the plasmids and patients are listed in Table 1. Hypothetical proteins are colored in grey; identical colors indicate same coding proteins
Conclusion
As the characterization of AMR plasmids is not part of routine surveillance efforts in hospitals, it is still unclear how AMR-specific plasmids are disseminated in the health care setting. Our results suggest that the plasmid diversity of bacterial isolates may be part of the local epidemiology of MDRB. As lrWGS becomes more common in molecular diagnostics and surveillance, analyzing AMR plasmids in more detail in the hospital setting will uncover horizontal transmission events and might occasionally give us clues to the travel history of their hosts or connect isolates with each other. So far, little is known about the function of small non-AMR coding plasmids, yet small non-AMR-coding plasmids may give us more information about epidemiologic links.
Availability of data and materials
All samples are submitted to NCBI in BioProject PRJNA972998.
References
Sandfort M, Hans JB, Fischer MA, Reichert F, Cremanns M, Eisfeld J, et al. Increase in NDM-1 and NDM-1/OXA-48-producing Klebsiella pneumoniae in Germany associated with the war in Ukraine, 2022. Euro Surveill. 2022. https://doi.org/10.2807/1560-7917.ES.2022.27.50.2200926.
Zwittink RD, Wielders CC, Notermans DW, Verkaik NJ, Schoffelen AF, Witteveen S, et al. Multidrug-resistant organisms in patients from Ukraine in the Netherlands, March to August 2022. Euro Surveill. 2022. https://doi.org/10.2807/1560-7917.ES.2022.27.50.2200896.
Junemann S, Sedlazeck FJ, Prior K, Albersmeier A, John U, Kalinowski J, et al. Updating benchtop sequencing performance comparison. Nat Biotechnol. 2013;31:294–6. https://doi.org/10.1038/nbt.2522.
Feldgarden M, Brover V, Haft DH, Prasad AB, Slotta DJ, Tolstoy I, et al. Validating the AMRFinder tool and resistance gene database by using antimicrobial resistance genotype-phenotype correlations in a collection of isolates. Antimicrob Agents Chemother. 2019. https://doi.org/10.1128/AAC.00483-19.
Robertson J, Nash JHE. MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom. 2018. https://doi.org/10.1099/mgen.0.000206.
Tanizawa Y, Fujisawa T, Nakamura Y. DFAST: a flexible prokaryotic genome annotation pipeline for faster genome publication. Bioinformatics. 2018;34:1037–9. https://doi.org/10.1093/bioinformatics/btx713.
Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. https://doi.org/10.1186/1471-2164-12-402.
Potron A, Poirel L, Nordmann P. Derepressed transfer properties leading to the efficient spread of the plasmid encoding carbapenemase OXA-48. Antimicrob Agents Chemother. 2014;58:467–71. https://doi.org/10.1128/AAC.01344-13.
Ondov BD, Treangen TJ, Melsted P, Mallonee AB, Bergman NH, Koren S, et al. Mash: fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016;17:132. https://doi.org/10.1186/s13059-016-0997-x.
Barry KE, Wailan AM, Sheppard AE, Crook D, Vegesana K, Stoesser N, et al. Don’t overlook the little guy: an evaluation of the frequency of small plasmids co-conjugating with larger carbapenemase gene containing plasmids. Plasmid. 2019;103:1–8. https://doi.org/10.1016/j.plasmid.2019.03.005.
Funding
Open Access funding enabled and organized by Projekt DEAL. This study was supported by Interdisciplinary Centre for Clinical Research (IZKF), Faculty of Medicine, University of Muenster. Funding ID: SEED/019/23.
Author information
Authors and Affiliations
Contributions
V.A. and V.S. wrote the main manuscript text. A.M. made substantial contributions to the conception and design of the work. N.S. made substantial contributions to the analysis and interpretation of data. F.S. made substantial contributions to the aquisition of data. A.M., N.S. and F.S. revised the work critically for important intellectual contentand. V.A. and A.S. drafted the work and prepared the figures. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
None.
Ethical approval
Not applicable.
Supplementary Information
Below is the link to the electronic supplementary material.

15010_2023_2136_MOESM1_ESM.jpg
Supplementary file1 (JPG 257 KB) Supplementary_Figure: Comparisons using BLAST analysis and BRIG software for imaging of IncL/M plamids. A) The IncL/M blaOXA-48 positive plasmids associated with patients from the Ukraine are colored in red, all other IncL/M plasmids found in our hospital in 2022 until April 2023 are colored in blue. As comparison, a reference plasmid from the NCBI database (GenBank accession number KX523901.1) was added (yellow)

15010_2023_2136_MOESM2_ESM.png
Supplementary file2 (JPG 243 KB) Supplementary_Figure: Comparisons using BLAST analysis and BRIG software for imaging of IncL/M plamids. B) Blast comparisons of all detected plasmids. Color-coding was done according to Table 1: The IncL/M blaOXA-48 positive plasmids are colored red. An IncF/IncR plasmid is colored blue (PatA_KpnST11_p1 and PatB_KpnST11_p2). The four remaining plasmid groups colored in yellow, purple, orange and green belong to the SCP clusters (also see Figure 1)
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

{kind=link}
{kind=link}
Cite this article
van Almsick, V.F., Sobkowiak, A., Scherff, N. et al. Characterization of blaOXA-48-carrying plasmids and small non-AMR-coding plasmids collected from Ukrainian patients. Infection 52, 661–665 (2024). https://doi.org/10.1007/s15010-023-02136-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s15010-023-02136-2